

Abstract
Mutations in BRCA1 account for 45% of families with high incidence of breast cancer and for 80–90% of families with both breast and ovarian cancer. BRCA1 protein includes an amino-terminal zinc finger motif as well as an excess of negatively charged amino acids near the C terminus. In addition, BRCA1 contains two nuclear localization signals and localizes to the nucleus of normal cells. While these features suggest a role in transcriptional regulation, no function has been assigned to BRCA1. Here, we show that the C-terminal region, comprising exons 16–24 (aa 1560–1863) of BRCA1 fused to GAL4 DNA binding domain can activate transcription both in yeast and mammalian cells. Furthermore, we define the region comprising exons 21–24 (aa 1760–1863) as the minimal transactivation domain. Any one of four germ-line mutations in the C-terminal region found in patients with breast or ovarian cancer (Ala-1708 → Glu, Gln-1756 C+, Met-1775 → Arg, Tyr-1853 → Stop), had markedly impaired transcription activity. Together these data underscore the notion that one of the functions of BRCA1 may be the regulation of transcription.
Keywords: breast cancer, tumor suppressor gene
Breast cancer is one of the most common diseases affecting women in the United States. Genetic factors contribute to an estimated 5% of all breast cancer cases, and up to 36% of the cases diagnosed before age 30 (1). In 1990, human BRCA1 was mapped by genetic linkage to the long arm of chromosome 17 (2), firmly establishing the existence of a breast cancer susceptibility gene. Mutations in BRCA1 alone account for ≈45% of the families with high incidence of breast cancer and 80% of families with high incidence of both breast and ovarian cancer (3). Identification of human BRCA1 by positional cloning techniques revealed an ORF coding for 1863 aa (4). No homology with any known protein was identified with the exception of a zinc finger domain located in its N-terminal region. This zinc binding RING finger domain (C3HC4) is found in several proteins that have their functions mediated through DNA binding (5). Other features in the sequence were also recognized, including two putative nuclear localization signals (aa 500–508 and 609–615), a leucine zipper (aa 1209–1231), and an excess of negative charged residues in the C-terminal region of BRCA1. In many eukaryotic transcription activators, the presence of an acidic region in the C terminus correlates with the transactivation domain (6). With the exception of the leucine zipper, which is imperfectly conserved, the other features are highly conserved in human and mouse Brca1, suggesting that these regions might be significant for its functions (7, 8, 9). Although reports have been published showing that BRCA1 localizes to the cytoplasm and secretory granules (10, 11), other investigators have shown that BRCA1 localizes to the nucleus of normal cells (12, 13). Taken together these features suggest a function of BRCA1 in transcription activation.
We have investigated whether the C-terminal region of BRCA1 is able to activate transcription. We found that BRCA1 C terminal can act as a transactivation domain when fused to a heterologous DNA binding domain, and that germ-line mutations found in patients impair this activity, suggesting that loss of transcription activation function by BRCA1 may predispose the carriers to cancer.
MATERIALS AND METHODS
Yeast Strains.
Two Saccharomyces cerevisiae strains, HF7c [MATa, ura3-52, his3-200, lys2-801, ade2-101, trp1-901, leu2-3, 112, gal4-542, gal80-538, LYS::GAL1-HIS3, URA3::(GAL4 17-mers)3-CYC1-lacZ] (14) and SFY526 (MATa, ura3-52, his3-200, lys2-801, ade2-101, trp1-901, leu2-3, 112, canr, gal4-542, gal80-538, URA3::GAL1-lacZ) (15), were used (CLONTECH). HF7c has a HIS3 reporter gene under the control of the GAL1 upstream activating sequence (UAS), responsive to GAL4 transcriptional activation. The HF7c transformants were inoculated in liquid medium lacking either tryptophan or tryptophan and histidine. The vectors used for expression confer growth in the absence of tryptophan (see below). If the fusion proteins activate transcription, yeast transformants are able to grow in medium lacking histidine. HF7c also has a lacZ reporter gene under the control of GAL4 17-mers UAS, responsive to GAL4 transcriptional activation. The SFY526 strain has lacZ under the control of GAL1 UAS. If the fusion proteins activate transcription, yeast transformants will produce β-galactosidase.
Yeast Expression Constructs.
All BRCA1 fragments were amplified by PCR using the plasmid F3 (a kind gift from Robert Bogden, Myriad Genetics, Salt Lake City) as a template, which contains the C-terminal domain of human BRCA1 (4). The following oligonucleotide primers were used: exons 16–24 (S9503101 and Z9503098, both containing an EcoRI site 5′ and a BamHI site 3′), exons 16–23 (S9503101 and L23), exons 16–22 (S9503101 and LX22), exons 16–21 (S9503101 and Z9403097) and exons 22–24 (Z9503100 and Z9503098), exons 21–24 (S9600330 and Z9503098), exons 21–22 (S9600330 and LX22) (S9503101, 5′-CGGAATTCGAGGGAACCCCTTACCTG-3′; Z9503098, 5′-GCGGATCCGTAGTGGCTGTGGGGGAT-3′; L23, 5′-GCGGATCCTGCATGGAAGCCATTGTCC-3′; LX22, 5′-CGGGATCCACCTGTGCCAAGG-3′; Z9403097, 5′-GCGGATCCATCTGTGGGCATGTTGGT-3′; Z9503100, 5′-CGGAATTCCAACTGGAATGGATGGTA-3′; S9600330, 5′-CGGAATTCCAGGACAGAAAGATC-3′). PCR products were gel purified, digested with BamHI and EcoRI, and ligated into the yeast expression vector pGBT9 (CLONTECH) (16) similarly digested. Fragments were ligated in frame to the GAL4 DNA binding domain. The junctions were sequenced to verify the reading frame. pGBT9 has TRP1 as a selectable marker, allowing growth in the absence of tryptophan. For the TA 16/24, the construct 16/24 pGBT9 was digested with BamHI and EcoRI, and the insert subcloned in-frame to the GAL4 transactivation domain in pGAD424 (CLONTECH) (16) similarly digested. pGAD424 has LEU2 as a selectable marker, allowing growth in the absence of leucine.
Mutations were made using splicing by overlap extension PCR (17) with the following oligonucleotide primers: for the Ala-1708 → Glu mutation S9503101 and LAE; UAE and Z9503098 in two separate reactions. Both products were combined and used as a template for a final round of PCR using S9503101 and Z9503098. All subsequent mutations used the same oligonucleotides (S9503101 and Z9503098) for the final round of PCR. For the first round of PCR the following oligonucleotides were used: for the Gln-1756 C+ mutation Z9600205 and S9503101; Z9600204 and Z9503098. For the Met-1775 → Arg mutation S9503101 and S9600207; S9600206 and Z9503098. For the Met-1775 → Lys, Met-1775 → Thr, Met-1775 → Glu, and Met-1775 → Val the S9600207 was replaced by primers LMK, LMT, LME, and LMV, respectively; and S9600206 was replaced by primers UMK, UMT, UME, and UMV, respectively. The Tyr-1853 → Stop mutation was obtained by direct PCR using S9503101 and L1853 (containing a BamHI site) (LAE, 5′-CCCATTTTCCTCCCTCAATTCCTAGAAAA-3′; UAE, 5′-TTTTCTAGGAATTGAGGGAGGAAAATGGG-3′; Z9600205, 5′-GATCTTTCTGTCGCTGGGATTCTCTTGCTC-3′; Z9600204, 5′-GAGCAAGAGAATCCCAGCGACAGAAAGATC-3′; S9600207, 5′-CAGTTGATCTGTGGGCCTGTTGGTGAAGG-3′; S9600206, 5′-CCTTCACCAACAGGCCCACAGATCAACTG-3′; LMK, 5′-CAGTTGATCTGTGGGCTTGTTGGTGAAGG-3′; UMK, 5′-CCTTCACCAACAAGCCCACAGATCAACTG-3′; LMT, 5′-CAGTTGATCTGTGGGCGTGTTGGTGAAGG-3′; UMT, 5′-CCTTCACCAACACGCCCACAGATCAACTG-3′; LME, 5′-CAGTTGATCTGTGGGCTCGTTGGTGAAGG-3′; UME, 5′-CCTTCACCAACGAGCCCACAGATCAACTG-3′; LMV, 5′-CAGTTGATCTGTGGGCACGTTGGTGAAGG-3′; UMV, 5′-CCTTCACCAACGTGCCCACAGATCAACTG-3′; L1853, 5′-GCGGATCCAGGTTAGGTGTCC-3′). The constructs were sequenced to confirm the mutations.
Yeast Transformation.
Competent yeast cells were obtained using the yeast transformation system (CLONTECH) based on lithium acetate, and cells were transformed according to manufacturer’s instructions.
Interference Assay.
Levels of expression were assessed by transforming the HF7c strain with pCL1 (wild-type full-length GAL4) (18) and measuring reduction in transcription activation by pCL1 upon cotransformation with the pGBT9-based constructs. Three independent clones from each construct were assayed for β-galactosidase production in liquid culture as described (19).
Growth Curves.
HF7c transformants containing different pGBT9 constructs were grown overnight in synthetic medium plus dextrose (SD medium) lacking tryptophan. The saturated cultures were used to inoculate fresh medium lacking tryptophan or tryptophan and histidine to an initial OD600 of 0.0002. Cultures were grown at 30°C in a shaker and the optical density was measured at different time intervals starting at 12 hr, then every 4 hr up to 36 hr after inoculation.
Growth on Solid Medium.
HF7c transformants were streaked on solid SD/agar medium lacking tryptophan or tryptophan and histidine. Growth was scored after 2 days.
Filter β-Galactosidase Assay.
SFY526 and HF7c transformants were streaked on a filter overlaid on top of medium lacking tryptophan and allowed to grow overnight. Cells growing on the filter were lysed by freeze thawing in liquid nitrogen, and each filter was incubated in 2.5 ml of Z buffer (16.1 g/liter Na2HPO4·7H2O/5.5 g/liter NaH2PO4·H2O/0.75 g/liter KCl/0.246 g/liter MgSO4·7H2O, pH 7.0) containing 40 μl of 5-bromo-4-chloro-3-indolyl β-d-galctopyranoside (X-Gal) solution (20 mg/ml of X-Gal in N,N-dimethylformamide) at 30°C for up to 24 hr.
Mammalian Expression Constructs:
pGBT9-based constructs containing the wild-type 16/24 sequences as well as the mutants were digested with EcoRI and XbaI, and the fragments including vector sequences downstream of the insert were ligated into similarly digested pSG424 (20) (a gift of Mark Ptashne, Harvard University). The fragments were subcloned in frame to GAL4 binding domain similar to the yeast expression vectors.
Transient Transfections.
Cos-7 cells were grown until confluent in DMEM (GIBCO/BRL) supplemented with 10% fetal bovine serum. Cells were transfected using Lipofectamine (GIBCO/BRL). Briefly, confluent cells were washed with OptiMEM (GIBCO/BRL) and overlaid with a mixture containing the following plasmids and Lipofectamine: 0.4 μg of pSG424 or pSG424 containing 16/24 wild-type, 16/24 (Gln-1756 C+) and 16/24 (Met-1775 → Arg), all together with 1 μg of reporter pG5E1bLuc (a gift of Roger Davis, University of Massachusetts) (21). Cells were exposed to the transfection mixture for 4 hr, then washed and subjected to a 2-min 15% glycerol shock. After the shock, fresh medium was added and cells were harvested 24 and 48 hr after transfection. Cells were lysed and extracts were assayed for luciferase activity as described (21).
RESULTS
Transcription Activation by BRCA1 C-Terminal Region.
The vast majority of reported mutations in BRCA1 lead invariably to truncation of the C-terminal region. The few missense mutations reported localize either to the zinc finger motif at the N terminus or to the C-terminal acidic region (22). Moreover, the C-terminal region includes a highly conserved stretch between mouse and human BRCA1 (aa 1636–1795) (8). We therefore investigated whether the region of a reported cluster of mutations adjacent to the C-terminal end (22), comprising exons 16–24 (aa 1560–1863), is involved in transcriptional activation.
Because BRCA1 DNA binding consensus has not been defined and increasing evidence suggests that many cellular processes, including transcription, are conserved among eukaryotic species, a yeast GAL4 DNA binding domain fusion system was used (16). We fused the GAL4 DNA binding domain with human BRCA1 fragments containing either exons 16/24, 16/23, 16/22, 16/21, 22/24, 21/24, or 21/22. The 16/24 fragment fused to transactivation domain of GAL4, which is unable to bind DNA (TA-16/24) was used as a negative control (Fig. 1). These constructs were transformed into yeast and ability to activate the reporter genes was tested. As shown in Fig. 2, only the fusions 16/24 and 21/24 were able to activate transcription of the HIS3 reporter gene. The 21/24 construct however, activated transcription somewhat less efficiently than the larger 16/24. Scoring HF7c transformants for growth on solid media gave identical results (Table 1). In agreement with the results obtained with HF7c, only 16/24 and 21/24 were able to activate transcription of lacZ in SFY526 (Table 1). No construct was able to activate β-galactosidase production in HF7c cells (data not shown). The 16/24 construct is thus able to function as a transactivation domain and sequences contained within exons 21 to 24 of BRCA1 have sufficient information for activation of transcription, although sequences contained in adjacent exons may enhance this activity.
Figure 1.
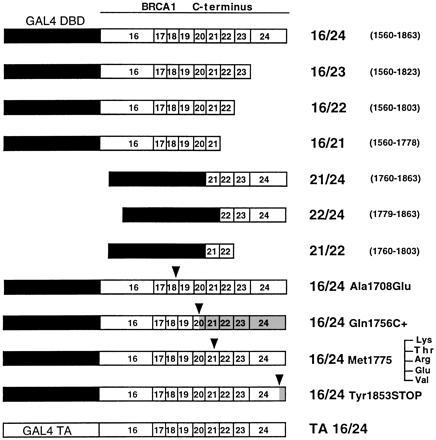
Structure of fusion proteins. Fragments 16/24, 16/23, 16/22, 16/21, 21/24, 22/24, and 21/22 contain exons 16–24 (aa 1560–1863, top bar), 16–23, 16–22, 16–21, 21–24, 22–24, and 21–22, respectively, of the BRCA1 gene fused to the DNA binding domain of GAL4 (black box). Fragments 16/24 (Gln-1756 C+) and 16/24 (Tyr-1853 → Stop) carry insertions reported to truncate BRCA1. Fragment 16/24 (Ala-1708 → Glu) is a substitution reported in patients. Fragments 16/24 (Met-1775 → Lys, Met-1775 → Thr, Met-1775 → Arg, Met-1775 → Glu, and Met-1775 → Val) are each individual mutations at codon 1775, with the Met-1775 → Arg mutant reported in patients with breast cancer. TA 16/24 represents exons 16–24 fused to the activation domain of GAL4. Arrowheads indicate the positions of the mutations, and shaded areas represent truncated parts of the mutant protein.
Figure 2.
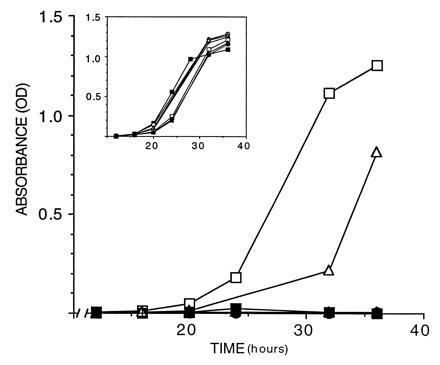
Transcriptional activation by BRCA1 C terminus. Growth curves of S. cerevisiae (HF7c) carrying the following fragments of BRCA1 in pGBT9 grown in medium lacking tryptophan and histidine: 16/24 (□), 16/23 (▪), 16/22 (○), 16/21 (•), 21/24 (▵), 22/24 (▴), and 21/24 (×). (Inset) Growth in medium lacking tryptophan only. OD600 of the cultures (y axis) is plotted against time in culture (x axis).
Table 1.
Transcriptional activation by BRCA1 C terminus
| HF7c
|
SFY526 β-gal‡ | COS-7 luciferase | ||
|---|---|---|---|---|
| Liquid* | Solid† | |||
| Wild type | ||||
| 16/24 | +(1.0) | + | + | +(1.0) |
| 16/23 | −(0.0) | − | − | ND |
| 16/22 | −(0.0) | − | − | ND |
| 16/21 | −(0.0) | − | − | ND |
| 21/24 | +(0.6) | + | + | ND |
| 22/24 | −(0.0) | − | − | ND |
| 21/22 | −(0.0) | − | − | ND |
| 16/24-TA | ND | − | − | ND |
| 16/24 mutants | ||||
| Ala-1708 → Glu | −(0.0) | − | − | ND |
| Gln-1756 C+ | −(0.0) | − | − | −(0.0) |
| Met-1775 → Arg | −(0.0) | − | − | −(0.0) |
| Tyr-1853 → Stop | −(0.0) | − | − | ND |
| Met-1775 → Lys | −(0.0) | − | − | ND |
| Met-1775 → Val | +(1.0) | + | + | ND |
| Met-1775 → Thr | −(0.0) | − | + | ND |
| Met-1775 → Glu | −(0.0) | − | − | ND |
ND, not determined.
Cultures were grown as described in Materials and Methods. Relative activity shown in parenthesis.
Several independent colonies were streaked on SD agar lacking tryptophan and histidine and scored for growth after 3 days at 30°C.
Several independent clones were assayed for β-galactosidase (β-gal) production on filters. To ensure that the negatives were not producing β-galactosidase, the assay was also scored after 18 h with the same results.
Germ-Line Mutations Impair Transcription Activation.
It has been shown that retroviral transfer of wild-type BRCA1 gene inhibits growth in vitro of breast and ovarian cancer cell lines, thus establishing that BRCA1 acts as a tumor suppressor (23). Loss or reduction of function of BRCA1 would therefore confer susceptibility to tumor progression. The fact that a portion of BRCA1 protein was able to drive expression of the reporter genes raised the possibility that impairment of this function could be related to the development of breast and ovarian cancer.
To test this hypothesis directly we introduced four mutations reported in afflicted patients, generating the constructs 16/24 (Ala-1708 → Glu, Gln-1756 C+, Met-1775 → Arg, and Tyr-1853 → Stop) shown in Fig. 1. Ala-1708 → Glu is associated with very early onset of breast cancer (24). Gln-1756 C+ is an insertion of a cytosine in codon 1756 resulting in a frameshift and is the most frequent mutation found in the C-terminal (4, 25, 26, 27, 28, 29). Met-1775 → Arg is the only recurring missense mutation in this region (4, 22, 24, 26). Moreover, this residue lies in a highly conserved stretch between mouse and human BRCA1 (8). Tyr-1853 → Stop is an insertion of an adenine in codon 1853 leading to a truncated protein only 11 aa from the C-terminal end that is associated with very early-onset breast cancer (30). None of the constructs carrying mutations found in patients was able to activate transcription of the HIS3 promoter in HF7c (Fig. 3), nor lacZ expression in SFY526 (Table 1). The difference in activation between the wild type and the mutants cannot be accounted for different levels in expression and stability as all constructs were expressed at roughly the same levels as measured by interference assays (data not shown).
Figure 3.
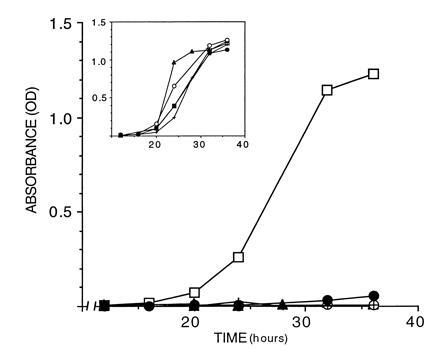
Mutations occurring in patients impair activity. Shown are growth curves of S. cerevisiae (HF7c) carrying the following fragments and mutants in pGBT9 grown in medium lacking tryptophan and histidine: 16/24 (□), 16/24 (Ala-1708 → Glu) (▴), 16/24 (Gln-1756 C+) (○), 16/24 (Met-1775 → Arg) (•), and 16/24 (Tyr-1853 → Stop) (×). (Inset) Growth in medium lacking tryptophan only. The OD600 of the cultures (y axis) is plotted against time in culture (x axis).
Transcription Activation in Mammalian Cells.
To rule out the possibility that BRCA1 activates transcription only in the yeast system, we examined the transcriptional activity of the wild-type 16/24 and two mutants (Gln-1756 C+ and Met-1775 → Arg) using a mammalian cell line, Cos-7. In this system we used GAL4 DNA binding domain fusions (20) and the luciferase gene as a reporter under the control of GAL4 responsive elements (21). In Fig. 4 the wild-type fusion activates transcription 10-fold, while the mutants were not able to activate transcription to a detectable level. Therefore, BRCA1 transcriptional activation function is not restricted to the yeast system. Similarly to the experiments performed in yeast, the difference cannot be accounted for different levels of expression, since all constructs were expressed, with the Met-1775 → Arg expressed at higher levels than the others, as determined by Western blotting (data not shown).
Figure 4.
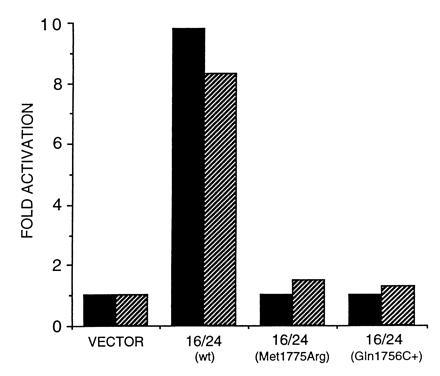
Transcription activation by BRCA1 C terminus in mammalian cells. Luciferase activity of the following fragments and mutants in pSG424, transfected along with luciferase reporter plasmid into Cos-7 cells plotted as fold activation over control: vector, pSG424 alone; 16/24, wild-type exons 16/24; 16/24 (Met-1775 → Arg), Met-1775 → Arg mutation; 16/24 (Gln-1756 C+), cytosine insertion at position 1756. Cells were harvested at 24 hr (black bars) and 48 hr (shaded bars).
Hydrophobic Residues Are Major Determinants.
Acidic regions of proteins that are not necessarily transcription activators can sometimes activate transcription when fused to heterologous DNA binding domains (31). To exclude this possibility in our assay using BRCA1 C terminus, we introduced a negative charge at position 1775 (Met-1775 → Glu). As shown in Table 1 this construct was not able to activate transcription, eliminating the possibility that a nonspecific cluster of negative charges in BRCA1 was spuriously activating transcription.
As has been suggested in other acidic domain-containing transcription factors, addition of positively charged residues irrespective of the position results in impairment of the transcriptional activation function (32). The mutant Met-1775 → Arg described above is a change from a hydrophobic to that of a positively charged residue, which resulted in drastically impaired activity of BRCA1 C terminus. We examined the effects of three other possible mutations at codon 1775: from the hydrophobic methionine to (i) a positively charged residue change similar to that seen in the reported patients (Met-1775 → Lys), (ii) a polar residue (Met-1775 → Thr), and (iii) a hydrophobic residue (Met-1775 → Val), for both growth in liquid and solid media, as well as in β-galactosidase assays. Changing the methionine at position 1775 to a conservative residue valine, resulted in wild-type transcriptional activity, while a change to a positively charged residue (Met-1775 → Lys) or to a polar residue (Met-1775 → Thr) drastically diminished transcriptional activation by BRCA1 C terminus (Table 1). Interestingly, the construct carrying the Met-1775 → Thr mutation does not change the net charge of the molecule and is still inactive in HF7c. These data suggest that BRCA1 may function analogously to herpes virus protein VP16 where negative charges are not the only major determinants but that hydrophobic residues are also crucial for activity (33).
DISCUSSION
The presence of an acidic region, nuclear localization signals and a zinc finger domain suggest that BRCA1 may regulate transcription (4). By using fusion proteins with a heterologous DNA binding domain (GAL4), we show that the C-terminal portion of BRCA1 (aa 1560–1863) is able to activate transcription of the HIS3 gene in HF7c (but not lacZ), the lacZ gene in SFY526, as well as the luciferase gene in mammalian cells, all driven by GAL4 responsive elements. Although HIS3 and lacZ in HF7c share GAL4-responsive elements, the rest of the promoter sequences are different (14, 15). The lack of transcriptional activation of lacZ in HF7c may therefore reflect the fact that the ability of transcriptional activators to interact with the basal transcription apparatus varies with different promoter contexts (34, 35).
Our results also indicate that the smaller C-terminal fragment 21/24 (aa 1760–1863) is also able to activate transcription, although less efficiently than the larger fragment 16/24 (aa 1560–1863). Therefore, exons 21/24 have sufficient information to activate transcription and represent the minimal transactivation domain of BRCA1. Moreover, adjacent exons, 16 to 20, contribute to full activity. Interestingly, most mutations found in patients generate proteins lacking all or part of the minimal transactivation domain defined above. According to our data, truncated proteins lacking even small regions of the minimal transactivation domain (e.g., Tyr-1853 → Stop) would be predicted to have no function in transcriptional activation, a situation borne out by our data.
While the wild-type BRCA1 C terminus fused to a heterologous DNA binding protein activates transcription, the proteins carrying missense or nonsense mutations found in patients with tumors fail to do so. The fact that fusion proteins carrying mutations known to confer breast cancer susceptibility abolish activity strongly supports the idea that BRCA1 protein is involved in transcription. We tested four reported mutations (Ala-1708 → Glu, Gln-1756 C+, Met-1775 → Arg, and Tyr-1853 → Stop) with association with disease (4, 22, 24, 25, 26, 27, 28, 29). Thus we suggest that mutations in BRCA1 which impair the ability to activate transcription may predispose the carrier to tumors. These results indicate that BRCA1 may function to activate transcription of genes involved in suppressing transformation.
Interestingly, a mutation that falls outside the minimal transactivation domain also abolished function (Ala-1708 → Glu). The observation that a construct lacking this region is active suggests that it may be involved in proper folding of the C-terminal region, which may either stabilize or destabilize the structure. When the region is deleted, the conformation may be conserved as in the wild type. An alternative explanation is that this amino acid is involved in nonessential sites of protein–protein interactions and that absence of these sites or nondisruptive mutations allow interaction. Other mutations such as Ala-1708 → Glu on the other hand, may destabilize this interaction.
During the preparation of this manuscript, Chapman and Verma (36) reported similar results. Using a mammalian transfection system the authors defined the fragment 16/24 as a transactivation domain and suggested that the fragment 21/24 represents the minimal transactivation domain, in agreement with our data. Taken together, these two studies analyze five different mutations of BRCA1, and all the mutations which have shown disease association have impaired ability to activate transcription. We believe that a detailed analysis of BRCA1 C-terminal region may be instrumental to understanding the etiology of familial breast cancer.
The simple assay used here, testing the ability of the BRCA1 fusion proteins to activate transcription in yeast is a direct way to assess the functional significance of documented mutations that localize to the transactivation domain. Furthermore, our system could be instrumental in discriminating between cancer predisposing mutations and neutral polymorphisms. The assay could also be used to screen for therapeutic compounds capable of restoring the normal function to the mutant proteins.
Definite confirmation that BRCA1 acts as a transcription activator, however, must await the demonstration of its DNA binding activity or identification of its target genes.
Acknowledgments
We thank Alison Egan for help with the constructs. We also thank Robert Roeder, Alexander Hoffmann, and Curt Horvath for helpful discussions and Shinya Tanaka and Raymond Birge for critical comments on the manuscript. A.N.A.M. is a Pew Fellow in the Biomedical Sciences and on leave from The Institute of Chemistry, Federal University of Rio de Janeiro. A.A. is a recipient of a post-doctoral fellowship from the National Science Foundation. This work was supported by Grant CA44356 from the National Cancer Institute.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: X-Gal, 5-bromo-4-chloro-3-indolyl β-d-galctopyranoside; SD, synthetic medium plus dextrose.
References
- 1.Claus E B, Risch N, Thompson W D. Am J Hum Genet. 1991;48:232–242. [PMC free article] [PubMed] [Google Scholar]
- 2.Hall J F, Lee M K, Newman B, Morrow J E, Anderson L A, Huey B, King M-C. Science. 1990;250:1684–1689. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 3.Easton D F, Bishop D T, Ford D, Crockford G P Breast Cancer Linkage Consortium. Am J Hum Genet. 1993;52:678–701. [PMC free article] [PubMed] [Google Scholar]
- 4.Miki Y, Swensen J, Shattuck-Eidens D, Futreal P A, Harshman K, et al. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 5.Saurin A J, Borden K L B, Boddy M N, Freemont P S. Trends Biochem Sci. 1996;21:208–214. [PubMed] [Google Scholar]
- 6.Ptashne M. Nature (London) 1988;335:683–689. doi: 10.1038/335683a0. [DOI] [PubMed] [Google Scholar]
- 7.Lane T F, Deng C, Elson A, Lyu M S, Kozak C A, Leder P. Genes Dev. 1995;9:2712–2722. doi: 10.1101/gad.9.21.2712. [DOI] [PubMed] [Google Scholar]
- 8.Abel K J, Xu J, Yin G-Y, Lyons R H, Meisler M H, Weber B L. Hum Mol Genet. 1995;4:2265–2273. doi: 10.1093/hmg/4.12.2265. [DOI] [PubMed] [Google Scholar]
- 9.Sharan S K, Wims M, Bradley A. Hum Mol Genet. 1995;4:2275–2278. doi: 10.1093/hmg/4.12.2275. [DOI] [PubMed] [Google Scholar]
- 10.Jensen R A, Thompson M E, Jetton T L, Szabo C I, van der Meer R, Helou B, Tronick S R, Page D L, King M-C, Holt J T. Nat Genet. 1996;12:303–308. doi: 10.1038/ng0396-303. [DOI] [PubMed] [Google Scholar]
- 11.Jensen R A, Thompson M E, Jetton T L, van der Meer R, Helou B, Arteaga C L, Page D L, Holt J T, Tronick S R, Gown A M, Skelly M, Ostermeyer B, Schieltz D, Szabo C I, King M-C. Nat Genet. 1996;13:269–272. doi: 10.1038/ng0396-303. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Chen C-F, Riley D J, Allred D C, Chen P-L, von Hoff D, Osborne C K, Lee W-H. Science. 1995;270:789–791. doi: 10.1126/science.270.5237.789. [DOI] [PubMed] [Google Scholar]
- 13.Scully R, Ganesan S, Brown M, De Caprio J A, Cannistra S A, Feunteun J, Schnitt S, Livingston D M. Science. 1996;272:123–125. doi: 10.1126/science.272.5258.123. [DOI] [PubMed] [Google Scholar]
- 14.Bartel P L, Chien C-T, Sternglanz R, Fields S. BioTechniques. 1993;14:920–924. [PubMed] [Google Scholar]
- 15.Feilotter H E, Hannon G J, Ruddel C J, Beach D. Nucleic Acids Res. 1994;22:1502–1503. doi: 10.1093/nar/22.8.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartel P L, Chien C-T, Sternglanz R, Fields S. In: Cellular Interactions in Development: A Practical Approach. Hartley D A, editor. Oxford: Oxford Univ. Press; 1993. pp. 153–179. [Google Scholar]
- 17.Higuchi R. In: PCR Protocols: A Guide to Methods and Applications. Innis M A, Gelfand D H, Sininski J J, White T J, editors. San Diego: Academic; 1990. pp. 177–183. [Google Scholar]
- 18.Fields S, Song O. Nature (London) 1989;340:245–247. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 19.Brent R, Ptashne M. Nature (London) 1984;312:612–615. doi: 10.1038/312612a0. [DOI] [PubMed] [Google Scholar]
- 20.Sadowski I, Ptashne M. Nucleic Acids Res. 1989;17:7539. doi: 10.1093/nar/17.18.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seth A, Gonzalez F A, Gupta S, Raden D L, Davis R J. J Biol Chem. 1992;267:24796–24804. [PubMed] [Google Scholar]
- 22.Breast Cancer Information Core, http://www.nchgr.nih.gov/dir/lab_transfer/bic/.
- 23.Holt J T, Thompson M E, Szabo C, Robinson-Benion C, Arteaga C L, King M-C, Jensen R A. Nat Genet. 1996;12:298–302. doi: 10.1038/ng0396-298. [DOI] [PubMed] [Google Scholar]
- 24.Futreal P A, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, et al. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 25.Simard J, Tonin P, Durocher F, Morgan K, Rommens J, Gingras S, Samson C, Leblanc J-F, Belanger C, Dion F, Liu Q, Skolnick M, Goldgar D, Shattuck-Eidens D, Labrie F, Narod S A. Nat Genet. 1994;8:392–398. doi: 10.1038/ng1294-392. [DOI] [PubMed] [Google Scholar]
- 26.Szabo C I, King M-C. Hum Mol Genet. 1995;4:1811–1817. doi: 10.1093/hmg/4.suppl_1.1811. [DOI] [PubMed] [Google Scholar]
- 27.Langston A A, Malone K E, Thompson J D, Daling J R, Ostrander E A. N Engl J Med. 1996;334:137–142. doi: 10.1056/NEJM199601183340301. [DOI] [PubMed] [Google Scholar]
- 28.Gayther S A, Warren W, Mazoyer S, Russel P A, Harrington P A, Chiano M, Seal S, Hamoudi R, van Rensburg E J, Dunning A M, Love R, Evans G, Easton D, Clayton D, Stratton M R, Ponder B A J. Nat Genet. 1995;11:428–433. doi: 10.1038/ng1295-428. [DOI] [PubMed] [Google Scholar]
- 29.Serova O, Montagna M, Torchard D, Narod S A, Tonin P, Sylla B, Lynch H T, Feunteun J, Lenoir G M. Am J Hum Genet. 1996;58:42–51. [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman L S, Ostermeyer E A, Szabo C, Dowd P, Lynch E D, Rowell S E, King M-C. Nat Genet. 1994;8:399–403. doi: 10.1038/ng1294-399. [DOI] [PubMed] [Google Scholar]
- 31.Ma J, Ptashne M. Cell. 1987;51:113–119. doi: 10.1016/0092-8674(87)90015-8. [DOI] [PubMed] [Google Scholar]
- 32.Bushman F D, Ptashne M. Cell. 1988;54:191–197. doi: 10.1016/0092-8674(88)90551-x. [DOI] [PubMed] [Google Scholar]
- 33.Cress W D, Triezenberg S J. Science. 1991;251:87–90. doi: 10.1126/science.1846049. [DOI] [PubMed] [Google Scholar]
- 34.Estojak J, Brent R, Golemis E A. Mol Cell Biol. 1995;15:5820–5829. doi: 10.1128/mcb.15.10.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seneca S, Punyammalee B, Bailly M, Ghysdael J, Crabeel M. Oncogene. 1991;6:357–360. [PubMed] [Google Scholar]
- 36.Chapman M S, Verma I M. Nature (London) 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]