

Abstract
The myelodysplastic syndromes (MDS) are a heterogeneous group of disorders that manifest as bone marrow failure with the risk of life threatening infections and bleeding. A third of these patients may transform to acute leukemia. Age and co-morbidities have limited treatment in the majority to supportive care with a minority of patients eligible for the only curative modality to date, allogeneic stem cell transplantation. The advent of targeted therapy has increased the repertoire of therapeutic options. In particular the methyl transferase inhibitor 5 Azacytidine, that targets epigenetic changes in MDS, has been shown to be effective in up to 60% of patients in a Phase III randomized controlled trial comparing it with best supportive care and has been licensed by the US Food and Drug Administration for use in all subtypes of MDS. It has been shown to prolong time to leukemic transformation (21 vs 12 months with 3% transforming to leukemia p=0.0001) and is the only disease-modifying drug. Patients with monosomy 7, trisomy 8, and diploid chromosomes appear to particularly benefit with the former deriving sustained remissions. As an outpatient therapy, with an acceptable side effect profile, treatment with Azacytidine needs to be considered in all MDS patients who are eligible for treatment.
Keywords: azacytidine, myelodysplastic syndromes, demethylation, methyltransferase inhibitor
Introduction
The myelodysplastic syndromes (MDS) are an acquired form of clonal stem cell disorders that manifest heterogeneously but are unified clinically by progressive bone marrow failure, susceptibility to life threatening infections, and a risk of transforming to leukemia. Although a third of patients transform to acute myeloid leukemia (AML), infection and bleeding are the commonest cause of morbidity and mortality (Heaney and Golde 1999). MDS occurs with a frequency of 15–50 new cases per 100 000 per year and this incidence appears to be increasing (Aul et al 1998) making it the commonest hematological malignancy along with chronic lymphocytic leukemia (CLL).
Therapeutically the management of cytopenia’s and infections predominates early MDS. Treatment options such as AML induction type chemotherapy followed by an allogeneic stem cell transplantation is an option for only a minority of patients, but is the only modality that offers a cure (Ho et al 2004). In the majority of patients the standard care is supportive in the form of blood and platelet transfusions, treatment of infective complications and administration of growth factors such as granulocyte-colony stimulating factor (G-CSF) and erythropoietin. There has been a dearth of suitable therapies for the elderly that bridged the gap between the extremes of supportive care on the one hand and stem cell transplantation on the other. The emergence of targeted therapies has rekindled interest in their application to MDS. Candidates have included the farnesyl transferase inhibitors, thalidomide and its immunomodulatory drug (IMiD) analogs such as lenalidomide and the methyltransferase inhibitors (MTI) such as Azacytidine and its analogue 5 Aza 2′ deoxycytidine in MDS. Potentially reversible silencing of genes (such as CDKN2B) by promoter methylation has been shown to occur in MDS and increase with disease progression. The MTIs have been shown to be demethylating agents both in vivo and in vitro and could target these changes in MDS. The MTIs are administered as outpatient therapies with proven efficacy. 5-Azacytidine, in particular, has been shown to be effective in all subtypes of MDS to prolong the time to disease progression and is the only drug in this group that has been licensed by the US Food and Drug Administration (FDA) for all subtypes of MDS (Kaminskas et al 2005).
This review aims to summarize the rationale for the use of Azacytidine and the current clinical evidence supporting its use in the MDS.
Classification of MDS
Since the risk of leukemic transformation is varied between various subgroups of MDS, various classification systems and prognostic scores have attempted to identify patients with a poor prognosis and an increased risk of leukemic transformation. The widely used French American British (FAB) classification of MDS identifies five morphological subgroups of prognostic importance (Table 1) (Bennett et al 1982). Patients with refractory anemia (RA) with excess blasts (RAEB) and RA with excess blasts in transformation (RAEB-t) are at a high risk of myeloid leukemic transformation (AML) and also have a shorter median survival whereas patients with RA and RARS (RA with ringed sideroblasts) have a longer survival and lower incidence of developing leukemia. The World Health Organisation (WHO) classification of MDS identifies 7 groups (Table 2) (Vardiman 2002). Patients with RARS and RA have only unilineage (erythroid) dysplasia with or without 15% ringed sideroblasts respectively and experience a protracted clinical course with median survivals of 5–6 years. The propensity for leukemic transformation is low in both RA (7%) and RARS (1%–2%). Refractory cytopenia with multilineage dysplasia (RCMD) and RCMD with ringed sideroblasts are characterized by bi or pancytopenia and dysplasia in ≥10% of the cells in 2 or more myeloid cell lines. Both these groups have a shorter median survival of approximately 33 months, and a frequency of leukemic transformation of 10%–13%. However patients in the RCMD group with complex karyotypes have survivals similar to those with RAEB. The WHO classification redefined AML as a group with bone marrow (BM) blasts >20% and RAEB as between 5%–20% blasts with RAEB being further classified into RAEBI (5%–9% blasts) and RAEBII (10%–19% blasts). Approximately 21% of patients with RAEBI and 34% of patients with RAEBII progress to AML. The rest succumb to the effects of bone marrow failure. The median survival is approximately 18 months for RAEBI and 10 months for RAEBII (Germing et al 2000). Patients with 5q-syndrome have a long survival and a low leukemic transformation rate. The International Prognostic Scoring System (IPSS) (Table 3) (Greenberg et al 1997) based on bone marrow blast percentage, cytogenetics, and number of cytopenias also identifies patients at high risk of transformation (IPSS score 1.5). These scoring systems help to individualize therapeutic decisions such as treatment with chemotherapy, allogeneic transplantation, or use of drugs like lenalidomide or methyltransferase inhibitors.
Table 1.
The FAB classification of MDS
| FAB subgroup | Peripheral blood criteria | BM criteria |
|---|---|---|
| RA | Blasts <1%, monocytes <1×109/L | Blasts <5%, ringed sideroblasts <15% of erythroblasts |
| RARS | Blasts <1%, monocytes < 1×109/L | Blasts <5%, ringed sideroblasts >15% of erythroblasts. |
| RAEB | Blasts>1%, <5%, monocytes <1×109/L | Blasts 5%–19% |
| RAEB-t | Blasts >5% or Auer rods or | Blasts 20%–29% or Auer rods |
| Chronic myelomonocytic leukemia | Blasts<5%, | Blasts up to 20%, promonocytes/monocytes often increased. |
Abbreviations: BM, bone marrow; FAB, French, American, British classification of MDS; MDS, myelodysplastic syndromes; RA, refractory anemia; RAEB, refractory anemia with excess blasts; RAEB-t, refractory anemia with excess blasts in transformation; RARS, refractory anemia with ringed sideroblasts.
Table 2.
The WHO Classification of MDS
| Disease | Peripheral blood criteria | BM Criteria |
|---|---|---|
| RA | Anemia, no or rare blasts | Erythroid dysplasia alone, <5% blasts, <15% ringed sideroblasts |
| RARS | Anemia, no blasts | Erythroid dysplasia alone, <5% blasts, >15% ringed sideroblasts |
| Refractory cytopenia with multilineage dysplasia | Cytopenias (bicytopenia or pancytopenia), no or rare blasts, no Auer rods, <1×109/L monocytes | Dysplasia in >10% of cells in >2 myeloid cell lines, <5% blasts, no Auer rods, <15% ringed sideroblasts |
| Refractory cytopenia with multilineage dysplasia and ringed sideroblasts | Cytopenias (bicytopenia or pancytopenia), no or rare blasts, no Auer rods, <1×109/L monocytes | Dysplasia in >10% of cells in >2 myeloid cell lines, <5% blasts, no Auer rods, >15% ringed sideroblasts |
| RAEB type 1 | Cytopenias, <5% blasts, no Auer rods, <1×109/L monocytes | Unilineage or multilineage dysplasia, 5%–9% blasts, no Auer rods |
| RAEB type 2 | Cytopenias, 5%–19% blasts, no Auer rods, <1×109/L monocytes | Unilineage or multilineage dysplasia, 10%–19% blasts, occasional Auer rods |
| MDS unclassified | Cytopenias, no or rare blasts, no Auer rods | Unilineage dysplasia in granulocytes or megakaryocytes, <5% blasts, no Auer rods |
| MDS associated with isolated deletion (5q) | Anemia, <5% blasts, platelet count normal to increased | Normal-to-increased megakaryocytes with hypolobated nuclei, <5% blasts, no Auer rods, isolated del (5q) |
Abbreviations: BM, bone marrow; FAB, French, American, British classification of MDS; MDS, myelodysplastic syndromes; RA, refractory anemia; RAEB, refractory anemia with excess blasts; RAEB-t, refractory anemia with excess blasts in transformation; RARS, refractory anemia with ringed sideroblasts; WHO,World Health Organisation.
Table 3.
The IPSS scoring system for MDS
| Prognostic variable | Score value | ||||
|---|---|---|---|---|---|
| 0 | 0.5 | 1.0 | 1.5 | 2.0 | |
| BM blasts % | <5 | 5–10 | – | 11–20 | 21–30 |
| Karyotypea | Good | Intermediate | Poor | ||
| Cytopeniasb | 0 or 1 | 2 or 3 | |||
Note: Scores for risk groups are as follows: Low: 0, Intermediate-1: 0.5–1.0, Intermediate-2: 1.5–2.0, High >2.5.
Karytotype: Good: normal, -Y, del(5q), del(20q); Poor: complex (>3 abnormalities), or chromosome 7 anomalies; Intermediate: other abnormalities;
Cytopenias: defined as Hb<10g/dl, Absolute neutrophil count <1.5×109/L and platelet count <100×109/L.
Abbreviations: BM, bone marrow; INT, intermediate; IPSS, International Prognostic Scoring System; MDS, myelodysplastic syndromes.
Epigenetic changes in MDS
Epigenetics is the heritable change in gene expression without DNA sequence alteration (Bird 2002). The primary epigenetic modifications are DNA methylation and histone modifications, both of which are potentially reversible. DNA methylation plays an important role in tissue and stage-specific gene regulation (Jaenisch and Bird 2003) and increases with age (Ahuja et al 1998). DNA methyltransferases (DNMT) mediate methylation by incorporating a methyl group into position 5 of the cytosine ring resulting in 5-methyl cytosine. This modification occurs most frequently in cytosines that precede guanosine in the DNA sequence called cytosine phosphodiester guanine (CpG) dinucleotides that occur in asymmetric clusters called CpG islands (Gardiner-Garden and Frommer 1987; Frommer et al 1992). These islands are often associated with the promoter regions of genes (Delgado et al 1998) and are usually unmethylated irrespective of whether the gene is being transcribed (Bird et al 1979). However aberrant methylation of such promoter regions can occur in disease, particularly cancers and correlates with gene silencing (Antequera et al 1990). Maintenance methylation is carried out by DNMT1 whereas de novo methylation is mediated by DNMT 3a and 3b although these functions are not exclusive (Okano et al 1999). Cancer is characterized by global DNA hypomethylation and regional promoter hypermethylation of genes. This hypermethylation occurs in a non-random, tumor specific manner and hypermethylation of tumor suppressor genes may act as the second inactivating hit invoked in Knudson’s hypothesis (Jones et al 1998). Several tumor suppressor genes such as VHL in renal cancer (Herman et al 1994), p16INK4A in solid tumors and lymphomas (Esteller et al 2001; Garcia et al 2002), E-Cadherin in breast, thyroid, gastric, and colorectal cancers (Wheeler 2005) are inactivated by promoter hypermethylation. Promoter hypermethylation of CDKN2B (encoding p15INK4b) has been shown to be restricted to the hematological malignancies (Esteller et al 2001).
Epigenetic studies in MDS
In MDS epigenetic studies to date have focused on the methylation of cell cycle regulatory genes such as the tumor suppressor gene CDKN2B that encodes the cyclin dependent kinase inhibitor p15INK4b. p15INK4b inhibits quiescent cells from entering the cell cycle and is important in preventing the uncontrolled proliferation of human hemopoietic stem cells (Dao et al 1998). Induction of p15INK4b by cytokines leads to myeloid progenitors being retained in G0 and differentiating into granulocytes and macrophages (Amanullah et al 2000; Teofili et al 2000). It is postulated that silencing of CDKN2B by promoter methylation in MDS leads to sustained progenitor proliferation without differentiation resulting in peripheral cytopenias and an excess of blasts, which are a hallmark of the disease (Drexler 1998; Cameron et al 1999).
Hypermethylation of other genes such as the calcitonin gene occurs in 65% of MDS (Dhodapkar et al 1995). Other genes such as HIC (Hypermethylated in cancer), E cadherin (Corn et al 2000) and ER (Estrogen Receptor) are also hypermethylated in MDS–AML. SOCS (Suppressor of Cytokines) methylation occurs in 31% of patients with MDS suggesting that activation of the Janus kinase – Signal transducer activation of transcription (JAK-STAT) pathway plays an important role in the progression of the disease in some patients (Johan et al 2004).
In AML numerous genes are concurrently hypermethylated suggesting that normal DNA methylation mechanisms are disrupted. Methylation profiling of the calcitonin, estrogen receptor, E-Cadherin, p15, p16, Rb, GST-Pi, and HIC1 gene promoters in AML showed that 95% had an abnormal methylation pattern in at least one gene whereas 75% had abnormal methylation of two or more genes (Melki et al 1999). In AML 160 of 261 (61%) patients (age less than 65) had estrogen receptor methylation (ERM). ERM decreased with increasing age (p=0.0001) and was significantly lower in patients with myelomonocytic and monocytic AML (FAB M4/M5) (p=0.0019). ERM positive patients had a significantly better overall survival (p=0.0044) (Li et al 1999). Furthermore, methylation profiling of a subset of the cases studied for ERM (36 cases of AML) found ER to be frequently hypermethylated (47%) as were MYOD1, PITX2, GPR37, and SDC4. Importantly the methylation density of each of the CpG islands correlated with ERM density suggesting the presence of a methylator phenotype in AML (Toyota et al 2001) and a means of identifying patients in whom demethylating therapy may be appropriate. Rush et al (2001) further identified a predisposition for chromosome 11 methylation, in patients with de-novo AML. Several fold increases in DNMT1, DNMT3a, and DNMT3b that correlated with aberrant p15 methylation also occurred in AML (Mizuno et al 2001).
However, the precise relationship between hypermethylation of the promoters of these genes and their contribution to the pathogenesis and progression of MDS remains unclear except perhaps for CDKN2B.
Progression to AML occurs frequently in patients with RAEB (28%) and RAEBt (45%) but less often in RA (10%) and RARS (8%) (Germing et al 2000). The IPSS score identifies chromosome abnormalities (chromosome 7 and complex cytogenetics) and increasing blast percentage as being important predictors of leukemic transformation. Hypermethylation of the CDKN2B promoter appears important in MDS evolution. p15INK4b is upregulated during in vitro granulocytic and megakaryocytic differentiation of normal CD34+ hemopoietic progenitors (Teofili et al 2001). Deletions or mutations of CDKN2B are uncommon in MDS (Nakamaki et al 1997). The frequency of CDKN2B methylation is low (23%) in early MDS however its presence at diagnosis is predictive of progressive disease and its acquisition also accompanies disease progression (Tien et al 2001). Furthermore CDKN2B hypermethylation occurs frequently in RAEB, RAEBt particularly in patients with more than 10% blasts (Quesnel et al 1998; Uchida et al 1998). The incidence of CDKN2B methylation is 58% in chronic myelomonocytic leukemia (CMML) (Tessema et al 2003) and 60%–75% in MDS–AML (Tien et al 2001). Therapy related MDS (t-MDS, MDS occurring in individuals previously treated with either cytotoxic chemotherapy and or radiotherapy) is also accompanied by CDKN2B promoter methylation and CpG methylation density has both been shown to increase significantly with advanced MDS subtypes (p=0.004 and p=0.0002) and deletion or loss of 7q (Christiansen et al 2003). Thus CDKN2B appears important in disease progression and in the development of high risk MDS.
5-Azacytidine in MDS
Rationale for its use
Azacytidine is a DNMT inhibitor that has in vitro and in vivo demethylating effects (Jones et al 1983). Since methylation of CDKN2B occurs at a high frequency in MDS and is acquired during disease progression, reactivation by demethylation may halt disease progression. At high doses Azacytidine is cytotoxic whereas at lower doses it induces differentiation and demethylation (Christman 2002). Clinical evidence for its efficacy in acute myeloid leukemia comes from 8 trials involving approximately 200 patients, treated with cytotoxic doses of Azacytidine (intravenously at doses ranging from 60–750 mg/m2 once daily or as a repeated dose for up to 10 days) resulting in an overall response rate of 36% (20% complete remission [CR] and 16% partial response [PR]). These results were considerably better than the results achieved in patients with chronic myeloid leukemia (CML), acute lymphoblastic leukemia (ALL), and solid tumors demonstrating the anti-leukemic activity of Azacytidine in AML (Glover et al 1987).
Chemical structure and pharmacokinetics of 5-Azacytidine
5-Azacytidine (Vidaza®) is a chemically synthesized nucleoside analogue that was manufactured in the 1960s. Its role as a DNMT inhibitor (MTI) that can potentially reverse pathogenic epigenetic events such as methylation of genes has led to renewed interest and enthusiasm in the drug. 5 Azacytidine is a pyrimidine ring analogue in which the ring carbon 5 is replaced by nitrogen (Figure 1). It is incorporated into both RNA and DNA (Figure 2). In DNA it binds DNMT, irreversibly leading to loss of their activity (Creusot et al 1982; Christman 2002). Additionally DNA cannot be methylated resulting in almost complete demethylation of genomic DNA (Jones and Taylor 1981). De-novo methylation is therefore inhibited, resulting in DNA hypomethylation (Figure 3). The DNMT and Azacytidine adducts are also toxic and mutagenic (Bhagwat and Roberts 1987). In vitro Azacytidine leads to decondensation of chromatin, chromosomal instability, and extends the replication time of normally late replicating heterochromatin (reviewed in Haaf 1995).
Figure 1.
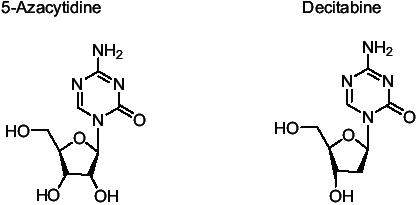
The ring structure of 5 Azacytidine and 5 Aza-2-deoxycytidine showing substitution of N at position 5. Azacytidine is attached to a ribose sugar whereas 5 Aza-2-deoxycytidine is attached to a deoxyribose.
Figure 2.
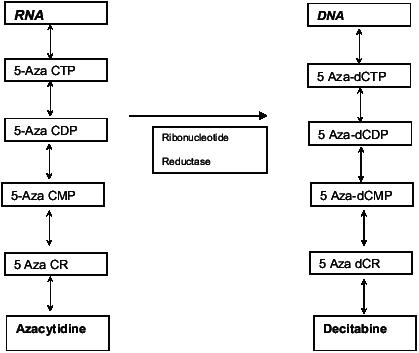
Incorporation of Azacytidine into RNA and its metabolite 5 Aza 2 deoxycytidine (Decitabine) into DNA.
Abbreviations: CMP,cytidine monophosphate ;CDP, Cytidine diphosphate, CTP, Cytidine triphosphate, CR, azacytidine; dCR, decitabine; dCMP, deoxyctidine monophosphate; dCDP, deoxycytidine diphosphate ; dCTP,deoxy cytidine triphosphate ; DNA, deoxyribonucleic acid; RNA, ribonucleic acid.
Figure 3.
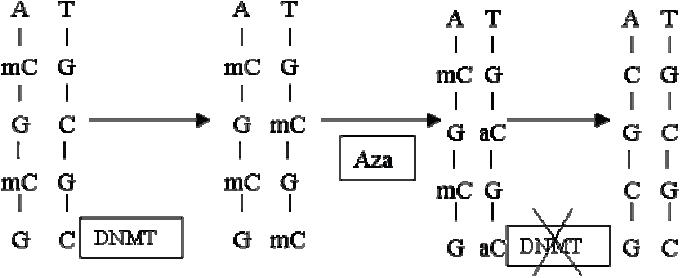
Demethylation by MTI’s. Hemimethylated DNA (mC) attracts DNMT that mediate copying of methylation onto the unmethylated sister strand once DNA replicates.When Azacytidine is incorporated into DNA it binds DNMT irreversibly resulting in its degradation.Thus replication of methylation is prevented resulting in passive demethylation of DNA when DNA replicates. Adapted with permission from Silverman LR, Demakos EP, Peterson BL, et al. 2002. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol, 20:2429–40.
Abbreviations: A, adenosine; aC, azacytidine, ; DNA, deoxyribonucleic acid; DNMT, DNA methyl transferase; G, guanosine ; mC, methylated cytosine; MTI, methyltransferase inhibitors;T, thymidine.
Azacytidine is chemically unstable due to the chemical substitution of nitrogen at position 5. This resulted in it being manufactured as a lyophilized powder. Each single use vial contains 100 mg of Azacytidine and 100 mg of Mannitol that is reconstituted to 25 mg/ml in sterile water. It is stable refrigerated for up to 8 hours but otherwise has to be administered within an hour of preparation.
Azacytidine may be used intravenously or subcutaneously with a similar bioavailability. It is rapidly absorbed subcutaneously achieving a maximal concentration within half an hour, and the concentration achieved is proportional to the dose administered (range 10–75 mg/m2). It has a half-life of 1.5–2.3 hours and is undetectable 8 hours following administration (Rudek et al 2005).
Azacytidine is phosphorylated by uridine-cytidine kinase and is incorporated as a monophosphate into newly synthesized RNA. It is metabolized by cytidine deaminase resulting in the production of 5 azauridine that is mainly eliminated by the kidneys.
Clinical trials with Azacytidine
Three successive Cancer and Leukemia Group B (CALGB) studies culminating in the phase III randomized controlled trial comparing 5 Azacytidine with best supportive care have demonstrated its effectiveness (summarized in Table 4). The dose of Azacytidine in all these studies was 75 mg/m2. In the first study CALGB 8421 the drug was administered intravenously initially and later converted to subcutaneous use, which was continued in CALGB 8921 and 9221. CALGB 8421 was an uncontrolled phase 2 study that enrolled patients with RAEB, RAEBt (excluded CMML) who had not received prior radiotherapy or chemotherapy although prior treatment with low dose cytarabine was permitted. Patients were treated with 75 mg/m2 intravenous (IV) continuous infusion and a dose escalation up to 150mg/m2 was permitted if no response was evident by the second cycle of treatment and no toxicity had occurred. Patients achieving CR were treated with 3 further cycles of treatment. Patients showing hematological improvement (HI) continued the treatment until disease progression or relapse was evident. 43/49 (88%) patients enrolled were evaluable, with 5 (12%) achieving a CR, 11 (25%) PR and 5 (12%) showed an improvement in blood counts. A median of 3.8 cycles of treatment was necessary prior to a response. This important observation has been confirmed in trials with other MTI and implies that at least four cycles of treatment be administered before removing patients from this treatment for non-response. No difference in response (survival or median duration of remission) based on age or FAB subtype was evident.
Table 4.
CALGB Azacytidine trials
| Trial 8421 Phase I | Trial 8921 Phase II | Trial 9221 RCT | ||
|---|---|---|---|---|
| Intravenous | Subcutaneous | Supportive care | Azacytidine | |
| Patient evaluated | 43 | 68 | 92 | 99 |
| CR | 5 (12%) | 8 (12%) | 0 (0%) | 7(7%)* |
| PR | 11 (25%) | 10 (15%) | 0 (0%) | 16 (16%)+ |
| Improved | 5 (12%) | 18 (27%) | 5 (5%) | 37 (37%)+ |
| Total response | 21 (49%) | 36 (53%) | 5 (5%) | 60 (60%)+ |
Note: p=0.01,
p<0.0001
Abbreviations: CALGB, Cancer And Leukemia Group B; CR, complete response; PR, partial response, RCT, randomized controlled trial.
The CALGB 8921 was also a phase 2 study in which a maximal permissible dose of 100 mg/m2 was administered subcutaneously. Sixty-seven patients were enrolled (included CMML). Overall response was 52% with CR observed in 12%, PR in 12% and 28% HI. In the phase three randomized controlled trial, CALGB 9221, (Silverman et al 2002) patients were randomized to 5 Azacytidine or a supportive care arm. Crossover was permitted 4 months later if the disease worsened. The patients received 5-Azacytidine at a dosage of 75 mg/m2/day subcutaneously for 7 days of a 28-day cycle with a minimum of four courses planned. 3 additional cycles were administered to responding patients, whereas those with partial responses continued 5 Azacytidine till complete response or relapse. Overall responses occurred in 60% with 7% achieving CR, 16% PR, and 37% HI (HI). Responses of 47% were seen in the crossover patients compared with 5% HI only in the supportive care arm. Additionally, time to progression to AML was significantly delayed (21 versus 12 months). After 6 months of treatment significantly less (3%) patients treated with Azacytidine transformed to AML when compared to those in the supportive care arm (3% vs 24%, p<0.0001). The median duration of response was 15 months. Median survival for patients on study for 6 months was 18 months (from initial randomisation) compared to 14 months for those who crossed over and 11 months for those who did not crossover. The main side effect was worsening of cytopenias (grade 3–4 hematological toxicity with 43% leucopenia and 58% granulocytopenia, 52% thrombocytopenia) although most patients recovered their blood counts by 4 weeks. Notably no case of bone marrow aplasia following Azacytidine occurred. Infection related to treatment occurred in 20% of patients (Silverman et al 2002). Despite this, the responses were accompanied by an improved quality of life (Kornblith et al 2002). Sequential cytogenetic studies carried out on a subgroup of these patients showed that 8% of patients with clonal cytogenetics achieved cytogenetic remissions. 13% of patients with initial abnormal cytogenetics developed additional clones, however 60% of patients with clonal abnormalities developed HI despite the presence of an abnormal clone. Additional cytogenetic abnormalities developed in those with normal cytogenetics in 36% of patients (the same rate as for MDS being treated with other modalities) (Najfeld et al 2002). This study remains the only randomized controlled study, however a confirmatory trial comparing patients with MDS (IPSS score 1.5 or greater) treated with Azacytidine versus conventional care or best supportive care is ongoing and due to report in 2006.
Cytogenetics and response to Azacytidine
Najfeld et al (2004) reported that patients with normal cytogenetics appeared to have a superior response to Azacytidine. Amongst 26 evaluable patients treated at King’s College Hospital, 7 patients achieved CR. 5 of these had a chromosome 7 abnormality and 2 a trisomy 8 (Raj et al 2005). The response rate for patients with a sole chromosome 7 abnormality was 80% (4/5 patients treated) and patients with monosomy 7 have durable remissions whereas patients with Trisomy 8 experienced an early relapse in their cytopenias. Interestingly this was not accompanied by an increase in BM blasts. These observations suggest that patients with chromosome 7 abnormalities may particularly benefit from Azacytidine therapy.
Clinical response
The clinical response usually follows a typical pattern with an increase in platelet counts being the first response. Increases of platelet counts above 100 × 109/L (usually 200– 700) are followed by an increase in the hemoglobin levels. These responses are usually accompanied by a reduction in BM blasts to less than 5%. Patients often achieve blast reduction to less than 5% even if this is not accompanied by an improvement in blood counts. In 8 patients BM aspirates were carried out at Day 7 of the first cycle of treatment and were often hypocellular with an increased apoptosis (unpublished data). BM biopsies following Azacytidine retain the dyplastic features of MDS despite the normal distribution of erythroid islands and a reduction in blasts. An increase in lymphoid follicles and lymphocytes may occur and a marked eosinophilia is also evident (Figure 4).
Figure 4.
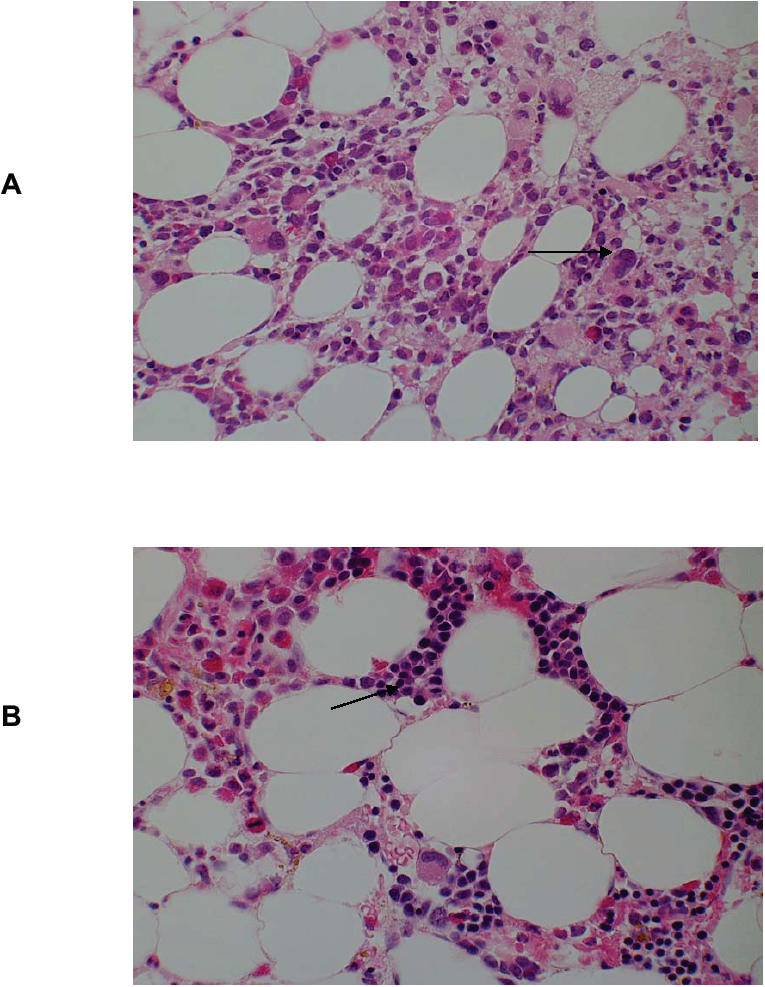
Bone marrow responses to 5-Azacytidine.The bone marrow trephine biopsies from a patient with monosomy 7 were stained with hamatoxylin and eosin.
(A) prior to 5-Azacytidine, showing a normocellular marrow with dysplastic megakaryocytes (arrow) and no erythropoiesis. (B) Post 5-Azacytidine in cytogenetic remission the bone marrow was normocellular and erythroid islands (arrow) were present. Marked eosinophilia post Azacytidine is evident.
Relapses following Azacytidine
Patients who relapse following complete remission are often at a different stage of the disease at relapse (Cheson et al 2000). For example, in our cohort of patients with Trisomy 8, relapses were manifest by neutropenia, followed by thrombocytopenia and then anemia. These relapses were unaccompanied by an increase in BM blast percentage or cytogenetic abnormality, however cytopenias prevented further administration of Azacytidine then resulting in a frank relapse at a median of 4 months after stopping therapy. These cytopenic relapses may represent an earlier stage of the disease ie, refractory cytopenia or a manifestation of dysplastic relapse.
Toxicity
Myelosuppression is the commonest toxicity but is difficult to evaluate due to antecedent cytopenias. Silverman et al (2002) assessed myelosuppression on the relative changes in peripheral blood counts compared with those at study entry (Silverman et al 2002). Grade 3 toxicity was defined as a decrease of 50%–74% whereas grade 4 constituted 75% or greater reduction in peripheral blood counts. Grade 3 or 4 leucopenia occurs in 43%, granulocytopenia in 58%, and thrombocytopenia in 52% of patients. Nausea or vomiting occured in 4% and constipation (either due to the drug or concomitant anti-emetics) occurred in 31% of patients. Injection site erythrema/reaction is common, occurring in 27% in our experience. Rarely gout and acute renal failure may ensue. Serum sickness like illness occurs rarely (Gryn et al 2002). Abnormal liver function tests occur in 7% and generalised weakness, muscle tenderness and lethargy occur in 3% (Azacytidine Investigators brochure)
Mechanism of action in MDS
Although the various mechanisms by which the drug works are known, definitive proof that its effects in MDS are either due to demethylation of several key genes or to its cytotoxic action remain unclear. Whilst CDKN2B may be an important gene in the pathogenesis of progression of early MDS to more aggressive phases of the disease, it is often not hypermethylated in the CD34+ or CD33+ BM cells but appears to be hypermethylated in the lymphocytes of some patients with MDS (John et al 2005). Moreover some of the patients who achieved CR are unmethylated at the CDKN2B promoter suggesting demethylation of this gene is not critical in the responses to Azacytidine in at least a subset of patients (Issa et al 2004; Raj et al 2004).
Previous studies have shown that apoptosis was treatment with Azacytidine. Increased apoptosis correlated increased in early MDS (RA, RARS) but decreased with with best response in patients whereas non-responders did disease progression (RAEB-t) (Parker et al 2000). not show an increase in apoptosis. Therefore in advanced Preliminary results show that patients with high risk MDS MDS, reversal of the low apoptosis by Azacytidine may had low apoptosis prior to treatment that increased following cause disease regression to an earlier stage (Raj et al 2005).
Duration of therapy
The CALGB 9221 scheduled Azacytidine for 4 cycles, if CR was achieved a further 3 cycles were administered. For patients showing PR/HI further courses were allowed till either CR or relapse occurred. However relapses in responses achieved may occur once the drug is stopped. The median duration of response was 15 months from randomization. The current recommendation is to use the drug at the same schedule of dosing indefinitely once CR is achieved. There is limited data on the precise role of maintenance therapy. In our experience patients often develop neutropenia accompanied by reduction in the bone marrow cellularity that necessitates increasing the interval between the courses of treatment. Re-treatment of 22 patients previously treated with decitabine led to one complete remission, 2 PR’s and 7 HI. The median duration of response was 4 months (1–16 months) suggesting that the quality and length of response to re-treatment was poorer than the original response. Thus patients probably need to continue treatment for prolonged periods of time, the dose and scheduling being guided by BM cellularity and neutrophil recovery
Other MTIs in MDS
Azacytidine is the prodrug for decitabine that is 10 times more potent in inhibiting DNMT. Decitabine is incorporated only into DNA whereas 5-Azacytidine is incorporated into both RNA and DNA. In phase II studies, continuous infusion of 40–50 mg/m2/day for 3 days was effective in inducing responses in 15/29 (52%) patients with MDS. Grade III/IV neutropenia and associated complications were common. A multicenter phase II study of decitabine at a dosage of 45 mg/m2 over 3–4 hours daily for 3 days at 6 weekly intervals for 4 courses in patients who had a CR in the first two courses or a maximum of 6 courses for partial responders. Overall responses were higher for the IPSS high-risk group with 64% versus 49% overall. The median duration of response was 31 weeks (Wijermans et al 2000). 19/61 (31%) patients with cytogenetic abnormalities obtained a complete cytogenetic response that was associated with improved survival (Lubbert et al 2001). Low dose decitabine 5 mg/m2/day, 10 mg/m2/day, 15 mg/m2/day, or 20 mg/m2/day intravenously over one hour for 5/7 days a week for 2 weeks was well tolerated with best responses observed in the 15 mg/m2/day (Issa et al 2004). Whilst the criteria for responses varied in the initial trials, reanalysis of the responses of all 177 patients treated in the three European phase II studies (median age 70 years) by the International Working Group (IWG) criteria confirmed the efficacy of decitabine in achieving a CR in 24% of patients and an overall response rate of 49%. The median duration of response was 36 weeks with a median survival of 15 months. 18% of patients progressed to leukemia during treatment. These results are comparable with those for 5-Azacytidine. Age, sex, FAB subgroup, IPSS risk group, lactate dehydrogenase (LDH) and cytogenetics were not predictive of response although patients with high risk cytogenetics according to the IPSS score showed a better overall survival than those with intermediate risk abnormalities. As the IPSS score of patients at diagnosis was not available, no comparison between predicted survival and actual survival following decitabine was possible to see if a particular subgroup benefited from this therapy (Wijermans et al 2005). 61/115 patients treated with decitabine had clonal cytogenetics and 31% of these achieved a cytogenetic response at a median of 3 cycles of decitabine therapy (18 weeks). The median duration of response was 7.5 months (3–15 months). 60% (3/5) of patients with a low IPSS score, 20% (6/30) with intermediate risk and 38% (10/26) with high-risk cytogenetics achieved cytogenetic remissions. These translated into a longer survival if complete cytogenetic remission was achieved (p=0.02) (Lubbert et al 2001). Cytogenetic remissions in response to decitabine preceded genome wide demethylation of genes suggesting that a cytotoxic effect or cell death of the abnormal clone led to remissions (Mund et al 2005). Increased platelet counts after the first course of decitabine therapy occurred in 58% of thrombocytopenic patients and significantly (p<0.0001) predicted overall survival (van den Bosch et al 2004). Myelosuppression leading to fever and sepsis occurred in 20% of patients in the phase II randomized study with the treatment related mortality being 7% (Wijermans et al 2000). Abnormal liver function tests were seen in 16% of patients, grade III cardiovascular problems occurred in 8% of patients and were mostly unrelated to decitabine.
Combination therapy
Although the DNMT inhibitors are effective in myeloid leukemia’s and MDS, they are limited by low response rates (23% CR and PR) and a short median duration of response (15 months for Azacytidine) (Silverman et al 2002).
Combination chemotherapy has led to improved CR rates in AML. Similarly, targeting complementary biological pathways with synergistic drugs, could improve the response rates and duration with MTI’s.
Other therapies being developed for MDS include, histone deacetylase inhibitors (HDACi), tumour necrosis factor (TNF) antagonists including etanercept and infliximab, farnesyl transferase inhibitors (tipifarnib and lonafarnib) arsenic trioxide, retinoids, lenalidomide, signal transduction inhibitors bevacizumab, SCIO469 and PTK787, and the glutathione S-transferase PI inhibitor TLK199. The combination of Azacytidine with HDACi is the furthest in clinical trials and is discussed below.
HDAC inhibitors
DNA is packaged in chromatin that is made up of an octamer of histones. Acetylation of lysine residues on histone tails adds a net negative charge that results in chromatin being in an open configuration (euchromatin) whereas removal of acetyl groups by histone deacetylases (HDAC‘s) results in chromatin being held in a closed configuration (heterochromatin), preventing transcription of genes. Methylation of gene promoters acts in conjunction with histone deacetylation in the silencing of genes. Methylated promoters recruit transcriptional repression complexes that include HDAC’s via specific methyl-binding proteins leading to heterochromatin being associated with that gene. Therefore inducing re-expression of epigenetically silenced genes could be achieved by either demethylation of DNA or acetylation of chromatin. In vitro studies have shown that HDACi are unable to reactivate the expression of heavily methylated genes. However the addition of HDACi following exposure to DNMT inhibitors leads to synergistic reactivation of methylated genes (Cameron et al 1999). There are five main classes of HDAC inhibitors (reviewed by Gore in Mufti et al 2003). Of these, Valproic acid and MS275 in combination with MTI’s decitabine and Azacytidine respectively are in phase I clinical trials (Gore 2005). Azacytidine combined sequentially with sodium phenylbutyrate (a HDACi) when used at 50 mg/m2 for 10 days (cohort A n=8), or 14 days (cohort B n=3) or 25 mg/m2 for 14 days (n=5) followed by 375 mg/kg daily continuous infusion of phenylbutyrate for 7 days led to clinical responses in all three cohorts. Cohort B experienced dose limiting toxicity of prolonged myelosuppression (greater than 14 days) in 2/3 patients however even this cohort had HI (1 HI-platelet and neutrophil major, and 1 HI-platelet major). Cohort A experienced CR in 3/8 and PR in 1/8 patients whereas cohort C developed HI-neutrophil major in 2 patients and HI-platelet major in 1 patient (Gore et al 2004). This combination appears promising with doses of Azacytidine equivalent to or lower than that used in the CALGB trials. Further studies are necessary to confirm whether the addition of HDACi result in increased response rates and/or prolongation of responses.
Future direction
Azacytidine induces prolonged complete remissions in up to 25% of patients. Overall, half the patients treated derive a clinically meaningful response in terms of transfusion independence and or blast cell reduction. This is associated with an improved quality of life. Patients with monosomy 7 are particularly responsive to treatment but this need confirmation in larger trials to enable selection of patients who would benefit maximally. Despite its universal demethylating and cytotoxic effects some patients do not respond. Factors responsible for this may include inadequate plasma levels of drug. Additionally some patients achieving CR relapse with cytopenias and identifying these patients and the reasons for this will enable modifications in the dosing regime and rational synergistic combination therapy with drugs such as HDACi. The identification of target genes and mechanisms by which the drug act are important as a means of monitoring responses to this therapy. Azacytidine as salvage therapy for relapsed AML with chromosome 7 abnormalities, maintenance chemotherapy for AML and in pre-transplant conditioning regimes are areas that are being explored in clinical trials. Epigenetic therapies such as Azacytidine have ushered in a new era in the management of MDS. We are at the beginning of understanding how to optimally use this group of agents in order to achieve maximal benefit.
References
- Ahuja N, Li Q, Mohan AL, et al. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–94. [PubMed] [Google Scholar]
- Amanullah A, Hoffman B, Liebermann DA. Deregulated E2F-1 blocks terminal differentiation and loss of leukemogenicity of M1 myeloblastic leukemia cells without abrogating induction of p15(INK4B) and p16(INK4A) Blood. 2000;96:475–82. [PubMed] [Google Scholar]
- Antequera F, Boyes J, Bird A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. 1990;62:503–14. doi: 10.1016/0092-8674(90)90015-7. [DOI] [PubMed] [Google Scholar]
- Aul C, Germing U, Gattermann N, et al. Increasing incidence of myelodysplastic syndromes: real or fictitious? Leuk Res. 1998;22:93–100. doi: 10.1016/s0145-2126(97)00089-1. [DOI] [PubMed] [Google Scholar]
- Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–99. [PubMed] [Google Scholar]
- Bhagwat AS, Roberts RJ. Genetic analysis of the 5-azacytidine sensitivity of Escherichia coli K-12. J Bacteriol. 1987;169:1537–46. doi: 10.1128/jb.169.4.1537-1546.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Bird AP, Taggart MH, Smith BA. Methylated and unmethylated DNA compartments in the sea urchin genome. Cell. 1979;17:889–901. doi: 10.1016/0092-8674(79)90329-5. [DOI] [PubMed] [Google Scholar]
- Cameron EE, Bachman KE, Myohanen S, et al. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–7. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- Cheson BD, Bennett JM, Kantarjian H, et al. Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood. 2000;96:3671–4. [PubMed] [Google Scholar]
- Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Methylation of p15INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2003;17:1813–19. doi: 10.1038/sj.leu.2403054. [DOI] [PubMed] [Google Scholar]
- Christman JK. 5-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–95. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- Corn PG, Smith BD, Ruckdeschel ES, et al. E-cadherin expression is silenced by 5' CpG island methylation in acute leukemia. Clin Cancer Res. 2000;6:4243–8. [PubMed] [Google Scholar]
- Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2'-deoxycytidine. J Biol Chem. 1982;257:2041–8. [PubMed] [Google Scholar]
- Dao MA, Taylor N, Nolta JA. Reduction in levels of the cyclindependent kinase inhibitor p27(kip-1) coupled with transforming growth factor beta neutralization induces cell-cycle entry and increases retroviral transduction of primitive human hematopoietic cells. Proc Natl Acad Sci U S A. 1998;95:13006–11. doi: 10.1073/pnas.95.22.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado S, Gomez M, Bird A, et al. Initiation of DNA replication at CpG islands in mammalian chromosomes. Embo J. 1998;17:2426–35. doi: 10.1093/emboj/17.8.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhodapkar M, Grill J, Lust JA. Abnormal regional hypermethylation of the calcitonin gene in myelodysplastic syndromes. Leuk Res. 1995;19:719–26. doi: 10.1016/0145-2126(95)00019-k. [DOI] [PubMed] [Google Scholar]
- Drexler HG. Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia. 1998;12:845–59. doi: 10.1038/sj.leu.2401043. [DOI] [PubMed] [Google Scholar]
- Esteller M, Corn PG, Baylin SB, et al. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–9. [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–31. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MJ, Martinez-Delgado B, Cebrian A, et al. Different incidence and pattern of p15INK4b and p16INK4a promoter region hypermethylation in Hodgkin’s and CD30-Positive non-Hodgkin’s lymphomas. Am J Pathol. 2002;161:1007–13. doi: 10.1016/S0002-9440(10)64261-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–82. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Germing U, Gattermann N, Strupp C, et al. Validation of the WHO proposals for a new classification of primary myelodysplastic syndromes: a retrospective analysis of 1600 patients. Leuk Res. 2000;24:983–92. doi: 10.1016/s0145-2126(00)00088-6. [DOI] [PubMed] [Google Scholar]
- Glover AB, Leyland-Jones BR, Chun HG, et al. Azacitidine: 10 years later. Cancer Treat Rep. 1987;71:737–46. [PubMed] [Google Scholar]
- Gore SD. Combination therapy with DNA methyltransferase inhibitors in hematologic malignancies. Nat Clin Pract Oncol. 2005;2(Suppl 1):S30–5. doi: 10.1038/ncponc0346. [DOI] [PubMed] [Google Scholar]
- Gore SD, Baylin SB, Dauses T, et al. Changes in promoter methylation and gene expression in patients with MDS and MDS-AML treated with 5-Azacitidine and sodium phenylbutyrate [abstract] Blood. 2004;104(11):469. [Google Scholar]
- Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88. [PubMed] [Google Scholar]
- Gryn J, Zeigler ZR, Shadduck RK, et al. Treatment of myelodysplastic syndromes with 5-azacytidine. Leuk Res. 2002;26:893–7. doi: 10.1016/s0145-2126(02)00028-0. [DOI] [PubMed] [Google Scholar]
- Haaf T. The effects of 5-azacytidine and 5-azadeoxycytidine on chromosome structure and function: Implications for methylation-associated cellular processes. Pharmacol Ther. 1995;65:19–46. doi: 10.1016/0163-7258(94)00053-6. [DOI] [PubMed] [Google Scholar]
- Heaney ML, Golde DW. Myelodysplasia. N Engl J Med. 1999;340:1649–60. doi: 10.1056/NEJM199905273402107. [DOI] [PubMed] [Google Scholar]
- Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. PNAS. 1994;91:9700–4. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AY, Pagliuca A, Kenyon M, et al. Reduced-intensity allogeneic hematopoietic stem cell transplantation for myelodysplastic syndrome and acute myeloid leukemia with multilineage dysplasia using fludarabine, busulphan, and alemtuzumab (FBC) conditioning. Blood. 2004;104:1616–23. doi: 10.1182/blood-2003-12-4207. [DOI] [PubMed] [Google Scholar]
- Issa JP, Garcia-Manero G, Giles FJ, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–40. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Johan MF, Goodeve AC, Bowen DT, et al. Promoter methylation of RASSFIA, SHP1 and SOCS1 genes in acute myeloid leukaemia (AML) and myelodysplastic syndromes (MDS) [abstract] Blood. 2004;104:2999. [Google Scholar]
- John A, Raj K, Mufti GJ, et al. Lymphocytes but not CD34+ or CD33+ myeloid cells, of MDS have a hypermethylated CDKN2B promoter. Br J Haematol. 2005;129(Suppl 1):6. [Google Scholar]
- Jones PA, Taylor SM. Hemimethylated duplex DNAs prepared from 5-azacytidine-treated cells. Nucleic Acids Res. 1981;9:2933–47. doi: 10.1093/nar/9.12.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Taylor SM, Wilson VL. Inhibition of DNA methylation by 5-azacytidine. Recent Results Cancer Res. 1983;84:202–11. doi: 10.1007/978-3-642-81947-6_15. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–91. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Kaminskas E, Farrell AT, Wang YC, et al. FDA drug approval summary: Azacitidine (5-azacytidine, VidazaTM) for injectable suspension. Oncologist. 2005;10:176–82. doi: 10.1634/theoncologist.10-3-176. [DOI] [PubMed] [Google Scholar]
- Kornblith AB, Herndon JE, 2nd, Silverman LR, et al. Impact of azacytidine on the quality of life of patients with myelodysplastic syndrome treated in a randomized phase III trial: a Cancer and Leukemia Group B study. J Clin Oncol. 2002;20:2441–52. doi: 10.1200/JCO.2002.04.044. [DOI] [PubMed] [Google Scholar]
- Li Q, Kopecky KJ, Mohan A, et al. Estrogen receptor methylation is associated with improved survival in adult acute myeloid leukemia. Clin Cancer Res. 1999;5:1077–84. [PubMed] [Google Scholar]
- Lubbert M, Wijermans P, Kunzmann R, et al. Cytogenetic responses in high-risk myelodysplastic syndrome following low-dose treatment with the DNA methylation inhibitor 5-aza-2'-deoxycytidine. Br J Haematol. 2001;114:349–57. doi: 10.1046/j.1365-2141.2001.02933.x. [DOI] [PubMed] [Google Scholar]
- Melki JR, Vincent PC, Clark SJ. Concurrent DNA hypermethylation of multiple genes in acute myeloid leukemia. Cancer Res. 1999;59:3730–40. [PubMed] [Google Scholar]
- Mizuno S, Chijiwa T, Okamura T, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97:1172–9. doi: 10.1182/blood.v97.5.1172. [DOI] [PubMed] [Google Scholar]
- Mufti G, List AF, Gore SD, et al. Myelodysplastic syndrome. Hematology. 2003;2003:176–99. doi: 10.1182/asheducation-2003.1.176. [DOI] [PubMed] [Google Scholar]
- Mund C, Hackanson B, Stresemann C, et al. Characterization of DNA demethylation effects induced by 5-Aza-2'-deoxycytidine in patients with myelodysplastic syndrome. Cancer Res. 2005;65:7086–90. doi: 10.1158/0008-5472.CAN-05-0695. [DOI] [PubMed] [Google Scholar]
- Najfeld V, Scalise A, Odchimer-Reissig R, et al. The modulating effect of Azacitidine (Aza C) on the cytogenetically tracked MDS clone impact survival [abstract] Blood. 2004;104:1429. [Google Scholar]
- Najfeld V, Silverman LR, Scalise A, et al. Modulation of the cytogenetically abnormal myelodysplastic (MDS) clone by Azacitidine (Aza C) [abstract] Blood. 2002;100:97a. [Google Scholar]
- Nakamaki T, Bartram C, Seriu T, et al. Molecular analysis of the cyclin-dependent kinase inhibitor genes, p15, p16, p18 and p19 in the myelodysplastic syndromes. Leuk Res. 1997;21:235–40. doi: 10.1016/s0145-2126(96)00115-4. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Parker JE, Mufti GJ, Rasool F, et al. The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood. 2000;96:3932–8. [PubMed] [Google Scholar]
- Quesnel B, Guillerm G, Vereecque R, et al. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood. 1998;91:2985–90. [PubMed] [Google Scholar]
- Raj K, John A, Ho A, et al. 5-Azacytidine in the treatment of high risk myelodysplastic syndromes: clinical response can occur without demethylation of the CDKN2B, CDKN1A or TP73 gene promoters [abstract] Blood. 2004;104:2369. [Google Scholar]
- Raj KK, John AM, Ho A, et al. Early and sustained response to azacytidine in high-risk MDS patients with monosomy 7 correlates with increased apoptosis and not CDKN2B demethylation [abstract] Blood. 2005;106:2530. [Google Scholar]
- Rudek MA, Zhao M, He P, et al. Pharmacokinetics of 5-Azacitidine given with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J Clin Oncol. 2005;23:3906–11. doi: 10.1200/JCO.2005.07.450. [DOI] [PubMed] [Google Scholar]
- Rush LJ, Dai Z, Smiraglia DJ, et al. Novel methylation targets in de novo acute myeloid leukemia with prevalence of chromosome 11 loci. Blood. 2001;97:3226–33. doi: 10.1182/blood.v97.10.3226. [DOI] [PubMed] [Google Scholar]
- Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–40. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- Teofili L, Martini M, Di Mario A, et al. Expression of p15(ink4b) gene during megakaryocytic differentiation of normal and myelodysplastic hematopoietic progenitors. Blood. 2001;98:495–7. doi: 10.1182/blood.v98.2.495. [DOI] [PubMed] [Google Scholar]
- Teofili L, Morosetti R, Martini M, et al. Expression of cyclindependent kinase inhibitor p15(INK4B) during normal and leukemic myeloid differentiation. Exp Hematol. 2000;28:519–26. doi: 10.1016/s0301-472x(00)00139-9. [DOI] [PubMed] [Google Scholar]
- Tessema M, Langer F, Dingemann J, et al. Aberrant methylation and impaired expression of the p15(INK4b) cell cycle regulatory gene in chronic myelomonocytic leukemia (CMML) Leukemia. 2003;17:910–18. doi: 10.1038/sj.leu.2402891. [DOI] [PubMed] [Google Scholar]
- Tien HF, Tang JH, Tsay W, et al. Methylation of the p15(INK4B) gene in myelodysplastic syndrome: it can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation. Br J Haematol. 2001;112:148–54. doi: 10.1046/j.1365-2141.2001.02496.x. [DOI] [PubMed] [Google Scholar]
- Toyota M, Kopecky KJ, Toyota MO, et al. Methylation profiling in acute myeloid leukemia. Blood. 2001;97:2823–9. doi: 10.1182/blood.v97.9.2823. [DOI] [PubMed] [Google Scholar]
- Uchida T, Kinoshita T, Hotta T, et al. High-risk myelodysplastic syndromes and hypermethylation of the p15Ink4B gene. Leuk Lymphoma. 1998;32:9–18. doi: 10.3109/10428199809059242. [DOI] [PubMed] [Google Scholar]
- van den Bosch J, Lubbert M, Verhoef G, et al. The effects of 5-aza-2'-deoxycytidine (Decitabine) on the platelet count in patients with intermediate and high-risk myelodysplastic syndromes. Leuk Res. 2004;28:785–90. doi: 10.1016/j.leukres.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Vardiman JW. The World Health Organisation (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- Wheeler JM. Epigenetics, mismatch repair genes and colorectal cancer. Ann R Coll Surg Engl. 2005;87:15–20. doi: 10.1308/1478708051423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijermans P, Lubbert M, Verhoef G, et al. Low-dose 5-aza-2'-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956–62. doi: 10.1200/JCO.2000.18.5.956. [DOI] [PubMed] [Google Scholar]
- Wijermans PW, Lubbert M, Verhoef G, et al. An epigenetic approach to the treatment of advanced MDS; the experience with the DNA demethylating agent 5-aza-2'-deoxycytidine (decitabine) in 177 patients. Ann Hematol. 2005;84(Suppl 13):9–17. doi: 10.1007/s00277-005-0012-1. [DOI] [PubMed] [Google Scholar]