

Short abstract
BLAST-based computational heuristics were used to identify large and recent segmental duplications in the mouse genome sequence. Here a database of recently duplicated regions of the mouse genome is presented.
Abstract
Background
The high quality of the mouse genome draft sequence and its associated annotations are an invaluable biological resource. Identifying recent duplications in the mouse genome, especially in regions containing genes, may highlight important events in recent murine evolution. In addition, detecting recent sequence duplications can reveal potentially problematic regions of the genome assembly. We use BLAST-based computational heuristics to identify large (≥ 5 kb) and recent (≥ 90% sequence identity) segmental duplications in the mouse genome sequence. Here we present a database of recently duplicated regions of the mouse genome found in the mouse genome sequencing consortium (MGSC) February 2002 and February 2003 assemblies.
Results
We determined that 33.6 Mb of 2,695 Mb (1.2%) of sequence from the February 2003 mouse genome sequence assembly is involved in recent segmental duplications, which is less than that observed in the human genome (around 3.5-5%). From this dataset, 8.9 Mb (26%) of the duplication content consisted of 'unmapped' chromosome sequence. Moreover, we suspect that an additional 18.5 Mb of sequence is involved in duplication artifacts arising from sequence misassignment errors in this genome assembly. By searching for genes that are located within these regions, we identified 675 genes that mapped to duplicated regions of the mouse genome. Sixteen of these genes appear to have been duplicated independently in the human genome. From our dataset we further characterized a 42 kb recent segmental duplication of Mater, a maternal-effect gene essential for embryogenesis in mice.
Conclusion
Our results provide an initial analysis of the recently duplicated sequence and gene content of the mouse genome. Many of these duplicated loci, as well as regions identified to be involved in potential sequence misassignment errors, will require further mapping and sequencing to achieve accuracy. A Genome Browser database was set up to display the identified duplication content presented in this work. This data will also be relevant to the growing number of investigators who use the draft genome sequence for experimental design and analysis.
Background
The evolutionary trajectory of duplicated genes has been an active area of investigation since gene duplication was first recognized as an important force in species evolution [1]. The availability of new sequence data and analyses has challenged the hypothesis suggesting most duplicated genes are destined to lose their function and become pseudogenes, with a few exceptions establishing new biological roles (reviewed in [2]). It is believed that the occurrence of gene duplication would result in relaxed selection of redundant copies permitting genes to evolve specialized sub-functions [3,4]. Moreover, nearly identical genomic regions provide important substrates for chromosomal rearrangements that permit rapid evolutionary changes to occur in a short period of time [5].
An estimated 3.5-5% of the human genome has undergone recent duplication [6-9], and these segmental duplications (also termed duplicons or low-copy repeats) are found to be hot spots, or predisposition sites, for the occurrence of nonallelic homologous recombination. This recombination can lead to genomic mutations such as deletion, duplication, inversion, or translocation, resulting in human disease [10]. Many mouse strains with chromosomal aberrations are known [11], and it remains to be seen whether segmental duplications have a role in any of these genomic mutations.
A high-quality draft genome sequence not only makes it possible to expand the known set of duplicate genes, but also reveals their genomic context. This genomic context contains the regulatory and structural elements responsible for gene expression that need to be interrogated for a better understanding of the mechanisms and consequences of gene duplication. Furthermore, an accurate and well-annotated mouse genome is an essential resource for many in the biomedical research community, especially those who use the sequence to design and interpret transgenic, mutagenesis, microarray, and proteomic studies.
Several lines of evidence show that the whole-genome shotgun (WGS) approach yielded a high-quality draft sequence that covers roughly 96% of the euchromatic genome excluding chromosome Y (a female C57BL/6J mouse was used in the sequencing project) [12]. The WGS sequence reads were assembled into sequence contigs using sequence-assembly programs to produce the February 2002 MGSCv3 working draft [12,13]. The newly released February 2003 assembly was a hybrid assembly comprising 705 megabases (Mb) of finished bacterial artificial chromosome (BAC) sequences incorporated into the MGSCv3 assembly.
We previously analyzed several versions of the human genome draft assemblies (NCBI Builds 28, 29, and 30) [9], and found substantial potential genome assembly errors in all builds, including approximately 40 Mb of sequence in Build 30. These assembly errors probably arose from difficulties in merging finished sequence or from incorrectly assigning sequence contigs into the genome assembly. In such cases, completely identical or nearly identical sequences (due to allelic differences or sequencing errors) would be present at distinct regions in the genome sequence. These sequence misassignment errors would yield near-perfect duplication artifacts, detected as having extremely high sequence identities (exceeding 99.5% and over 5 kilobase (kb) in length), in genome assemblies. However, a small subset of such results could represent duplications that arose from very recent evolutionary events and will require further experimental analysis.
A number of web-based resources, specifically those provided by the National Center for Biotechnology Information (NCBI), Ensembl (at the European Bioinformatics Institute and Sanger Centre), and the University of California Santa Cruz (UCSC), make the genome sequence and associated annotations readily accessible. Because of the success of the mouse genome sequencing consortium (MSGC), investigators worldwide are utilizing the draft 'as is' in both medical and evolutionary studies. In this paper we show that even though the genome assembly is still in draft form, an initial analysis of the sequence can reveal novel genomic duplications and demarcate regions of the genome that require additional examination.
Results and discussion
We performed a search for all recent segmental duplications that were larger than 5 kb in size and showed greater than 90% sequence identity from both the February 2002 (numerical results for February 2002 assembly are presented at our web site [14]) and the February 2003 mouse genome sequence assemblies [15]. Our method was based on pairwise (mega-) BLAST2 [16] sequence comparisons between entire chromosome sequences. From our analysis of the February 2003 assembly, a total of 33.6 Mb (1.2%) of the genome sequence (2,695 Mb) was found to be involved in recent segmental duplications (Table 1) and 8.9 Mb of this sequence was unmapped data (found in the unmapped chromosome sequence). On the basis of the 20 mapped chromosomes, more than 712 distinct intrachromosomal segmental duplications, comprising 19.9 Mb of sequence (Figure 1), and 475 distinct interchromosomal duplications, comprising 7.1 Mb of sequence, were identified. We also found that 57% of the duplications were in tandem, which we defined as two related intrachromosomal duplicons located within 200 kb of one another.
Table 1.
Recent segmental duplication in the mouse genome
| Chromosome | Chromosome length | Intrachromosomal duplication | % | Interchromosomal duplication | % | Total | % |
| 1 | 195,869,683 | 1,392,568 | 0.7 | 238,739 | 0.1 | 1,552,908 | 0.8 |
| 2 | 181,423,755 | 1,106,879 | 0.6 | 173,602 | 0.1 | 1,184,304 | 0.7 |
| 3 | 160,674,399 | 790,500 | 0.5 | 158,011 | 0.1 | 948,511 | 0.6 |
| 4 | 152,921,959 | 1,743,027 | 1.1 | 647,795 | 0.4 | 1,921,970 | 1.3 |
| 5 | 149,719,773 | 1,102,772 | 0.7 | 761,950 | 0.5 | 1,560,683 | 1.0 |
| 6 | 149,950,539 | 2,042,585 | 1.4 | 562,415 | 0.4 | 2,339,839 | 1.6 |
| 7 | 134,401,573 | 1,655,438 | 1.2 | 713,287 | 0.5 | 2,038,845 | 1.5 |
| 8 | 128,923,138 | 738,203 | 0.6 | 331,970 | 0.3 | 1,005,575 | 0.8 |
| 9 | 124,467,299 | 437,352 | 0.4 | 188,427 | 0.2 | 623,089 | 0.5 |
| 10 | 130,738,012 | 345,768 | 0.3 | 258,429 | 0.2 | 604,197 | 0.5 |
| 11 | 122,862,689 | 900,355 | 0.7 | 127,774 | 0.1 | 1,012,479 | 0.8 |
| 12 | 114,462,600 | 1,139,786 | 1.0 | 374,365 | 0.3 | 1,404,279 | 1.2 |
| 13 | 116,242,670 | 855,835 | 0.7 | 547,462 | 0.5 | 1,349,974 | 1.2 |
| 14 | 115,844,145 | 450,161 | 0.4 | 451,782 | 0.4 | 748,465 | 0.6 |
| 15 | 104,111,694 | 443,805 | 0.4 | 43,937 | 0.0 | 487,742 | 0.5 |
| 16 | 98,986,639 | 389,255 | 0.4 | 67,290 | 0.1 | 456,545 | 0.5 |
| 17 | 93,529,596 | 1,329,664 | 1.4 | 660,440 | 0.7 | 1,760,982 | 1.9 |
| 18 | 91,041,441 | 162,916 | 0.2 | 58,996 | 0.1 | 210,422 | 0.2 |
| 19 | 61,093,376 | 328,909 | 0.5 | 193,387 | 0.3 | 479,687 | 0.8 |
| X | 149,996,094 | 2,592,361 | 1.7 | 574,950 | 0.4 | 3,018,682 | 2.0 |
| chrUn* | 117,911,829 | 6,049,538 | 5.1 | 5,710,057 | 4.8 | 8,885,604 | 7.5 |
| Total | 2,695,172,903 | 25,997,677 | 1.0 | 12,845,065 | 0.5 | 33,594,782 | 1.2 |
The analysis is based on the February 2003 mouse genome assembly. *chrUn, unmapped chromosome sequence.
Figure 1.

Intrachromosomal segmental duplications identified in the mouse genome (chromosomes 1-X; results are based on the February 2003 assembly). Each line represents a duplicated module and connects a paralogous duplicon pair. Red, 99-100% sequence identity; purple, 96-98%; green, 93-95%; and blue, 90-92%. Correspondences to chromosome ideograms (obtained from Ensembl) are only crude. Graphics were produced using GenomePixelizer [34].
Duplications can be found in all chromosomes analyzed, with chromosomes 6, 7, 17, and X having the highest, and chromosome 18 having the least, duplicated content (Table 1, Figure 1). Substantial amounts (8.9 Mb) of the duplicated content are found in the unmapped chromosome (ChrUn) sequence, suggesting that the correct chromosomal assignment of these segments remains a major assembly challenge. It is possible that small subsets of these duplications are due to chimeric reads and other sequencing artifacts and thus should not be part of the finished genome sequence. On the other hand, these unmapped duplicated sequences represent true duplications that have been excluded from the assembly. One example of this occurs with a member of the mouse Bcl2 family of apoptosis regulators, Bcl2a1. Bcl2a1 contains four highly similar genes (> 97% identical at the nucleotide level) that have been mapped together on chromosome 9 of the C57BL/6 and 129SV genomes [17,18]. Currently, the Bcl2a1 genes are not assembled on the mapped chromosome and are found in three distinct unmapped contigs. In the human genome only one copy of BCL2A1 is found, although a recent, independent 8.5 kb tandem duplication containing the last exon of BCL2A1 has occurred, forming a novel BCL2A1-related transcript (AF249277). An example of a region that has changed between assemblies is the Amy2 locus. Amy2 is known to vary in copy number between inbred strains of mice [19]. In the February 2002 assembly, only one copy of the Amy2 gene resided on chromosome 3 in addition to a second copy found on a large 10 kb unmapped contig. In addition, partial high identity matches (> 95%) to four distinct unmapped contigs were found (note that these partial copies were not detected in our analysis as they are less than 5 kb long). In the February 2003 assembly, six Amy2 genes exist, which is close to the five Amy2-like genes that were detected in the genome of strain A/J mice using quantitative densitometry of Southern blots [20]. It is, however, important to note that a gap, not bridged by a clone, still exists between the Amy2 locus and the Amy1 gene, and so the copy number in the C57BL/6J genome assembly may still vary.
We analyzed the distribution of segmental duplication content by sorting the duplications into six different sequence-similarity categories: 90-92%, 92-94%, 94-96%, 96-98%, 98-99.5%, and 99.5-100%, for both the February 2002 and 2003 assembly builds (Table 2). The amount of duplication content appears to be unevenly distributed across these categories, with a distinct rise in the 94-96% category. This might suggest recent duplicative events in the mouse genome have not occurred at a steady rate. However, it is unclear at this point how these results were affected by the draft status of the genome assemblies. Between the 2002 and the 2003 assembly builds we found that the amount of duplication content is nearly the same within each percent category except for the 99.5-100% category, which contained 4.8 Mb of sequence in 2002 and 18.5 Mb in 2003 (Table 2). Furthermore, we determined that the majority (88%) of the duplicated sequence in the 99.5-100% category occurred intrachromosomally, within 200 kb of each other. Using the assembly component tables (provided by UCSC [21]), which contain information about the underlying makeup of the February 2003 genome assembly (shotgun-assembled scaffolds and BAC sequences), we found that 215/216 (99.5%) of these duplications involved a BAC sequence. Hence, we suspect that the large increase in near-identical duplications could be the result of sequence misassignment errors arising from the inherent difficulty of merging finished BAC sequence with shotgun sequence contigs.
Table 2.
Comparison between genome assemblies
| Sequence identity level | February 2002 assembly* | February 2003 assembly |
| Duplication content (bp) | ||
| 90-92% | 4,966,470 | 3,543,429 |
| 92-94% | 15,685,840 | 13,981,642 |
| 94-96% | 17,533,730 | 17,970,287 |
| 96-98% | 11,539,392 | 11,731,958 |
| 98-99.5% | 5,865,024 | 5,487,899 |
| †Potential sequence misassignment error detected (bp) | ||
| 99.5-100% | 4,832,594 | 18,456,096 |
The comparison is of duplication content by sequence identity and potential sequence misassignment errors between the February 2002 (MGSCv3) and February 2003 (a hybrid assembly of MGSCv3 with 705 Mb finished BAC sequence) genome assemblies. *Analysis of the duplication content for February 2002 assembly can be found at [14].†Sequences detected to show extremely high percent identity duplications are likely to be genome assembly artifacts and were not included in the duplication content shown in Table 1.
We previously observed that the human genome sequence assembled by Celera's WGS method [22] showed poor quality in regions with near-identical segmental duplications [23]. To assess the finishing status of duplicated regions in the WGS mouse genome assembly (February 2002 MGSCv3 assembly), we calculated the amount of unfinished sequence (regions with gaps or Ns) within the immediate neighborhood (20 kb) of each duplicon (the unmapped chromosome sequence was excluded from this analysis). We observed substantially higher amounts of unfinished sequence (number of Ns) in these regions. Whereas 8.0% of the assembly is comprised of Ns, regions harboring duplications contain an average of 12.2%. This average rises to 16.6% for duplications with more than 98% sequence identity (statistics can be obtained from our website [14]). This suggests that the WGS assembler had difficulty assembling regions containing recent sequence duplication and that these regions are good candidates for finishing using clone resources.
Using the NCBI Refseq and Ensembl mouse gene annotation, we identified 675 genes that mapped to duplicated regions of the mouse genome (a full list of genes can be obtained from our website [14]); 414 of these genes were found to be fully contained within a segmental duplication, thus representing the best candidates for whole-gene duplication. While it is likely that some of these duplicate copies have become pseudogenes, others may have evolved specialized functions [3]. Moreover, we sought to use the identified gene sequences, which were expressed sequence tags (ESTs) and/or cDNAs, as experimentally derived resources to help validate the genomic duplication content presented in this study. We aligned duplicated gene sequences to each genomic region using UCSC BLAT [17] and determined their percent identity matches. Unambiguous gene-to-genomic identity matches were established for all 128 gene pairs we examined. Each gene sequence was mapped to their respective genomic region with at least 99.1% identity (examples are shown in Table 3; a full table is available at [14]). We also examined the identified duplicated genes using their InterPro protein-domain classification present in 608 Ensembl genes to see whether specific kinds of genes or protein domains have been preferentially duplicated. We found that genes containing protein domains related to signal transduction (rhodopsin-like G-protein-coupled receptor superfamily), olfaction (olfactory receptors, vomeronasal receptors) immunity (immunoglobulin/MHC, serine protease), and drug metabolism (cytochrome P450) are significantly enriched (by at least threefold) (Table 4).
Table 3.
Examples of recent mouse gene duplications
| Locus1* | Gene | Percent identity† | Annotation | Locus2* | Gene | Percent identity† | Annotation | Duplication % identity‡ |
| 1 F | NM_009888 | 99.6 | Cfh (Complement component factor h) | 1 F | M29010 | 99.0 | Complement factor H-related protein mRNA | 97.1 |
| 3 G1 | NM_009669 | 100 | Amy2 (Amylase 2, pancreatic) | 3 G1 | M11896 | 99.6 | Pancreatic amylase B-1 | 97.6 |
| 5 E2 | NM_053184 | 99.9 | Ugt2a1 (UDP glycosyltransferase 2 A1) | 5 E2 | BF144793 | 99.6 | cDNA clone IMAGE:4021939 | 95.7 |
| 5 E2 | NM_009467 | 100 | Ugt2b5(UDP-glucuronosyl-transferase 2b5) | 5 E2 | NM_053215 | 100 | RIKEN cDNA 0610033E06 gene | 93.3 |
| 5 E4 | NM_008620 | 99.9 | Mpa2 (macrophage activation 2) | 5 E4 | BC007143 | 99.5 | Similar to macrophage activation 2 | 90.6 |
| 5 G1 | NM_029693 | 100 | RIKEN cDNA 1700123K08 | 7 B2 | NM_027702 | 100 | RIKEN cDNA 4933421I07 gene | 91.1 |
| 6 C1 | NM_053238 | 100 | V1rc8 (Vomeronasal 1 receptor, C8) | 6 C1 | NM_053239 | 99.7 | V1rc9 (Vomeronasal 1 receptor, C9) | 95.1 |
| 6 D1 | NM_011467 | 99.9 | Spr (sepiapterin reductase) | 6 D1 | BE862957 | 99.5 | EST sequence | 95.8 |
| 6 F1 | AI505330 | 100 | Similar to initiation factor eIF-4AI | 6 F1 | AI503670 | 99.8 | Similar to initiation factor eIF-4AI | 98.9 |
| 6 F2 | NM_008646 | 99.9 | Mug2 (Murinoglobulin 2) | 6 F2 | NM_008645 | 99.9 | Mug1 (Murinoglobulin 1) | 94.6 |
| 6 F3 | NM_020257 | 99.8 | Dcl1 (c-type lectin 1) | 6 F3 | NM_027562 | 99.9 | 4632413B12Rik (C-lectin related protein) | 90.8 |
| 6 F3 | NM_008463 | 99.7 | Klra5 (Killer cell lectin-like receptor, A5) | 6 F3 | NM_008464 | 99.5 | Klra6 (Killer cell lectin-like receptor, A6) | 90.0 |
| 6 F3 | NM_010649 | 99.8 | Klra4 (Killer cell lectin-like receptor A4) | 6 F3 | NM_016659 | 99.8 | Klra1 (Killer cell lectin-like receptor A1) | 91.4 |
| 6 F3 | NM_010737 | 99.8 | Klrb1b (Killer cell lectin-like receptor 1b) | 6 F3 | NM_008527 | 99.9 | Klrb1c (Killer cell lectin-like receptor 1c) | 90.8 |
| 7 A2 | NM_011860 | 100 | Mater (Maternal effect gene) | 7 A1 | AK016782 | 100 | Similar to Mater protein | 96.6 |
| 7 B1 | NM_032541 | 100 | Hamp hepcidin antimicrobial peptide | 7 B1 | AK007975 | 99.8 | Prohepcidin homolog | 92.8 |
| 7 B2 | NM_010115 | 99 | Klk13 (Kallikrein 13) | 7 B2 | NM_008454 | 99.9 | Klk16 (Kallikrein 16) | 92.2 |
| 8 D1 | L11333 | 99.9 | Carboxylesterase | 8 D1 | NM_144511 | 100 | Es31 | 95.2 |
| 9 F4 | NM_130864 | 99.6 | Acaa acetyl-Coenzyme A acyltransferase | 9 F4 | BC019882 | 100 | Similar to acetyl-CoA acyltransferase | 96.6 |
| 10 B3 | NM_013532 | 99.9 | Gp49a (Glycoprotein 49A) | 10 B3 | NM_008147 | 100 | Gp49b (glycoprotein 49B) | 96.7 |
| 10 D2 | NM_017372 | 100 | Lyzs (Lysozyme) | 10 D2 | NM_013590 | 99.8 | Lzp-s (P lysozyme structural) | 95.3 |
| 11 A3.2 | NM_172792 | 100 | hypothetical protein 4932414J04 | 17 D | AK03001 | 100 | Tyrosine protein kinase/cysteine-rich region | 94.0 |
| 11 B1.3 | NM_011396 | 99.9 | Slc22a5 (Solute carrier family 22) | 11 B1.3 | NM_019723 | 100 | Slc22a9 (solute carrier family 22) | 91.2 |
| 11 D | NM_021347 | 100 | Gsdm (Gasdermin) | 11 D | NM_029727 | 99.9 | 2200001G21Rik | 94.2 |
| 12 F1 | BC002065 | 99.6 | Serine protease inhibitor 2-1 | 12 F1 | BY761363 | 99.9 | EST sequence | 92.2 |
| 12 F1 | NM_013772 | 100 | Tcl1b3 (T-cell leukemia/lymphoma 1B, 3) | 12 F1 | NM_013776 | 100 | Tcl1b5 (T-cell leukemia/lymphoma 1B, 5) | 95.3 |
| 13 A1 | NM_013778 | 99.5 | Akr1c13 (Aldo-keto reductase 1, C13) | 13 A1 | NM_013777 | 99.5 | Akrc12 (Aldo-keto reductase 1, C12) | 96.1 |
| 13 A3 | NM_008864 | 99.2 | Csh1 (chorionic somatomammotrophin 1) | 13 A3.3 | AK082929 | 100 | Similar to placental lactogen 1 | 98.8 |
| 13 A4 | NM_011456 | 100 | Spi14 (Serine Protease Inhibitor 14) | 13 A4 | NM_011455 | 100 | Spi13 (serine protease inhibitor 13) | 95.2 |
| 13 D1 | NM_010872 | 100 | Birc1b (Neuronal apoptosis inhibitory 2) | 13 D1 | NM_008670 | 99.9 | Birc1a (Neuronal apoptosis inhibitory 1) | 90.0 |
| 14 C1 | NM_010373 | 99.7 | Gzme (Granzyme E) | 14 C1 | NM_010372 | 99.9 | Gzmd (Granzyme D) | 94.7 |
| 14 C2 | NM_172603 | 100 | 4933417L10Rik | 14 C3 | BE381578 | 100 | EST sequence | 94.0 |
| 15 E2 | NM_007781 | 100 | Csf2rb2 (Colony stimulating factor 2, β-2) | 15 E2 | NM_007780 | 99.8 | Csf2rb1 (Colony stimulating factor 2, β-1) | 95.0 |
| 15 E2 | NM_010005 | 100 | Cyp2d10 (Cytochrome P450, 2d10) | 15 E2 | NM_010006 | 100 | Cyp2d9 (cytochrome P450, 2d9) | 92.1 |
| 16 B1 | NM_023125 | 100 | Kng (Kininogen) | 16 B1 | BI330914 | 99.1 | EST sequence | 90.0 |
| 16 B3 | M92418 | 99.8 | MS2 (Cysteine proteinase inhibitor) | 16 B3 | BB654253 | 100 | EST sequence | 95.2 |
| 17 B2 | NM_009780 | 99.9 | C4 (Complement component 4) | 17 B2 | M21576 | 99.5 | Slp (MHC sex-limited protein) | 96.3 |
| X A2 | NM_008955 | 100 | Psx1 (Placenta specific homeobox 1) | X A2 | NM_023894 | 100 | Homeobox protein GPBOX | 91.6 |
*Locations of duplicons by mouse chromosome banding; locus 1 and 2 represent a duplication pair. †Alignment percent identity between gene and genomic sequences showing correct matches. ‡ % similarity: average DNA percent identity between paralogous gene/transcript sequences in locus 1 and 2 (duplicated pair)
Table 4.
Protein domain enrichment found in recently duplicated mouse genes*
| InterPro entry ID | Protein domain description | Number found in 608 duplicated genes | Number found in all 16,515 annotated genes in genome | Enrichment† |
| IPR000276 | Rhodopsin-like GPCR superfamily | 135 | 1229 | 3.0 |
| IPR000725 | Olfactory receptor | 103 | 861 | 3.3 |
| IPR003006 | Immunoglobulin/major histocompatibility complex | 46 | 372 | 3.4 |
| IPR004072 | Vomeronasal receptor, type 1 | 31 | 108 | 7.8 |
| IPR001909 | KRAB box | 23 | 103 | 6.1 |
| IPR001254 | Serine protease, trypsin family | 21 | 117 | 4.9 |
| IPR002401 | E-class P450, group I | 20 | 61 | 8.9 |
| IPR001128 | Cytochrome P450 | 20 | 68 | 8.0 |
| IPR007086 | Zn-finger, C2H2 subtype | 20 | 139 | 3.9 |
| IPR001314 | Chymotrypsin serine protease, family S1 | 19 | 108 | 4.8 |
| IPR002403 | E-class P450, group IV | 17 | 56 | 8.2 |
| IPR002397 | B-class P450 | 13 | 29 | 11.9 |
| IPR001304 | C-type lectin | 13 | 96 | 3.7 |
| IPR000215 | Serpin | 12 | 48 | 6.8 |
| IPR002402 | E-class P450, group II | 9 | 14 | 18.5 |
| IPR006046 | Glycoside hydrolase family 13 | 7 | 8 | 23.0 |
| IPR006047 | Alpha amylase, catalytic domain | 7 | 10 | 19.2 |
| IPR001400 | Somatotropin hormone | 7 | 32 | 6.1 |
| IPR006048 | Alpha amylase, C-terminal all-beta domain | 6 | 7 | 24.7 |
| IPR002018 | Carboxylesterase, type B | 6 | 13 | 12.3 |
| IPR004073 | Vomeronasal receptor, type 2 | 6 | 13 | 12.3 |
| IPR001039 | Major histocompatibility complex protein, class I | 6 | 17 | 9.9 |
| IPR001828 | Extracellular ligand-binding receptor | 6 | 29 | 5.5 |
| IPR002213 | UDP-glucoronosyl/UDP-glucosyl transferase | 5 | 12 | 11.8 |
| IPR002448 | Odour-binding protein | 4 | 9 | 13.2 |
| IPR000068 | Extracellular calcium-sensing receptor | 4 | 10 | 11.0 |
*Only Ensembl gene annotation (608 genes) was used in this analysis. †All results shown are statistically significant with p-values < 10-5 (chi2 test).
From this list of genes, we performed a detailed analysis of Mater, a maternal-effect gene of potential medical importance. Mater encodes an autoantigen in a mouse model for human autoimmune premature ovarian failure [24]. Knockout studies have shown that it is essential for early embryonic development in mice [25]. Mater encodes a protein of 1,111 amino acids from a 3.5 kb transcript that spans 57 kb on mouse chromosome 7. A 42 kb segmental duplication involving two duplicons (DUP1, where Mater is located; DUP2, where a novel Mater2 is located) are situated about 5 Mb apart and in an inverted orientation (Figure 2). DUP1 and DUP2 are on average 91.1% identical over the entire 42 kb genomic region, with a 96.6% average in the exonic regions. Furthermore, we identified an intron-less Mater pseudogene (MaterP), which shares 87% DNA sequence identity to Mater, at a location 10 Mb proximal to Mater (Figure 2; see Additional data files for a detailed comparative genomic analysis of the Mater locus). The mapping locations of these duplications have been confirmed by fluorescence in situ hybridization (FISH) (Figure 3). Thus, Mater serves as one example of a gene that has been knocked out in mice but for which there is a second, highly similar transcript whose biological role is not yet known.
Figure 2.
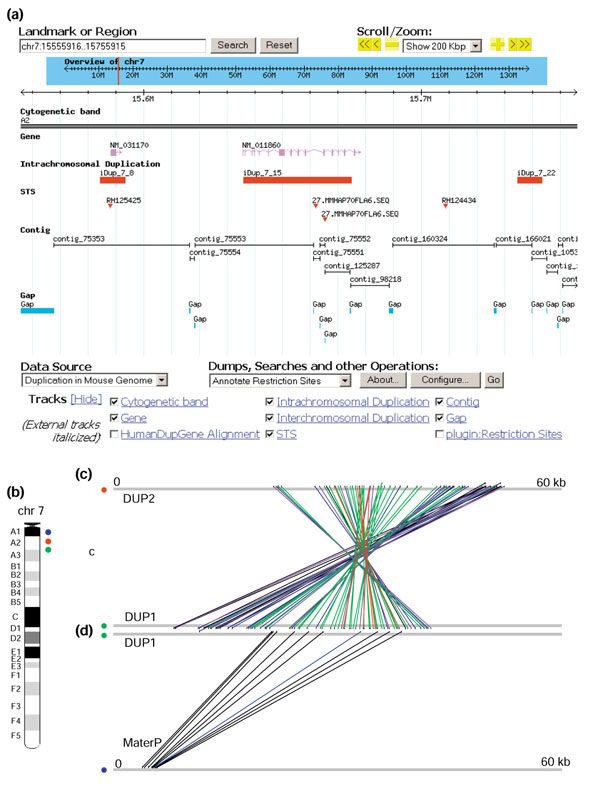
The genomic organization of the Mater duplication. (a) Location of the Mater duplication. A snapshot view of GMOD browser (details can be found at [14]). (b) Chromosomal view (mouse chromosome 7) of the three Mater duplication locations (DUP1, DUP2, MaterP). (c) Graphical view of the sequence similarity between DUP1 and DUP2 shown by GenomePixelizer. DUP2 is situated in an inverse orientation with respect to DUP1. Red, 99-100% sequence identity; purple, 96-98%; green, 93-95%; blue, 90-92%; black, 85-89%. (d) Graphical view of the sequence similarity between DUP1 and the MaterP region. As shown, MaterP is an intron-less, retrotransposed pseudogene. Blue, 90-92% sequence identity; black, 85-89%.
Figure 3.
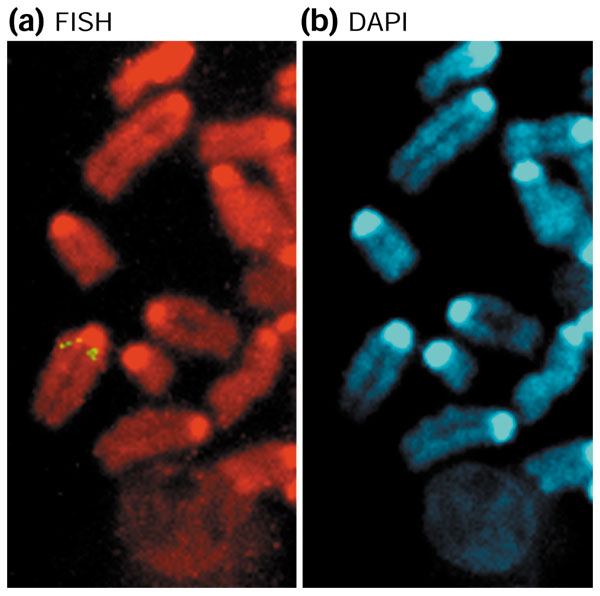
FISH detection of Mater duplication. (a) Metaphase FISH showing three pairs of signals (yellow) detected on mouse chromosome 7 using BAC clone RP23-225F5 (detection frequency of 70%) mapping to duplicated Mater regions. (b) DAPI banding of the same partial mitotic figures for the identification of mouse chromosome 7. A control probe RP23-464L20 was mapped to a single location in the F2 region (data not shown).
In addition, we were interested in determining whether any of the 675 genes have undergone recent (≥ 90% sequence identity over ≥ 5 kb) and independent duplication in the human genome. Some of these genes could be recently evolving via the 'birth and death model of evolution' which has been used to describe the evolution of the major histocompatibility complex (MHC) and immunoglobulin multigene families [26]. This model describes genes that are repeatedly created through duplication, with some genes becoming fixed while others are rendered nonfunctional by deleterious mutations [26].
We examined the 675 duplicated mouse genes using best reciprocal BLAST hits to identify their putative human orthologs. We subsequently analyzed regions containing these putative orthologs for recent sequence duplication in the human genome. Sixteen of the 675 genes were found to be involved in recent, independent gene duplication in mouse and human (see Table 5). Some of these regions containing whole-gene duplications are part of multigene families known to be evolving via duplication and are found in tandem duplicated arrays in both species (that is, the Amy2, H2-Q1, Gsta1, and Olfr54 genes). An interesting example of a recent and apparently independent whole-gene duplication that occurred in mouse and human involves Bmp8a and a second intronic transcript Oxct2. Of the partial gene duplications, the recent duplication within the Tnxb gene and its human ortholog TNXB (found at the MHC III locus of mouse chromosome 17 and human 6p21) is particularly intriguing. In humans, this locus consists of a tandem array of genes (RP, C4, CYP21, and TNXB (RCCX)), which through gene duplication, can exist as mono-, di- and tri-modular forms in the caucasian population [27]. Recent studies have also shown the presence of a deletion haplotype in one individual, leading to a fusion of the TNXA/TNXB gene on one chromosome and a duplication of CYP21 on the other chromosome [28]. Furthermore, complex haplotypes of the complement genes (C4A and C4B) residing in the RCCX module have been characterized and postulated to have a role in individual susceptibility to infection and autoimmune disease [29]. A closer inspection of the genomic region surrounding this recent duplication in the mouse reveals that the C57BL/6J duplication encompasses homologous genes (Tnxb, Slp (a C4 paralog), Cyp21a1, and C4). Similarly, in humans, this orthologous region of the mouse genome has been shown to undergo multiple recombination events, giving rise to a variety of haplotypes [30]. Overall, many of the genes that have recently experienced duplications in the mouse and human genomes are of biomedical and evolutionary interest. The complexity and polymorphic nature of these recent duplications underscores the need for, and the difficulty of, performing the detailed structural and functional analyses that will help discern their true genomic organization, evolutionary history, and biological implications.
Table 5.
Genes that have undergone recent duplication in both the mouse and human genome*
| Refseq† | Gene description |
| NM_007534 | B-cell leukemia/lymphoma 2 related protein A1b (Bcl2a1b) |
| NM_007558 | Bone morphogenetic protein 8a (Bmp8a) |
| NM_007812 | Cytochrome P450, 2a5 (Cyp2a5) |
| NM_008181 | Glutathione S-transferase, alpha 1 (Ya) (Gsta1) |
| NM_010390 | Histocompatibility 2, Q region locus 1 (H2Q1) |
| NM_009467 | UDP-glucuronosyltransferase 2 family, member 5 (Ugt2b5) |
| NM_009669 | Amylase 2, pancreatic (Amy2) |
| NM_009888 | Complement component factor h (Cfh) |
| NM_010856 | Myosin heavy chain, cardiac muscle, adult (Myhca) |
| NM_013778 | Aldo-keto reductase family 1, member C13 (Akr1c13) |
| NM_022033 | 3-Oxoacid CoA transferase 2 (Oxct2) (Imbedded in the Bmp8a gene) |
| NM_026419 | Elastase 3B, pancreatic (Ela3b) |
| NM_031176 | Tenascin XB (Tnxb) |
| NM_130864 | Acetyl-coenzyme A acyltransferase (peroxisomal 3-oxoacyl-Coenzyme athiolase) (Acaa) |
| NM_010997 | Olfactory receptor 54 (Olfr54) |
| NM_031170 | Keratin complex 2, basic, gene 8 (Krt2-8) |
*Six hundred and seventy-five duplicated mouse gene sequences were aligned to the June 2002 human genome assembly by BLAST (with an initial expected value cutoff of <10-10). The best aligned human genes were subsequently used for reciprocal BLAST alignments (against the mouse genome sequence) to establish a putative orthologous relationship between the mouse and human gene pairs. Using results from our human genome duplication analysis [9,37], we examined regions of the human genome where the human genes are involved in recent segmental duplication. †Italics represents genes that are entirely within a duplication in the mouse genome.
Conclusions
Our current analysis of the presence and organization of recent segmental duplications in the mouse genome has identified recent gene-duplication events and potentially problematic regions of the mouse genome assembly. At a practical level, identifying regions with segmental duplication will be useful in highlighting the most dynamic regions of any mammalian genome assembly. For the genome-sequencing community, these potential misassemblies/putative duplications can become initial targets for clone-based finishing; and for the biologist, they can serve as sentinels for regions of the genome most likely to change in subsequent assemblies. Additional hierarchical shotgun sequencing effort [12] will undoubtedly be critical to finish the mouse genome sequence and reveal additional duplicated regions that are incomplete at the moment.
Many of the duplicated genes are of evolutionary and medical importance (that is, genes involved in immune defense, olfaction, and drug metabolism). Knowledge of these duplicated regions could be important for accurately mapping mutants derived from ethylnitrosourea (ENU) mutagenesis, designing targeting vectors for embryonic stem cell alterations, and validating putative single-nucleotide polymrophisms (SNPs) that may have arisen from recently duplicated sequences rather than allelic variants [31]. The ability to create large, sophisticated targeting vectors, by engineering BACs using homologous recombination in Escherichia coli [32], should prove very useful for designing in vivo experiments aimed at dissecting the function of recently duplicated genes. Knowledge of all recent duplications in mouse may also highlight regions subject to chromosomal rearrangement and polymorphism within and between species, and provide an opportunity to model the stability of such genomic architecture in a mammalian genome.
Materials and methods
Genome sequence and chromosome-wide BLAST
We obtained the February 2002 (MGSCv3) and February 2003 mouse genome assemblies (lower-case repeat-masked sequences), as well as the assembly component tables, through the UCSC Human Genome Browser website [21]. For each assembly, detection of intrachromosomal segmental duplications involved comparing each of the 20 masked chromosome sequences (excluding the Y chromosome not targeted by the MGSC) and the masked unmapped chromosome sequence against itself by BLAST2 [16] (21 comparisons made). Interchromosomal analysis of segmental duplications involved pairwise comparisons between each of the 21 chromosomes (420 comparisons made). Analyses were repeated with the exclusion of the unmapped chromosome sequence to examine its contribution to the overall duplication content (results posted at [14]). All BLAST results were subsequently parsed to eliminate low-quality and fragmented alignments under the following criteria: BLAST results having ≥ 90% sequence identity, ≥ a length of 80 bp, and with expected value ≤ 10-30.
Parsing of BLAST results and duplication detection
Each BLAST report was sorted by chromosomal coordinates. All identical hits (same coordinate alignments), including suboptimal BLAST alignments recognized by multiple, overlapping alignments, as well as mirror hits (reverse coordinate alignments) from the BLAST results of the intrachromosomal set, were removed. Contiguous alignments separated by a distance of less than 3 kb, then 5 kb, and subsequently 9 kb, were joined stepwise into modules in order to traverse masked repetitive sequences and to overcome breaks in the BLAST alignments caused by insertions/deletions and sequence gaps. Such contiguous sequence-alignment modules represent sequence similarity between the subject and query chromosome sequence in question (at their respective positional coordinates) [9]. Potential sequence misassignment errors are results detected to have > 99.5% sequence identity with another region.
Online database for recent segmental duplications
We overlaid all duplication content and regions containing potential sequence misassignment errors onto the mouse genome sequence, which can be viewed using the interactive Generic Genome Browser [33] hosted at our website [14]. Results and analyses were presented for both the February 2002 and February 2003, each as a separate database. Results are also summarized in tables that include information on chromosomal coordinates, band locations, size of duplications, level of identity between duplicated copies, as well as genes mapped to these regions [14]. Graphical representation for intrachromosomal duplications was generated using the visualization tool GenomePixelizer (Figure 1) [34].
Identification of recent gene duplications
We obtained the NCBI Refseq gene annotation file (refGene.txt.gz) from the UCSC Downloads website [21] and the Ensembl gene annotation (Mus_musculus.cdna.fa.gz; Ensembl Known genes only) from the Ensembl website [35]. Genes that mapped to duplicated regions of the mouse genome were identified using their chromosome sequence coordinates. In total, 439 Refseq and 608 Ensembl annotated genes were found to be involved in duplicated regions; together these two datasets made up 675 unique gene annotations (372 overlapped annotations). To establish gene-pair relationships between duplicated gene sequences, each of the 238 NCBI RefSeq genes that were found to be fully contained within a segmental duplication was searched against the UCSC and NCBI GenBank database for spliced ESTs, full-length cDNAs and additional annotated genes. A total of 128 gene pairs were established, most of which are likely to be novel gene paralogs previously unknown in the literature. In the analysis of protein-domain enrichment in duplicated genes, InterPro annotation for duplicated genes (608 Ensembl genes) as well as for the entire gene set (16,515) was obtained from Ensembl EnsMart [36]. We counted the number of times a protein-domain class is found in each gene set and tabulated our results (see Table 4). To examine the subset of genes that had undergone recent duplication in the human genome, each of the duplicated gene sequences was aligned to the June 2002 human genome assembly by BLAST (with an initial expected value cutoff of <10-10). The best-aligned human genes were subsequently used for reciprocal BLAST alignments (against the mouse genome sequence) to establish a putative orthologous relationship between the mouse and human gene pairs. Using results from our human genome duplication analysis [37], we examined regions of the human genome where the human genes were involved in recent segmental duplication.
Fluorescence in situ hybridization (FISH)
Mouse lymphocytes were isolated from the spleen and cultured at 37°C in RPMI 1640 medium supplemented with fetal calf serum, concanavalin A and lipopolysaccharide. After 44 hours, the cultured lymphocytes were treated with bromodeoxyuridine for an additional 14 hours. The synchronized cells were washed and recultured at 37°C for 4 hours in a-minimal Eagle's medium with thymidine. Chromosome slides were made by conventional methods including hypotonic treatment, fixation and air-drying [38]. BAC probes RP23-225F5 (mapped to the Mater locus (DUP1) by BAC-end sequences) and RP23-464L20 (a control probe) were biotinylated respectively. Hybridization and detection were carried out according to [39]. FISH signals were observed under fluorescent microscopy using FITC and DAPI filters. Images were captured by CCD camera.
Additional data files
Further analysis of Mater duplication, including a figure showing a multiple percent identity plot of Mater versus Mater2, MATER (human), and MaterP, is available (additional data file 1).
Supplementary Material
Further analysis of Mater duplication
Acknowledgments
Acknowledgements
We thank John Taylor and Duane Martindale for critical comments on the manuscript. This work was supported by the Canadian Institutes of Health Research (CIHR) and Genome Canada to S.W.S. B.F.K. is supported by the CIHR and M.D.W. is supported by the Michael Smith Foundation for Health Research (MSFHR). S.W.S. is an Investigator of CIHR and International Scholar of the Howard Hughes Medical Institute.
References
- Ohno S. Evolution by Gene Duplication. New York: Springer; 1970. [Google Scholar]
- Prince VE, Pickett FB. Splitting pairs: the diverging fates of duplicated genes. Nat Rev Genet. 2002;3:827–837. doi: 10.1038/nrg928. [DOI] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA, Rogozin IB, Wolf YI, Koonin EV. Selection in the evolution of gene duplications. Genome Biol. 2002;3:research0008.1–0008.9. doi: 10.1186/gb-2002-3-2-research0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Newman T, Linardopoulou E, Trask BJ. Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions. Genome Res. 2002;12:1663–1672. doi: 10.1101/gr.338402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Cheung J, Estivill X, Khaja R, MacDonald JR, Lau K, Tsui LC, Scherer SW. Genome-wide detection of segmental duplications and assembly errors in the human genome sequence. Genome Biol. 2003;4:R25. doi: 10.1186/gb-2003-4-4-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel BS, Shaikh TH. Segmental duplications: an 'expanding' role in genomic instability and disease. Nat Rev Genet. 2001;2:791–800. doi: 10.1038/35093500. [DOI] [PubMed] [Google Scholar]
- The Jackson Laboratory JAX Strain Information http://jaxmice.jax.org/info/chromosomal_abberati.html
- Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Butler J, Gnerre S, Mauceli E, Lindblad-Toh K, Mesirov JP, Zody MC, Lander ES. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 2003;13:91–96. doi: 10.1101/gr.828403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCAG: mouse recent segmental duplication homepage http://chr7.ocgc.ca/mousedup
- NCBI Mouse Genome Resources http://www.ncbi.nih.gov/genome/guide/mouse
- Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- Lin EY, Kozak CA, Orlofsky A, Prystowsky MB. The bcl-2 family member, Bcl2a1, maps to mouse chromosome 9 and human chromosome 15. Mamm Genome. 1997;8:293–294. doi: 10.1007/s003359900418. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Hamasaki A, Negishi I, Loh DY, Sendo F, Nakayama K, Nakayama K. Multiple gene duplication and expression of mouse bcl-2-related genes, A1. Int Immunol. 1998;10:631–637. doi: 10.1093/intimm/10.5.631. [DOI] [PubMed] [Google Scholar]
- Gumucio DL, Wiebauer K, Dranginis A, Samuelson LC, Treisman LO, Caldwell RM, Antonucci TK, Meisler MH. Evolution of the amylase multigene family. YBR/Ki mice express a pancreatic amylase gene which is silent in other strains. J Biol Chem. 1985;260:13483–13489. [PubMed] [Google Scholar]
- Hagenbuchle O, Wellauer PK, Cribbs DL, Schibler U. Termination of transcription in the mouse alpha-amylase gene Amy-2a occurs at multiple sites downstream of the polyadenylation site. Cell. 1984;38:737–744. doi: 10.1016/0092-8674(84)90269-1. [DOI] [PubMed] [Google Scholar]
- UCSC Genome Bioinformatics http://genome.ucsc.edu
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Cheung J. Discovery of the human genome sequence in the public and private databases. Curr Biol. 2001;11:R808–R811. doi: 10.1016/s0960-9822(01)00490-0. [DOI] [PubMed] [Google Scholar]
- Tong ZB, Nelson LM. A mouse gene encoding an oocyte antigen associated with autoimmune premature ovarian failure. Endocrinology. 1999;140:3720–3726. doi: 10.1210/endo.140.8.6911. [DOI] [PubMed] [Google Scholar]
- Tong ZB, Gold L, Pfeifer KE, Dorward H, Lee E, Bondy CA, Dean J, Nelson LM. Mater, a maternal effect gene required for early embryonic development in mice. Nat Genet. 2000;26:267–268. doi: 10.1038/81547. [DOI] [PubMed] [Google Scholar]
- Nei M, Gu X, Sitnikova T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci USA. 1997;94:7799–7806. doi: 10.1073/pnas.94.15.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, Zipf WB, Rennebohm RM, Yung Yu C. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. J Exp Med. 2000;191:2183–2196. doi: 10.1084/jem.191.12.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaatinen T, Chung EK, Ruuskanen O, Lokki ML. An unequal crossover event in RCCX modules of the human MHC resulting in the formation of a TNXB/TNXA hybrid and deletion of the CYP21A. Hum Immunol. 2002;63:683–689. doi: 10.1016/s0198-8859(02)00416-0. [DOI] [PubMed] [Google Scholar]
- Chung EK, Yang Y, Rennebohm RM, Lokki ML, Higgins GC, Jones KN, Zhou B, Blanchong CA, Yu CY. Genetic sophistication of human complement components C4A and C4B and RP-C4-CYP21-TNX (RCCX) modules in the major histocompatibility complex. Am J Hum Genet. 2002;71:823–837. doi: 10.1086/342777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanakitsakul S, Nakayama K, Takahashi M, Nonaka M. Three extra copies of a C4-related gene in H-2w7 mice are C4/Slp hybrid genes generated by multiple recombinational events. Immunogenetics. 1990;32:431–439. doi: 10.1007/BF00241638. [DOI] [PubMed] [Google Scholar]
- Estivill X, Cheung J, Pujana MA, Nakabayashi K, Scherer SW, Tsui LC. Chromosomal regions containing high-density and ambiguous-mapped single nucleotide polymorphisms (SNPs) correlate with segmental duplications in the human genome. Hum Mol Genet. 2002;11:1987–1995. doi: 10.1093/hmg/11.17.1987. [DOI] [PubMed] [Google Scholar]
- Testa G, Zhang Y, Vintersten K, Benes V, Pijnappel WW, Chambers I, Smith AJ, Smith AG, Stewart AF. Engineering the mouse genome with bacterial artificial chromosomes to create multipurpose alleles. Nat Biotechnol. 2003;21:443–447. doi: 10.1038/nbt804. [DOI] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozik A, Kochetkova E, Michelmore R. GenomePixelizer-a visualization program for comparative genomics within and between species. Bioinformatics. 2002;18:335–336. doi: 10.1093/bioinformatics/18.2.335. [DOI] [PubMed] [Google Scholar]
- Ensembl Mouse Genome Server http://www.ensembl.org/Mus_musculus/
- Ensembl EnsMart http://www.ensembl.org/EnsMart/
- TCAG: human recent segmental duplication homepage http://chr7.ocgc.ca/humandup
- Heng HHQ, Tsui L-C. Modes of DAPI banding and simultaneous in situ hybridization. Chromosoma. 1993;102:325–332. doi: 10.1007/BF00661275. [DOI] [PubMed] [Google Scholar]
- Heng HHQ, Squire J, Tsui L-C. High resolution mapping of mammalian genes by in situ hybridization to free chromatin. Proc Natl Acad Sci USA. 1992;89:9509–9513. doi: 10.1073/pnas.89.20.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong ZB, Nelson LM, Dean J. Mater encodes a maternal protein in mice with a leucine-rich repeat domain homologous to porcine ribonuclease inhibitor. Mamm Genome. 2000;11:281–287. doi: 10.1007/s003350010053. [DOI] [PubMed] [Google Scholar]
- Tong ZB, Bondy CA, Zhou J, Nelson LM. A human homologue of mouse Mater, a maternal effect gene essential for early embryonic development. Hum Reprod. 2002;17:903–911. doi: 10.1093/humrep/17.4.903. [DOI] [PubMed] [Google Scholar]
- Loots GG, Ovcharenko I, Pachter L, Dubchak I, Rubin EM. rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res. 2002;12:832–839. doi: 10.1101/gr.225502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Riemer C, Martindale DW, Schnupf P, Boright AP, Cheung TL, Hardy DM, Schwartz S, Scherer SW, Tsui LC, et al. Comparative analysis of the gene-dense ACHE/TFR2 region on human chromosome 7q22 with the orthologous region on mouse chromosome 5. Nucleic Acids Res. 2001;29:1352–1365. doi: 10.1093/nar/29.6.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, Gibbs R, Hardison R, Miller W. PipMaker - a web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–586. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–1047. doi: 10.1093/bioinformatics/16.11.1046. [DOI] [PubMed] [Google Scholar]
- NCBI BLAST 2 Sequences http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Further analysis of Mater duplication