

Summary
Moenomycin A (MmA) is a member of the phosphoglycolipid family of antibiotics, which are the only natural products known to target directly the extracellular transglycosylases involved in peptidoglycan biosynthesis. The structural and biological uniqueness of MmA make it an attractive starting point for the development of new antibacterial drugs. In order both to elucidate the biosynthesis of this unusual compound, and to develop tools to manipulate its structure, we have identified the MmA biosynthetic genes in Streptomyces ghanaensis (ATCC14672). We show via heterologous expression of a subset of moe genes that the economy of the MmA pathway is enabled through the use of sugar-nucleotide and isoprenoid building blocks derived from primary metabolism. The work reported lays the foundation for genetic engineering of MmA biosynthesis to produce novel derivatives.
Introduction
The emergence of resistance to existing antibiotics represents a significant threat to public health. The increasing frequency of resistance to the cell wall inhibitor vancomycin is of particular concern because this antibiotic is the last line of defense against methicillin-resistant Gram-positive infections. New antibiotics with activity against resistant bacterial strains are desperately needed. Antibiotics that inhibit validated bacterial targets but are structurally and mechanistically unrelated to compounds in clinical use may be especially attractive candidates for further development. One example of such a compound is moenomycin A (MmA; Figure 1), a phosphoglycolipid antibiotic that is several orders of magnitude more potent than vancomycin against many Gram-positive pathogens, including the vancomycin-resistant pathogens [1]. MmA is a representative member of a family of compounds that inhibit the transglycosylases involved in bacterial peptidoglycan formation [2]. Vancomycin also inhibits these enzymes, but the mechanisms of vancomycin and MmA are completely unrelated. Whereas vancomycin binds to peptidoglycan intermediates, which are substrates for the transglycosylases [3], MmA binds to the transglycosylases themselves. The moenomycins are the only natural products known to have such a mechanism of action.
Figure 1.

Structures of MmA and proposed structures of moenomycins produced by recombinant S. ghanaensis and S. lividans strains described in this work. Capital letters A–H indicate the 8 “building blocks” of MmA.
Paralleling the unusual mechanism of action of the moenomycins is an unusual structure that contains an unprecedented phosphoglycerate-ether linkage to an irregular isoprenoid chain. This structural feature, which is of considerable interest from a biosynthetic standpoint, is also believed to be the primary cause of the moenomycins’ poor physicochemical properties, which have hampered their development as therapeutic agents for human use. In the hope of learning how the phospholipid of MmA is assembled and attached, and of acquiring tools to enable the production of MmA analogs with better properties, we sought to identify the genes for MmA production. Access to these genes could enable the preparation of a range of MmA derivatives to probe the mechanism of transglycosylase inhibition and the structural requirements for activity, work that may lead eventually to the discovery of transglycosylase inhibitors for therapeutic use. Here we report the identification of the MmA biosynthetic genes from Streptomyces ghanaensis (ATCC14672) using a whole genome scanning approach combined with gene knockouts and complementation experiments. Analysis of the gene cluster suggests that the MmA biosynthetic pathway differs from previously characterized carbohydrate-containing antibiotic gene clusters in that the nucleotide-sugar building blocks – as well as most of the other building blocks of the molecule - are derived exclusively from primary metabolism. We have confirmed this hypothesis by demonstrating that heterologous expression of a subset of the MmA biosynthetic genes in S. lividans TK24 leads to the production of a moenomycin pentasaccharide derivative. Thus, the MmA cluster provides an alternative paradigm for the biosynthesis of glycosylated secondary metabolites that may influence genome scanning approaches to identify other carbohydrate-rich natural products. The availability of a heterologous expression system for moenomycin production will facilitate analysis of gene function, and may yield intermediates that can be chemically modified to produce novel antibiotics or small molecule probes for bacterial transglycosylases.
Results
Identification of the genes for C5N subunit biosynthesis (moe cluster 2)
Many gene clusters for antibiotic production have been readily identified by degenerate primer-based PCR amplification of highly conserved genes [4]. Although structurally unique, MmA does contain some features found in other secondary metabolites. The most recognizable one is perhaps the C5N subunit (A ring; Figure 1). The biosynthesis of this subunit involves an aminolevulinate synthase (ALS) [5], and we were able to amplify an internal fragment of a putative ALS gene, moeC4, from the S. ghanaensis genome using degenerate primers based on sequences of ALS genes proposed to govern the production of asukamycin and the polyene ECO-02301 [5, 6]. Using this amplicon as a probe for screening a S. ghanaensis SuperCos1-based gene library, we retrieved a cosmid, moeno5, carrying the three-gene operon moeA4moeB4moeC4 (referred here to as moe cluster 2; Figure 2). Analysis of a S. ghanaensis moeA4 deficient strain LH1, prepared as described in Experimental Procedures, showed that it did not produce MmA. Instead, it accumulated an intermediate lacking the C5N chromophore and proposed to have the structure 2 (Figures 1 and 3B). Although interruption of unit A attachment to the pentasaccharide was expected to yield exclusively the galactopyranuronic acid form of MmA (which was observed in the mutant as a minor intermediate; data not shown), the exact mass of the major product supports the proposed structure (calculated mass of the negative ion: 1484.6329, found: 1484.6371; MS2 spectra provided in Supplemental Data).
Figure 2.
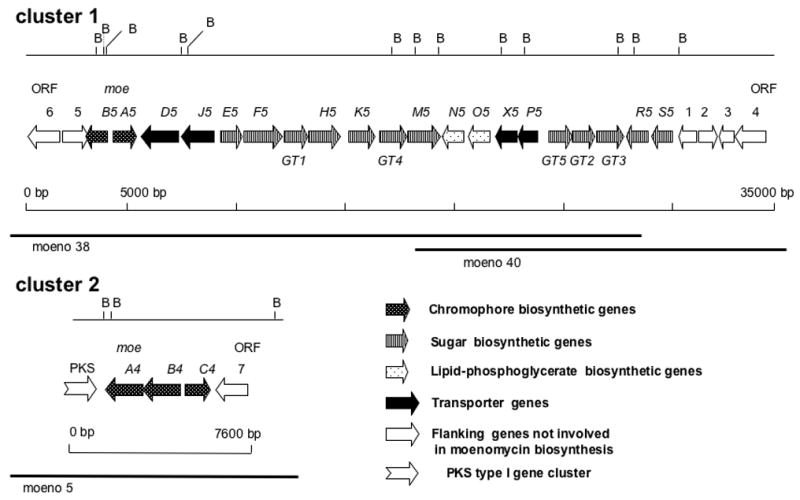
Genetic organization of MmA biosynthesis (moe) gene clusters 1 and 2. The moe genes are located within cosmids moeno5, 38 and 40. B = BamHI restriction sites.
Figure 3.

HPLC-MS analysis (ESI, negative mode) showing the moenomycins found in cell extracts of S. ghanaensis wild type (A), moeA4 mutant LH1 (B), moeGT1 mutant OB21e (C) and moeM5 mutant OB20a (D).
Compound 2 showed antibiotic activity, consistent with experiments suggesting that the C5N subunit is not critical for biological activity [8]. The introduction of plasmid pKC11395-8 (which carries the moeC4-moeB4 intergenic region and the entire moeB4moeA4 genes) into the LH1 strain restored MmA biosynthesis, as evident from LC-MS analysis. Expression of the moeB4 gene alone (plasmid pOOB12) in the LH1 strain did not lead to MmA production, implying that only moeA4 is affected in the mutant. Based on sequence analysis, MoeA4 is proposed to be an acyl-CoA ligase that cyclizes aminolevulinate, while MoeB4 is proposed to be an amide synthetase that couples the C5N unit to the C6 acid of the MmA B ring sugar (Figure 4). Accumulation of the galactopyranouronamide derivative 2 in MmA producers has been reported by other laboratories [7] and may be due to amidation of unit B by the same enzyme(s) that operate on unit F (Figure 1). Although the preceding results establish a role for moeA4moeB4moeC4 genes in MmA production, none of the genes flanking these three genes proved to be involved in MmA production. We concluded that the primary moe cluster must be elsewhere in the chromosome.
Figure 4.

Proposed biosynthetic scheme for the chromophore and pentasaccharide portions of MmA. Letters G-H in shaded rectangle mark lipid-phosphoglycerate moiety of MmA. MoeGTn represents any of 4 Moe Gtfs (MoeGT2-5). UDP-GlcNAc = UDP-N-acetyl glucosamine; UDP-Glc = UDP-glucose; UDP-GlcA/UDP-GalA = UDP-glucuronic acid/UDP-galacturonic acid.
Identification of the primary MmA biosynthetic gene cluster (moe cluster 1)
We attempted to clone the main MmA gene cluster by targeting genes proposed to be involved in the biosynthesis of the isoprenoid chain and some of the sugar subunits. The isoprenoid chain in moenomycin was shown using tracer experiments to be of nonmevalonate origin [9–11]. Nonmevalonate (also called DOXP) pathway enzymes are involved in the biosynthesis of all primary as well as some secondary isoprene-derived metabolites in streptomycetes. In one case, genes encoding DOXP-pathway enzymes have been found to be clustered with a natural product pathway [12]. Efforts to identify the primary moe cluster by targeting DOXP pathway enzymes were unsuccessful, however. Furthermore, all other attempts to clone moe genes via a degenerate primer-based PCR approach also failed: although a number of putative hexose 4,6-dehydratase, carbamoyltransferase and prenyltransferase genes were cloned, none of them proved to be involved in MmA production.
We considered several other strategies to identify the primary moe cluster, including in vivo approaches (transposon mutagenesis followed by screening for altered MmA production/sensitivity; complementation of MmA non-producing mutants) and in silico whole genome scanning. After some experimental work aimed at testing various in vivo approaches, we concluded that genome scanning would be the most direct and reliable way to identify the moe gene cluster. Advances in sequencing and software technologies have reduced both the cost and the time of the process to the point where whole genome sequencing is competitive with other approaches as a tool for discovery of novel metabolic pathways.
The genome of S. ghanaensis ATCC14672 (~8.6–9.0 Mb) was shotgun-sequenced to 6.6× coverage and partially assembled at the Broad Institute (Cambridge, MA), yielding 1018 contigs ranging from 1 to 95 kb in size. The contigs cover 7.4 Mb of the strain’s chromosome. They were analyzed for the presence of candidate moe genes using the BLASTX and FRAMEPLOT [13] online programs. Contig 908 contained the previously identified moe cluster 2. Four other neighboring contigs (70–73), located more than 1 Mb away from contig 908 in the assembled genome, contained groups of genes that appeared functionally capable of governing the biosynthesis of MmA. Two cosmids (moeno 38 and moeno40) covering contigs 70–73 were retrieved from a S. ghanaensis genomic library and fully sequenced to identify a cluster of 20 moe genes (moe cluster 1; Figure 2). The boundaries of cluster 1 are defined by regions of extensive synteny to the terminal region of the S. coelicolor chromosome [14]. The predicted functions of all moe genes are presented in the Table 1.
Table 1.
Deduced functions for genes in moenomycin A gene clusters 1 and 2*
| Protein | AA | Homologue | ID%/SI% | Accession No. | Proposed Function |
|---|---|---|---|---|---|
| MoeA4 | 516 | Putative acyl CoA ligase (Streptomyces aizunensis) | 63/73 | AAX98210.1 | Acyl CoA ligase |
| MoeB4 | 521 | SimL (Streptomyces antibioticus) | 45/62 | AAG34163.1 | Amide synthetase |
| MoeC4 | 412 | HemA-AsuA (Streptomyces asukaensis) | 70/83 | AY240962 | Aminolevulinate synthase |
| MoeB5 | 301 | Putative acyl CoA ligase (S. aizunensis) | 58/76 | AAX98210.1 | Nonfunctional acyl CoA |
| MoeA5 | 394 | HemA-AsuA (S. asukaensis) | 64/78 | AY240962 | Aminolevulinate synthase |
| MoeD5 | 591 | Putative ABC transporter (Symbiobacterium thermophilum) | 41/55 | YP075256.1 | ABC transporter |
| MoeJ5 | 564 | As above | 45/61 | YP075255.1 | ABC transporter |
| MoeE5 | 340 | Putative UDP-glucose 4-epimerase (Symbiobacterium thermophilum) | 46/58 | YP074610.1 | NDP-hexose 4-epimerase |
| MoeF5 | 645 | WbpS (Pseudomonas aeruginosa) | 29/43 | AAF24002.1 | Aminotransferase subunit 1 |
| MoeGT1 | 402 | Putative Gtf (Polaromonas sp) | 27/40 | EAM38951.1 | Glycosyltransferase |
| MoeH5 | 513 | AsnB-like amidotransferase (Azoarcus sp) | 32/48 | CAI08539.1 | Aminotransferase subunit 2 |
| MoeK5 | 407 | Putative methyltransferase (Pyrococcus horikoshii) | 34/52 | NP_142754.1 | Methyltransferase |
| MoeGT4 | 427 | Putative Gtf (Mycobacterium vahbaalenii) | 27/38 | EAS23724.1 | Glycosyltransferase |
| MoeM5 | 530 | GdmN (Streptomyces hygroscopicus) | 29/44 | AAO06921.1 | Carbamoyltransferase |
| MoeN5 | 260 | Putative prenyltransferase (Loktanella vestfoldensis) | 29/43 | EAQ07619.1 | Prenyltransferase |
| MoeO5 | 281 | GGGPS (Thermoplasma acidophilum) | 27/43 | JC7965 | Prenyl 3-phosphoglycerate synthase |
| MoeX5 | 226 | Putative membrane protein (Mycobacterium sp) | 26/40 | EAS99725.1 | ABC transporter membrane protein |
| MoeP5 | 223 | ABC transporter ATPase (Mycobacterium flavescens) | 43/58 | EAS11435.1 | ABC transporter ATP-binding protein |
| MoeGT5 | 312 | Gtf MoeGT4 (S. ghanaensis; see above) | 45/59 | Glycosyltransferase | |
| MoeGT2 | 286 | Putative Gtf (Methylococcus capsulatus) | 35/51 | AAU93096.1 | Glycosyltransferase |
| MoeGT3 | 414 | Putative Gtf (Kineococcus radiotolerans) | 44/56 | ZP_00616987.1 | Glycosyltransferase |
| MoeR5 | 374 | CapD (Nocardioides sp) | 53/68 | EAO07657.1 | Hexose-4,6-dehydratase |
| MoeS5 | 282 | SCO7194 (Streptomyces coelicolor) | 62/75 | CAC01594.1 | Hexose-4-ketoreductase |
We confirmed the involvement of moe cluster 1 in MmA biosynthesis through disruptions of genes moeGT1 and moeM5, which encode a putative glycosyltransferase (Gtf) and a carbamoyltransferase, respectively. Cell extracts from the mutants were analyzed for both antibiotic activity and the presence of MmA analogs. The moeGT1-deficient S. ghanaensis strain OB21e showed no antibiotic activity, and no peaks for MmA or its anticipated intermediates could be detected upon mass analysis of OB21e semipurified extracts (Figure 3C) or culture broths. Complementation of the OB21e mutant with plasmid pOOB41, which carries a PermE-moeGT1 fusion, restored MmA production as judged from bioassays and LC-MS analysis. The MmA trisaccharide degradation product (units C-E-F-G-H, Figure 1) is reported to have substantial antibacterial activity [15], suggesting that the moeGT1 knockout blocks transfer of the C, E or F rings. The moeM5 null strain, OB20a, produced a compound having greatly reduced antibiotic activity and a mass of 1537.6 Da ([M-H]−; Figure 3D), consistent with the loss of the NH2CO group from the C′-3 position of ring F of MmA (Figure 1). Complementation of OB20a with pOOB43, which carries a PermE-moeM5 fusion, restored production of MmA, as judged by the reappearance of a mass peak at 1580.6 Da. We suggest that MoeM5 acts as a late stage tailoring enzyme to install the carbamoyl group, which is an important element in the MmA pharmacophore [1].
Description of moe cluster 1
Cluster 1 contains genes encoding two prenyltransferases, five Gtfs, a 4,6-dehydratase/ketoreductase pair putatively involved in C ring biosynthesis, a possible epimerase, and several sugar tailoring genes (Figure 2 and Table 1) that are presumed to be involved in functionalization of the F ring following pentasaccharide assembly. It also contains a second ALS gene and a gene for ALA cyclization that contains an in-frame deletion and is presumed to be non-functional; however, there is no gene for attachment of the cyclized C5N unit. The biosynthetic moe cluster 2 most likely arose from duplication of a chromophore biosynthetic operon from moe cluster 1, whereupon the redundant chromophore operon in cluster 1 subsequently underwent deletion events. In addition to the MmA biosynthetic genes, four genes encoding components of two ATP-binding cassette (ABC) transport systems have been identified in moe cluster 1. ABC-type transporters are found in many antibiotic gene clusters and are typically thought to be involved in transport of natural products or precursors across bacterial cell membranes [16]. Full descriptions of the moe genes are provided in Supplemental Data.
The structures of the individual sugars in MmA and the paucity of sugar biosynthetic genes in the cluster led us to surmise that all of the NDP-sugars in MmA must be derived from building blocks produced by primary metabolism. For example, the D ring donor is predicted to be UDP-glucose; the E and C rings are predicted to share a common precursor, UDP-GlcNAc, which is used without further modification as the E ring donor and converted to UDP-chinovosamine by MoeR5 and MoeS5 to make the C ring donor (Figure 4). The B and F rings in MmA are both derived from C6 acid precursors, epimeric with respect to the C4 hydroxyls. The B and F rings are predicted to share a common precursor, however, because S. ghanaensis also produces a moenomycin variant in which the methyl substituent on the F ring is lacking [2], and in this compound the C4 hydroxyl has the same stereochemistry as the B ring. The common precursor is speculated to be either UDP-glucuronic acid or UDP-galacturonic acid, both of which are used in exopolysaccharide biosynthesis in streptomycetes. Uncertainty in the precise nature of the donor arises because moe cluster 1 contains a putative epimerase, MoeE5, that could modify UDP-glucuronic acid to UDP-galacturonic acid. Other sugar tailoring genes include moeM5 for the carbamoyltransferase described above, as well as moeF5/moeH5 and moeK5, which encode the putative amidotransferase subunits and a putative methylcobalamin-dependent radical SAM methyltransferase [17,18], respectively, that modify the F ring (Figure 4).
The hypothesis that the sugars in MmA are derived from primary metabolism is supported by sequence analysis of the moe Gtfs themselves. The vast majority of Gtfs involved in secondary metabolite biosynthesis are predicted to have a characteristic two-domain fold, known as the GT-B superfamily fold [19]. These enzymes share an easily recognizable sequence motif. Only one of the Gtfs in moe cluster 1 is predicted to resemble Gtfs typically found in antibiotic production. This Gtf, MoeGT1, shows strongest homology to MurG, an UDP-N-acetylglucosaminyl transferase involved in transferring GlcNAc to a glycolipid during peptidoglycan biosynthesis [20, 21]. Therefore, we suggest that MoeGT1 carries out a similar reaction during MmA biosynthesis, namely, the transfer of N-acetylglucosamine to a lipid-linked F ring precursor (Figure 4). The other Gtfs show homology to Gtfs proposed to be involved in exopolysaccharide biosynthesis in various organisms. Thus, the Gtfs in moe cluster 1 are more similar to enzymes known or thought to be involved in primary metabolic processes than to enzymes typically involved in natural product biosynthesis.
The use of building blocks from primary metabolism apparently extends to the biosynthesis of the phosphoglycerate-lipid chain of MmA. There are only two putative prenyltransferases in moe cluster 1, one of which, MoeN5, shows homology to other prenyltransferases involved in isoprenoid biosynthesis and is thus predicted to be involved in the synthesis of the C25 lipid chain. Arigoni, Welzel, and coworkers have proposed a mechanism for the formation of the irregular isoprenoid chain of MmA that involves the coupling of C15 and C10 subunits, both of which could be derived from precursors produced in primary metabolic pathways [1, 9–11]. Thus, a relatively complex isoprenoid structure is apparently governed by a single gene operating on available cellular metabolites (Figure 5).
Figure 5.

Proposed scheme for the biosynthesis of the lipid-phosphoglycerate moiety 4 of MmA. The letters PPO in the shaded rectangle represent pyrophosphate. 1 = farnesyl pyrophosphate; 2 = C10isoprenoid precursor. Intermediates on the pathway to moenocinol pyrophosphate 3 were postulated by Arigoni, Welzel and coworkers [10]. 3-R-PG = 3-phosphoglycerate. The formation of 1 with the cis configuration could be governed by a primary metabolism enzyme similar to M. tuberculosis Z-farnesyl diphosphate synthase [22].
The second prenyltransferase, MoeO5, is predicted to catalyze formation of the ether linkage between 3-R-phosphoglycerate, an abundant metabolite produced during glycolysis, and the isoprenoid chain (Figure 5). MoeO5 shows sequence homology to the archaeal enzymes that form the first ether linkage between isoprenoid precursors and glycerol phosphate in the biosynthesis of the unusual membrane lipids that distinguish Archaea from the other kingdoms of life [23]. These archaeal prenyltransferases have a TIM barrel fold, one of the most common folds in nature, but unknown in other prenyltransferases [24]. MoeO5 is also predicted to have a TIM barrel fold, making it the first bacterial homolog of the archaeal prenyltransferases to be putatively assigned a function. We note that MoeO5 homologues exist in a range of bacteria and may catalyze similar prenyltransfer reactions, which would suggest that bacteria produce other metabolites that resemble archaeal membrane lipids. Indeed, there is recent evidence that this is the case [25].
Heterologous expression of subset of moe cluster 1 in Streptomyces lividans TK24
We have proposed that moe cluster 1 encodes all of the genes required to assemble a bioactive MmA derivative lacking the chromophore unit (compound 2, Figure 1) from cellular pools of available metabolites. To test this hypothesis, we constructed cosmid moeno38-1 from cosmid moeno38 in which the neo gene marker has been replaced with the 5.3-kb hyg-oriTRK2-intφC31 PCR fragment of vector pOOB40 using the λ-RED recombination approach [26, 27]. In a similar manner, the neo gene was replaced in the SuperCos1 vector to produce SuperCos1Hy. Cosmid moeno38-1 contains all the moe cluster 1 genes except moeR5moeS5, which are proposed to convert UDP-GlcNAc to UDP-chinovosamine. This cosmid was transferred conjugally into S. lividans TK24 and the presence of moeno38-1 in the TK24 strain was confirmed by PCR using primers specific to moeGT1, moeGT3 and to attRϕC31 [28]. Biochromatography showed that the cell extract and culture broth from the moeno38-1+ TK24 transconjugant, but not from a strain harboring the empty vector SuperCos1Hy, contain a biologically active compound (Figure 6). HPLC analysis showed that the compound has the same mobility and UV spectrum as the MmA precursor 2 lacking the C5N chromophore unit. High-resolution MS analysis showed that the new compound has a mass of 1500.6278 Da ([M-H]−). MS/MS analysis of the compound revealed several characteristic mass peaks commonly observed during fragmentation of moenomycins (Supplemental Data). Based on HPLC mobility, exact mass and biological data, the compound is proposed to be a pholipomycin derivative lacking the chromophore unit (compound 4, Figure 1; calculated mass of the negative ion: 1500.6278 Da). Pholipomycin is a known member of the moenomycin family in which the C ring is N-acetylglucosamine rather than chinovosamine [2]. Exclusive production of a pholipomycin precursor by the recombinant S. lividans strain is consistent with the proposed roles of the moeR5moeS5 genes in the biosynthesis of UDP-chinovosamine from UDP-GlcNAc. These data also indicate that the Gtf responsible for unit C attachment can accept not only UDP-chinovosamine but also UDP-GlcNAc. This finding is consistent with recent studies showing that the moenomycin complex of antibiotics purified from S. ghanaensis also contains pholipomycin (or 6C-hydroxy-MmA) as a component [7]. Most important, the results show that cosmid moeno38-1 carries all of genes necessary for formation of the moenomycin pentasaccharide phospholipid scaffold, confirming that the building blocks of the molecule are supplied from cellular metabolite pools. Based on sequence analysis and the results of heterologous moe gene expression, we suggest that production of a fully active 1.4 kDa MmA precursor will take only 9 dedicated genes (roughly 12-kb DNA fragment), 6 of which govern the assembly of the phosphoglycolipid scaffold and 3 of which direct 2 biologically important sugar tailoring reactions (carbamoylation and carboxyamidation of unit F).
Figure 6.

Biochromatography of S. lividans TK24 moeno38-1+ cell extracts (A) and culture broth (B), S. lividans TK24 SuperCos1Hy+ cell extracts (C) and culture broth (D). The MmA standard (10 mcg) is in lane E.
Discussion
The cloning and analysis of the genes governing MmA production provides the first insight into the genetics of phosphoglycolipid antibiotic biosynthesis. Certain features of the MmA biosynthetic pathway merit further comment. First, the involvement of groups of genes at two widely separated loci in natural product biosynthesis is rare but not completely unprecedented [29, 30]. More unusual is the size of the primary moe cluster 1, which is remarkably small (<30 Kb) given the size of the molecule produced (1583 Da). The antibiotic calicheamicin, which has a mass similar to that of MmA (1364 Da) and contains four sugar subunits, is produced by a gene cluster that is more than three times larger and contains many more ORFs than moe cluster 1 [31]. A comparison of the clusters for the biosynthesis of MmA, calicheamicin and other antibiotics containing multiple sugar subunits reveals a striking difference: the moe cluster contains very few genes involved in the biosynthesis of the individual sugar subunits. With few exceptions [32, 33], gene clusters for glycosylated metabolites contain nucleotidyltransferases. However, there is not a single nucleotidyltransferase gene in the moe cluster and other sugar biosynthetic genes are sparse as well.
We have demonstrated via heterologous expression of a subset of genes from moe cluster 1 that all of the sugar building blocks (as well as the building blocks of the moenocinol-phosphoglycerate moiety) are derived from primary metabolism and used either directly or with minimal modification to produce the pentasaccharide scaffold of MmA, which is subsequently tailored on the B and F rings. Although many other types of antibiotics derive building blocks from cellular metabolite pools, carbohydrate-containing antibiotic gene clusters typically contain a great many genes devoted to sugar biosynthesis. In fact, it was recently suggested that the formation of NDP-sugar precursors by dedicated nucleotidyltransferases represents a crucial branching point of secondary carbohydrate metabolic pathways from primary metabolism [34]. The discovery of moe cluster 1 shows that there is another paradigm for the synthesis of carbohydrate-rich secondary metabolites.
MmA is produced late in the streptomycetes life cycle [35], consistent with its role as a secondary metabolite, but the building blocks used to assemble the molecule are expected to be present throughout cell growth. Apparently, they are used to construct MmA only when the moe genes are transcribed. The regulation of moe cluster gene expression is unclear because it appears to lack dedicated regulatory genes. However, the moeA5, moeO5, moeR5, and moeE5 genes all contain rare TTA leucine codons. In S. coelicolor, 5′-processed LeutRNAUUA is accumulated in significant quantities only in late stationary phase [36], thus setting an important regulatory switch for antibiotic production [37]. Fewer than 0.2% of genes in the S. coelicolor genome contain TTA codons [37]. Therefore, the appearance of this codon in 20% of the genes within moe cluster 1 seems significant and suggests that in S. ghanaensis MmA production is coincident with the increased accumulation of the mature tRNA for TTA codon expression late in growth.
It is worth asking whether the MmA biosynthetic pathway, with its heavy reliance on available primary metabolite pools, represents a special case or a common paradigm for the synthesis of glycosylated secondary metabolites. We favor the latter possibility, and suggest that an appropriate search of bacterial genomes will turn up previously unrecognized natural product gene clusters having a genetic architecture similar to that of moe cluster 1. For example, the genome of the enterobacterium Photorhabdus luminescens contains a cluster of genes (plu3353-plu3368) encoding 5 putative Gtfs (one shows homology to both MoeGT4 and MoeGT5), one MoeO5 homologue, a prenyltransferase, as well as set of hypothetical proteins [38]. The genetic capacity of this cluster is enough to direct the formation of a phosphoglycolipid molecule. This and other clusters may have been overlooked as potential determinants of antibiotics or other bioactive compounds both because of their economy and because many of the carbohydrate-processing genes, like those in the MmA cluster, bear a stronger resemblance to genes involved in primary metabolism than to those involved in secondary metabolism. Thus, besides providing us with genetic tools for manipulating MmA structure, the discovery of the MmA cluster provides new insights into the biosynthesis of carbohydrate-containing natural products that may be useful in the annotation of bacterial genomes.
Significance
MmA is a chemically and biologically unique phosphoglycolipid antibiotic with potent activity against Gram-positive bacteria, including vancomycin- and methicillin-resistant pathogens. Development of novel antibacterial drugs based on the MmA pharmacophore is hampered by our poor understanding of the mechanisms of MmA inhibition and limited chemical tools for manipulating MmA structure. The cloning of the MmA biosynthetic genes paves the way towards the combinatorial biosynthesis and chemoenzymatic synthesis of moenomycin derivatives for use as antibiotics and as chemical probes to understand better the mechanism of action of bacterial transglycosylases involved in peptidoglycan biosynthesis and remodeling. The genetic organization of MmA pathway itself is remarkable for the apparent absence of genes for the formation of activated sugars, isoprenoid metabolism, and regulation; and for the presence of such novel genes as moeO5, which is homologous to prenyltransferases involved in archaeal membrane biosynthesis. Thus, impressive streamlining of the moe cluster and the presence of genes for unusual biotransformations are two salient features of MmA biosynthetic pathway. The MmA biosynthetic pathway provides new insights into secondary metabolism that may lead to the discovery of previously unrecognized natural product clusters in other bacterial genomes.
Experimental Procedures
Antibiotics
Pure MmA was provided by M. Adachi and S. Fuse (Dept. of Chemistry and Chemical Biology, Harvard University). For recombinant strains selection following commercially available antibiotics were used (mcg/mL): ampicillin (100), chloramphenicol (35), kanamycin (50), apramycin (50), hygromycin (100), spectinomycin (200), streptomycin (100).
Strains and Vector DNAs
Streptomyces ghanaensis ATCC14672 and Bacillus cereus ATCC19637 were obtained from ATCC. S. lividans TK24 was kindly provided by M. Kobayashi (University of Tsukuba, Japan). E. coli XL1 Blue MR and cosmid SuperCos1 were purchased from Stratagene. The methylation-deficient conjugative strain E. coli ET12567 (pUB307) [39] was obtained from Prof. C. P. Smith (Manchester University, UK). E. coli BW25113 (pIJ790) was from John Innes Center (Norwich, UK). Strains S. ghanaensis LH1, OB20a and OB21e with disrupted moeA4, moeM5 and moeGT1 genes, respectively, were constructed in this work. The vector pKC1139 with temperature-sensitive pSG5 replicon and integrative vector pSET152 [40] have been obtained from Prof. C.P. Smith. Vector pMKI9 was constructed by cloning of 0.2-kb HindIII-XbaI fragment carrying ermE promoter into respective sites of pKC1139 polylinker (provided by I. Ostash, L’viv University, Ukraine). To generate vector pOOB40, the central portion of the apramycin resistance gene aac(3)IV in vector pSET152 has been excised with SacI endonuclease, and the rest of the vector has been treated with Klenow fragment and ligated to the EcoRV hygromycin resistance cassette hyg [40]. Vector pOOB5 that which carries the spectinomycin resistance marker aadA instead of aac(3)IV was generated in the same way.
DNA Manipulations and Analysis
Standard molecular biology procedures were used throughout the work [40, 41]. The plasmid and fosmid libraries for S. ghanaensis ATCC14672 genome sequencing were created at the Broad Institute. Sequencing of cosmids moeno5, 38, 40 and their subclones was done at the Biopolymers Facility of Harvard Medical School using standard (M13, T7, T3) and custom designed primers. The generation, assembly and analysis of S. ghanaensis genomic sequences will be described separately. BLAST search tools (on the server of the National Center for Biotechnology Information, Bethesda, MD), FramePlot2.3.2 (13), CUPplot1.0 (at www.nih.go.jp/-jun/cgi-bin/frameplot.pl) and the Lasergene software package were used for S. ghanaensis sequence assembly, analysis and annotation. The CDD search engine (BLAST server) and a set of programs (HHPred, Pfam, TMHHM) on the ExPaSy proteomics server were utilized for identification of topology and conserved domains of Moe proteins.
The S. ghanaensis genomic library has been constructed using of SuperCos1. DIG-labeled internal fragments of genes moeC4, moeGT1 and moeGT3 amplified from the chromosome (primers are described in Supplemental Data) were used as hybridization probes for library screening. The 5.3-kb fragment of pOOB40 (hyg-oriT-int cassette) was engineered into cosmid moeno38 utilizing the primers and the strategy reported by Yanai et al. [42].
Generation of S. ghanaensis Disruption Mutants and Their Analysis
S. ghanaensis transconjugants (obtained according to described procedures [40]) carrying pKC1139-based disruption plasmids in replicative form were grown for 3 days in tryptic soy broth (TSB; Difco) at 30°C. The biomass was washed twice with water to remove apramycin used for plasmid selection, and approximately 105 cfu were inoculated into fresh TSB without antibiotic. The culture was incubated for 6 days at 40°C (to eliminate free plasmid), plated onto LB agar supplemented with apramycin and grown for 4–5 days at 37°C. Colonies with disruption plasmids integrated into the gene of interest were obtained with a frequency (3–4)× 10−5. No apramycin resistant colonies were detected in a control strain carrying the empty vector, implying that frequency of nonspecific integration of pKC1139 is below the level of detection (we usually plated 3×108 cfu), and that appearance of AmR colonies in the experimental strains is due to homologous integration. Moreover, 10 independent colonies of each disruption mutant were assayed for MmA production and in no case could we detect the MmA+ phenotype due to possible non-specific integration of the plasmid into the S. ghanaensis genome. We also did not detect reversions to the AmS and MmA+ phenotype when the strains were grown in presence of apramycin at 37°C, indicating that under the stated conditions the pKC1139-based plasmids are stably integrated into the homology region of generated mutants (3–4 thousands colonies have been assayed). Passage of wild type strains under the cultivation conditions used to generate moe mutants did not affect MmA production. The knockouts were verified by Southern analysis using the internal fragments of disrupted genes as probes. For complementation experiments, matings of S. ghanaensis mutants with donor E. coli strains were incubated at 37°C and overlaid after 8 hours of growth. Control conjugations with empty vectors (either pOOB5 (Spr) or pOOB40 (Hyr)) were performed and the obtained transconjugants were analyzed in parallel with experimental strains to ensure that workup conditions do not affect the stability of MmA-minus mutants.
Moenomycin Production Analysis
For all moenomycin extraction procedures equal amounts of biomass (2g, wet weight) and fermentation medium (30mL) were used. For MmA production, S. ghanaensis mutants with disrupted moe genes and S. lividans strains were grown at 37°C for 4–5 days in mTSB (TSB supplemented with trace elements solution [40], 0.8 mL per 1L). For antibiotic disc diffusion assays, fermentation medium and concentrated methanol extracts of MmA from mycelium of streptomycete strains were applied to antibiotic assay discs (10 mm, Sigma) and stacked onto LA plates overlaid with soft agar containing MmA-sensitive B. cereus. Zones of B. cereus growth inhibition were observed after 12–14h of incubation at 30°C. Moenomycins were partially purified (60% purity) from the cell extracts using 100 mg C18 SPE cartridge (Waters) and analyzed by LC-MS as described in [43] on an Agilent 1100 series LC/MSD machine. For the biochromatography, moenomycin extracts were separated on silica gel aluminum TLC plates (mobile phase -methanol:acetonitrile:water 35:40:25), which were dried and overlaid with soft agar containing B. cereus. Following overnight incubation at 30°C, the plates were visualized with visible and UV light (254 nm). Accurate mass spectra of purified moenomycins from recombinant S. ghanaensis LH1 and S. lividans TK24 (moeno38-1) strains were acquired using an Agilent LC/MSD TOF machine. For the LC analysis a Gemini C18 column (5 μ, 4.6 mm × 100 mm) from Phenomenex was used (mobile phase A: H2O and mobile phase B: acetonitrile, with 0.1% ammonium hydroxide as a solvent modifier). Samples were run in negative ionization mode with the capillary voltage set to 4.0 kV and the fragmentor voltage to 150 V. The drying gas temperature was 300 °C, the drying gas flow rate was 7 L/min, and the nebulizer pressure was 15 psi. (−)ESI-MS2 spectra of compounds 2 and 4 were acquired with Micromass qTOF2 machine. Cone and capillary voltages were set to 35 V and 3 kV, respectively, collision energy was 30 V.
Supplementary Material
Supplemental Data, including detailed description of moe genes, primer sequences, plasmid construction, mutant strains verification and MS/MS analysis are available online.
Acknowledgments
This work was supported by startup funds from Harvard Medical School and NIH grant AI50855 (to S.W.). We gratefully acknowledge the contributions of Dr. Bruce Birren and his staff at the Broad Institute. We thank Dr. M. Adachi for assistance with purification and analysis of MmA derivatives from mutant strains, and Wenjiang Zhu for assistance with manuscript preparation. Prof. M. Bibb (John Innes Center, UK) is thanked for the REDIRECT system.
Abbreviations
- MmA
moenomycin A
- moe
moenomycin biosynthetic genes
- Gtf(s)
glycosyltransferase(s)
- ALA
–5-aminolevulinic acid
Footnotes
Accession Numbers
The sequences reported in this paper have been deposited in the GenBank database (accession N DQ988993 and DQ988994 for moe clusters 2 and 1, respectively )
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ostash B, Walker S. Bacterial transglycosylase inhibitors. Curr Opin Chem Biol. 2005;9:459–466. doi: 10.1016/j.cbpa.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Welzel P. Syntheses around the transglycosylation step in peptidoglycan biosynthesis. Chem Rev. 2005;105:4610–4660. doi: 10.1021/cr040634e. [DOI] [PubMed] [Google Scholar]
- 3.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 4.Weber T, Welzel K, Pelzer S, Vente A, Wohlleben W. Exploiting the genetic potential of polyketide producing streptomycetes. J Biotechnol. 2003;106:221–232. doi: 10.1016/j.jbiotec.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Petricek M, Petrickova K, Havlicek L, Felsberg J. Occurrence of two 5-aminolevulinate biosynthetic pathways in Streptomyces nodosus subsp asukaensis is linked with the production of asukamycin. J Bacteriol. 2006;188:5113–5123. doi: 10.1128/JB.01919-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McAlpine JB, Bachmann BO, Piraee M, Tremblay S, Alarco AM, Zazopoulos E, Farnet CM. Microbial genomics as a guide to drug discovery and structural elucidation: ECO-02301, a novel antifungal agent, as an example. J Nat Prod. 2005;68:493–496. doi: 10.1021/np0401664. [DOI] [PubMed] [Google Scholar]
- 7.Zehl M, Pitternauer E, Rizzi A, Allmaier G. Characterization of moenomycin antibiotic complex by multistage MALDI-IT/RTOF-MS and ESI-IT-MS. J Am Soc Mass Spectrom. 2006;17:1081–1090. doi: 10.1016/j.jasms.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 8.Fehlhaber HW, Girg M, Seibert G, Hobert K, Welzel P, Heijenoort Y, Heijenoort JV. Moenomycin A: a structural revision and new structure-activity relations. Tetrahedron. 1990;46:1557–1568. [Google Scholar]
- 9.Schuricht U, Endler K, Hennig L, Findeisen M, Welzel P. Studies on the biosynthesis of the antibiotic moenomycin A. J Prakt Chem. 2000;342:761–772. [Google Scholar]
- 10.Schuricht U, Hennig L, Findeisen M, Endler K, Welzel P, Arigoni D. The biosynthesis of moenocinol, the lipid part of the moenomycin antibiotics. Tetrahedron Lett. 2001;42:3835–3837. [Google Scholar]
- 11.Neundorf I, Koehler C, Hennig L, Findeisen M, Arigoni D, Welzel P. Evidence for the combined participation of a C10 and a C15 precursor in the biosynthesis of moenocinol, the lipid part of the moenomycin antibiotics. ChemBioChem. 2003;4:1201–1205. doi: 10.1002/cbic.200300622. [DOI] [PubMed] [Google Scholar]
- 12.Durr C, Schnell HJ, Luzhetskyy A, Murillo R, Weber M, Welzel K, Vente A, Bechthold A. Biosynthesis of the terpene phenalinolactone in Streptomyces sp Tu6071: analysis of the gene cluster and generation of derivatives . Chem Biol. 2006;13:365–377. doi: 10.1016/j.chembiol.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa J, Hotta K. FramePlot: a new implementation of the Frame analysis for the predicting protein-coding regions in the bacterial DNA with a high G+C content. FEMS Microbiol Lett. 1999;174:251–253. doi: 10.1111/j.1574-6968.1999.tb13576.x. [DOI] [PubMed] [Google Scholar]
- 14.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, et al. Complete genome sequence of the model actinomyceteStreptomyces coelicolor A3(2) Nature . 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 15.Welzel P, Kunisch F, Kruggel F, Stein H, Scherkenbeck J, Hiltmann A, Duddeck H, Mueller D, Maggio JE, Fehlhaber HW, et al. Moenomycin A: minimum structural requirements for biological activity. Tetrahedron. 1987;43:585–598. [Google Scholar]
- 16.Mendez C, Salas JA. The role of ABC transporters in antibiotic-producing organisms: drug secretion and resistance mechanisms. Res Microbiol. 2001;152:341–350. doi: 10.1016/s0923-2508(01)01205-0. [DOI] [PubMed] [Google Scholar]
- 17.Kamigri K, Hidaka T, Imai S, Murakami T, Seto H. Studies on the biosynthesis of bialaphos (SF-1293). C-P bond formation mechanism of bialaphos: discovery of a P-methylation enzyme. J Antibiot. 1992;45:781–787. doi: 10.7164/antibiotics.45.781. [DOI] [PubMed] [Google Scholar]
- 18.Woodyer R, Li G, Zhao H, van der Donk W. New insight into the mechanism of methyl transfer during the biosynthesis of fosfomycin. Chem Commun. 2007 doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Walker S. Remarkable structural similarities between diverse glycosyltransferases. Chem Biol. 2002;9:1287–1296. doi: 10.1016/s1074-5521(02)00295-8. [DOI] [PubMed] [Google Scholar]
- 20.Ha S, Walker D, Shi Y, Walker S. The 1.9 A crystal structure of Escherichia coli MurG, a membrane-associated glycosyltransferase involved in peptidoglycan biosynthesis. Protein Sci. 2000;9:1045–1052. doi: 10.1110/ps.9.6.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, Chen L, Ha S, Gross B, Falcone B, Walker D, Mokhtarzadeh M, Walker S. Crystal structure of the MurG:UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases. Proc Natl Acad Sci USA. 2003;100:845–849. doi: 10.1073/pnas.0235749100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulbach MC, Mahapatra S, Macchia M, Barontini S, Papi C, Minutolo F, Bertini S, Brennan PJ, Crick DC. Purification, enzymatic characterization, and inhibition of the Z-farnesyl diphosphate synthase from Mycobacterium tuberculosis. J Biol Chem. 2001;276:11624–11630. doi: 10.1074/jbc.M007168200. [DOI] [PubMed] [Google Scholar]
- 23.Nemoto N, Oshima T, Yamagishi A. Purification and characterization of geranylgeranylglyceryl phosphate synthase from a thermoacidophilic archaeon, Thermoplasma acidophilum. J Biochem. 2003;133:651–657. doi: 10.1093/jb/mvg083. [DOI] [PubMed] [Google Scholar]
- 24.Payandeh J, Fujihashi M, Gillon W, Pai EF. The crystal structure of (S)-3-O-geranylgeranylglyceryl phosphate synthase reveals an ancient fold for an ancient enzyme. J Biol Chem. 2006;281:6070–6078. doi: 10.1074/jbc.M509377200. [DOI] [PubMed] [Google Scholar]
- 25.Weijers JW, Schouten S, Hopmans EC, Geenevasen JA, David OR, Coleman JM, Pancost RD, Sinninghe Damste JS. Membrane lipids of mesophilic anaerobic bacteria thriving in peats have typical archaeal traits. Environ Microbiol. 2006;8:648–657. doi: 10.1111/j.1462-2920.2005.00941.x. [DOI] [PubMed] [Google Scholar]
- 26.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stinchi S, Azimonti S, Donadio S, Sosio M. A gene transfer system for the glycopeptide producer Nonomuraea sp. ATCC. 39727. FEMS Microbiol Lett. 2003;225:53–57. doi: 10.1016/S0378-1097(03)00490-7. [DOI] [PubMed] [Google Scholar]
- 29.Yu TW, Bai L, Clade D, Hoffman D, Toelzer S, Trihn KQ, Xu J, Moss SJ, Leistner E, Floss HG. The biosynthetic gene cluster of the maytansinoid antitumor agent ansamitocin from Actinosynnema pretiosum. Proc Natl Acad Sci USA. 2002;99:7968–7973. doi: 10.1073/pnas.092697199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tahlan K, Park HU, Jensen SE. Three unlinked gene clusters are involved in clavam metabolite biosynthesis in Streptomyces clavuligerus. Can J Microbiol. 2004;50:803–810. doi: 10.1139/w04-070. [DOI] [PubMed] [Google Scholar]
- 31.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, et al. The calicheamicin gene cluster and its iterative type I enediyne PKS. Science. 2002;297:173–176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 32.Madduri K, Waldron C, Merlo DJ. Rhamnose biosynthesis pathway supplies precursors for primary and secondary metabolism in Saccharopolyspora spinosa. J Bacteriol. 2001;183:5632–5638. doi: 10.1128/JB.183.19.5632-5638.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gullon S, Olano C, Abdelfattah MS, Brana AF, Rohr J, Mendez C, Salas JA. Isolation, characterization, and heterologous expression of the biosynthesis gene cluster for the antitumor anthracycline steffimycin. Appl Environ Microbiol. 2006;72:4172–4183. doi: 10.1128/AEM.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kudo F, Kawabe K, Kuriki H, Eguchi T, Kakinuma K. A new family of glucose-1-phosphate/glucosamine-1-phosphate nucleotidylyltransferase in the biosynthetic pathways for antibiotics. J Am Chem Soc. 2005;127:1711–1718. doi: 10.1021/ja044921b. [DOI] [PubMed] [Google Scholar]
- 35.Wallhauesser KH, Nesemann G, Prave P, Steigler A. Moenomycin, a new antibiotic. I Fermentation and isolation. Antimicrob Agents Chemother. 1965:734–736. [PubMed] [Google Scholar]
- 36.Leskiw BK, Mah R, Lawlor EJ, Chater KF. Accumulation of bldA-specified tRNA is temporally regulated in Streptomyces coelicolor A3(2) J Bacteriol. 1993;175:1995–2005. doi: 10.1128/jb.175.7.1995-2005.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chater KF. Streptomyces inside-out: a new perspective on the bacteria that provide us with antibiotics. Phil Trans R Soc . 2006;B 361:761–768. doi: 10.1098/rstb.2005.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duchaud E, Rusniok C, Frangeul L, Buchrieser C, Givaudan A, Taourit S, Bocs S, Boursaux-Eude C, Chandler MM, Charles JF, et al. The genome sequence of the entomopathogenic bacterium Photorhabdus luminescens. Nat Biotechnol. 2003;21:1307–1313. doi: 10.1038/nbt886. [DOI] [PubMed] [Google Scholar]
- 39.Flett F, Mersinias V, Smith CP. High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol Lett. 1997;155:223–229. doi: 10.1111/j.1574-6968.1997.tb13882.x. [DOI] [PubMed] [Google Scholar]
- 40.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich, U.K.: 2000. [Google Scholar]
- 41.Sambrook J, Russel DW. Molecular Cloning: A Laboratory Manual. 3 Cold Spring Harbor Lab. Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 42.Yanai K, Murakami T, Bibb M. Amplification of the entire kanamycin biosynthetic gene cluster during empirical strain improvement of Streptomyces kanamyceticus. Proc Natl Acad Sci USA. 2006;103:9661–9666. doi: 10.1073/pnas.0603251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eichhorn P, Aga D. Characterization of moenomycin antibiotics from medicated chicken feed by ion-trap mass spectrometry with electrospray ionization. Rapid Commun Mass Spectrom. 2005;19:2179–2186. doi: 10.1002/rcm.2044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data, including detailed description of moe genes, primer sequences, plasmid construction, mutant strains verification and MS/MS analysis are available online.