

Abstract
Apolipoprotein AI (apoAI) is the principal protein constituent of high density lipoproteins and it plays a key role in human cholesterol homeostasis; however, the structure of apoAI is not clearly understood. To test the hypothesis that apoAI is organized into domains, three deletion mutants of human apoAI expressed in Escherichia coli were studied in solution and in reconstituted high density lipoprotein particles. Each mutant lacked one of three specific regions that together encompass almost the entire 243 aa sequence of native apoAI (apoAI Δ44-126, apoAI Δ139-170, and apoAI Δ190-243). Circular dichroism spectroscopy showed that the α-helical content of lipid-free apoAI Δ44-126 was 27% while the other mutants and native apoAI averaged 55 ± 2%, suggesting that the missing N-terminal portion contains most of the α-helical structure of lipid-free apoAI. ApoAI Δ44-126 exhibited the largest increase in α-helix upon lipid binding (125% increase versus an average of 25% for the others), confirming the importance of the C-terminal half of apoAI in lipid binding. Denaturation studies showed that the N-terminal half of apoAI is primarily responsible for α-helix stability in the lipid-free state, whereas the C terminus is required for α-helix stability when lipid-bound. We conclude that the N-terminal half (aa 44–126) of apoAI is responsible for most of the α-helical structure and the marginal stability of lipid-free apoAI while the C terminus (aa 139–243) is less organized. The increase in α-helical content observed when native apoAI binds lipid results from the formation of α-helix primarily in the C-terminal half of the molecule.
Epidemiological studies have shown that high density lipoprotein (HDL) and its major protein component, apolipoprotein AI (apoAI), are protective factors against atherosclerosis and coronary artery disease (1). It is generally accepted that HDL removes unesterified (free) cholesterol (FC) and other lipids from peripheral cells and delivers them to the liver for catabolism. Along this reverse cholesterol transport pathway, apoAI can exist in multiple conformations that are related to its degree of lipid association. However, the regions of the apoAI sequence where such lipid-induced structural changes occur remain unknown and are the focus of this work.
The 243 aa polypeptide chain of apoAI is organized into repeating 22-mer segments (see Fig. 1) that are predicted to form the amphipathic α-helical segments that are primarily responsible for lipid-binding (for a review, see ref. 2). Intensive study of reconstituted HDL (rHDL) particles has led to a model in which antiparallel segments are arranged around a disc-shaped phospholipid (PL) bilayer. The hydrophobic helical faces interact with the PL acyl chains while the hydrophilic faces are exposed to the aqueous medium (3). The number of apoAI helical segments varies between 6 and 9 per molecule, depending on the number of molecules per particle and on the amount of PL present (4). Much less is known about the structure of the lipid-free form of apoAI. Free apoAI is soluble in water to high concentrations and self-associates by an unknown mechanism (5). Initially, investigators placed little importance on lipid-free apoAI because it is present at low levels in plasma (6). However, recent reports have indicated that apoAI may dissociate from plasma HDL or other lipoproteins (7, 8) to form particularly efficient FC acceptor particles. Fielding et al. (9, 10) have proposed that lipid-poor pre-β1 particles, which are conformationally distinct from lipid-bound apoAI (9), account for a significant portion of FC transfer from the surface of peripheral cells (10). An understanding of why lipid-poor apoAI efficiently removes lipids requires knowledge of the conformational transitions that occur across the apoAI molecule as it binds lipids. These details have not yet been determined from studies of intact apoAI.
Figure 1.
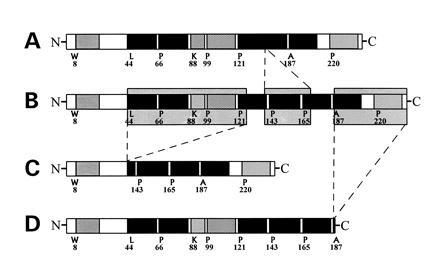
Organization of potential α-helical segments of human apoAI and three deletion mutants. The proteins shown are: A, apoAI Δ139-170 (211 aa, 24.4 kDa); B, native WT apoAI (243 aa, 28 kDa); C, apoAI Δ44-126 (160 aa, 18 kDa); D, apoAI Δ190-243 (190 aa, 22 kDa). The sites of potential α-helical structures are taken from Brouillette et al. (2, 26).
The x-ray crystal structure of a fragment of apolipoprotein E (apoE) has provided important clues to the solution structures of the exchangeable apolipoproteins. Lipid-free apoE contains a well-defined domain structure in which the N-terminal half of the protein resembles a water soluble, globular protein composed of an organized bundle of α-helices (11). The much less stable C-terminal domain is likely responsible for lipid binding. Given the sequence similarities and close evolutionary relationship (12) between apoE and apoAI, we hypothesized that apoAI exhibits a similar domain organization and that these domains play different roles in stabilizing the lipid-free and lipid-bound states of the protein. Previous findings from this laboratory have already suggested strong protein-protein interactions in the N terminus of lipid-free apoAI (3), consistent with an organized N-terminal domain. Furthermore, as with apoE, several groups have identified the C terminus of apoAI as being critical for lipid binding (2, 13, 14).
To determine in more detail the structural changes that occur throughout apoAI in response to lipid binding, we used three mutants of human apoAI lacking three regions that together encompass almost the entire length of the protein. The mutant proteins were designated apoAI Δ44-126, which lacked most of the N-terminal half of apoAI, apoAI Δ139-170, which lacked a central region, and apoAI Δ190-243, which lacked two C-terminal α-helices that are known to be important in lipid binding.
EXPERIMENTAL PROCEDURES
Materials.
Sodium cholate, FC, and 1-palmitoyl, 2-oleoyl-phosphatidylcholine (POPC) were purchased from Sigma (+99% grade). Bis(sulfosuccinimidyl) suberate (BS3) was purchased from Pierce. All other reagents were analytical grade.
Methods.
Purification of apoAI and construction of apoAI mutants. Human HDL was isolated from plasma of normal subjects from the Champaign County Blood Bank by sequential ultracentrifugation in the density range of 1.063 to 1.21 g/ml. ApoAI was isolated as described previously (3, 4) and stored in lyophilized form at −20°C. Before use, purified apolipoproteins were solubilized either in 3 M guanidine·HCl or in 7 M urea and dialyzed extensively against Tris buffer (10 mM Tris/150 mM NaCl/1.0 mM EDTA/0.02% NaN3, pH 8.0). The mutant forms of mature human apoAI were constructed, expressed in Escherichia coli, and purified in P. Holvoet’s laboratory as described (15).
Preparation and characterization of rHDL particles.
All particles were reconstituted using the Bio-bead/sodium cholate method as described (4, 16). Initial molar ratios of PL/FC/apolipoprotein were 100:5:1. The particles were isolated on a calibrated Superdex 200 HR (10 × 30 mm) gel filtration column (Pharmacia) eluted at 0.5 ml/min with Tris buffer. The hydrodynamic diameters of rHDL particles were estimated from the column elution volume and by native 8–25% polyacrylamide gradient gel electrophoresis (PAGGE) (Pharmacia) as described (17). Protein contents were determined by the Markwell/Lowry protein assay (18). Phospholipids were determined by the method of Sokoloff and Rothblat (19). The number of apoAI molecules per particle and the self-association properties of the lipid-free proteins were determined by cross-linking with BS3 (4).
Circular dichroism (CD) and isothermal denaturation studies.
The average α-helical contents of apoAI and the mutants were determined by CD spectroscopy using a Jasco (Easton, MD) J-720 spectropolarimeter at 222 nm. Spectra were measured at 25°C in a 0.1-cm quartz cuvette. Sample concentrations from 0.05 to 0.1 mg/ml were used so that all apolipoproteins were monomeric (see Fig. 2). The α-helix contents of the proteins after 72 h incubations with increasing concentrations of guanidine HCl (Gdn·HCl) at 4°C were used to obtain the free energy of unfolding of apoAI as proposed by Aune and Tanford (20) and modified by Sparks et al. (21).
Figure 2.
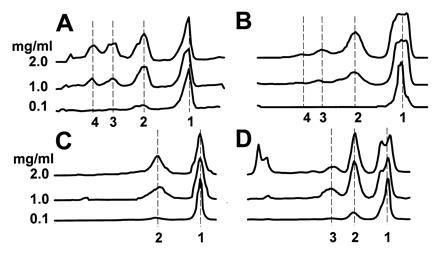
Densitometry scan of an SDS/PAGE analysis of the self association properties of apoAI and the deletion mutants studied by BS3 cross-linking. Samples were loaded onto the gels at the far left and the molecules migrated to the right. The numbers on the y axis that are next to individual lane scans show the concentration of each protein in 50 mM phosphate buffer at pH 7.4. The numbers on the x axis show the number of cross-linked apolipoprotein molecules associated with each band stained with Coomassie blue. (A) The concentration-dependent oligomerization of native, human plasma apoAI; (B) apoAI Δ190-243; (C) apoAI Δ139-170; (D) apoAI Δ44-126.
Fluorescence spectroscopy.
The concentration of protein was 0.1 mg/ml for all fluorescence studies. The wavelength of maximum fluorescence (WMF) of the Trp residues was determined from uncorrected spectra on a Perkin–Elmer MPF-66 fluorescence spectrophotometer using 4 nm excitation and emission band-passes. The samples were excited at 295 nm to avoid Tyr fluorescence. The emission was scanned from 305 to 375 nm at 25°C. Fluorescence quenching experiments were carried out on an ISS PC1 photon counting spectrofluorimeter at 25°C using increasing concentrations of KI (0–0.5 M) as described (22). After correction for buffer effects, the quenching parameters were calculated using two Stern-Volmer equations. The first assumes that all Trp residues are accessible to the quencher:
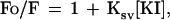 |
where Fo is the integrated fluorescence intensity in the absence of quencher, F is the intensity at each KI concentration, and Ksv is the Stern-Volmer constant. The second equation allows for a heterogeneous distribution of Trp environments in which a certain fraction (fa) is accessible to the quencher and the remainder is completely inaccessible (23):
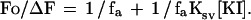 |
ΔF is the difference between Fo and the observed intensity at each concentration of KI. The model that best fit the data without unnecessary sacrifice in degrees of freedom was determined using the f statistic (P < 0.05) (24).
Tryptophan lifetime measurements were performed using a cross correlation phase and modulation fluorimeter at an excitation wavelength of 295 nm. The Trp lifetimes were observed under magic angle conditions of 0° excitation, 55° emission through a long wavelength pass filter (WG 320, Schott) to remove scattered light. P-terphenyl in absolute ethanol (lifetime, 1.05 ns) was used as a lifetime reference. The phase and modulation data were collected across a harmonic range of 3–300 MHz. Data were analyzed using globals unlimited software (25) for multiple lifetime components with a constant error of the modulation and phase data fixed at 0.004 and 0.2, respectively.
RESULTS
The mutant forms of mature apoAI purified from this E. coli system appeared homogeneous by SDS/PAGGE, and the calculated molecular weights were in agreement with those inferred from the sequences. A graphical representation of the three deletions examined in this study is shown in Fig. 1 with respect to elements of secondary structure as predicted from computer analysis of the primary sequence (2).
Self-Association of the Lipid-Free Apolipoproteins.
ApoAI in solution oligomerizes in a reversible and concentration-dependent manner (5). To assess the impact of the sequence deletions on protein self-association, the lipid-free apoAI and the deletion mutants were solubilized in phosphate buffer at three concentrations (0.1, 1.0, and 2.0 mg/ml) and treated with the homobifunctional cross-linking agent BS3 (span of 11 Å). Protein molecules in sufficient proximity in solution were covalently joined and visualized by SDS/PAGGE (Fig. 2). At 0.1 mg/ml, native apoAI and all mutants existed predominantly as monomers (>85% monomers). As the protein concentration was increased, the ratio of oligomers relative to monomers increased for all four proteins. At 2.0 mg/ml, native apoAI oligomerized extensively exhibiting monomeric (34% of total protein), dimeric (29%), trimeric (21%), and tetrameric (15%) forms (Fig. 2A). At the same concentration, the profile for apoAI Δ190-243 (Fig. 2B) showed a similar oligomerization pattern to that of native apoAI, although the self-association process was less efficient in the absence of the C-terminal segment (monomer, 56%; dimer, 30%; trimer, 10%; tetramer, 4%). The apoAI Δ139-170 (Fig. 2C) pattern was unique in that the largest oligomer formed was a dimer, even at 2.0 mg/ml (monomer, 62%; dimer, 38%). Despite the absence of a substantial portion of the N-terminal half of the protein, apoAI Δ44-126 retained the ability to form all of the oligomers found in native apoAI (monomer, 43%; dimer, 28%; trimer, 5%; tetramer, 3%) and much larger complexes that remained at the stacking/running gel interface (22%) (Fig. 2D).
Physical Properties of rHDL Particles.
The abilities of the three apoAI deletion mutants to form rHDL particles with POPC and FC were determined. In all cases, the reconstitutions were carried out with an initial POPC/FC/protein ratio of 100:5:1 (mol:mol), which has been shown to result primarily in a 96 Å particle containing two molecules of native apoAI per complex (3). The final particle compositions and hydrodynamic diameters are listed in the legend to Fig. 3.
Figure 3.
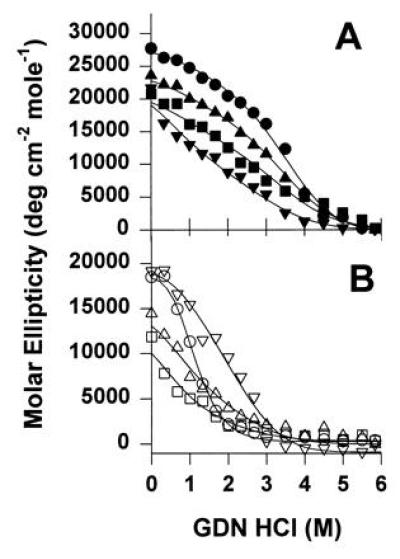
The effect of Gdn·HCl concentration on the molar ellipticity of native apoAI and the deletion mutants in both the lipid-bound (rHDL) (A) and lipid-free (B) states. Samples at 50 μg protein per ml were incubated with from 0 to 6 M Gdn·HCl in 0.05 M phosphate buffer (pH 7.4) for 72 h at 4°C. The molar ellipticity was measured at 25°C in a 0.1 cm path length quartz cell. The rHDL particles were: apoAI Δ190-243 (▾), native apoAI (•), apoAI Δ139-170 (▴), and apoAI Δ44-126 (▪). The molar composition of the the rHDL made with native apoAI (PL/FC/protein) was 168:10:2 with a diameter of 100 Å as measured by native PAGGE. rHDL Δ190-243 had a composition of 84:2:4, diameter of 79 Å; rHDL Δ139-170 had a composition of 132:6:3, diameter of 91 Å, and rHDL Δ44-126 had a composition of 168:8:4, diameter of 109 Å. The lipid-free proteins that were denatured were: apoAI Δ190-243 (▿), native apoAI (○), apoAI Δ139-170 (▵), and apoAI Δ44-126 (□). The curves were derived by nonlinear analysis of the data which represents two separate denaturation experiments performed on each of two independent reconstitutions.
Secondary Structure and Conformational Stability of Lipid-Free Apolipoproteins and rHDL Particles.
CD was used to determine the impact of the various deletions on the average α-helical content of the lipid-free proteins and the corresponding rHDL particles. Table 1 shows the α-helix contents of each apolipoprotein and the calculated number of amino acids that are involved in α-helices. Lipid-free native apoAI and wild-type apoAI exhibited similar α-helix contents of 53–57%, consistent with previous studies (17). Lipid-free apoAI Δ44-126 was strikingly less helical (27%), with only about 43 helical aa compared with 139 aa for native apoAI. The lower helicity suggests that the C-terminal half of apoAI is predominantly nonhelical when present in solution. Furthermore, the large portion of the N-terminal sequence deleted in this mutant must be responsible for most of the helical structure of native lipid-free apoAI. The sequence deletions in apoAI Δ139-170 and apoAI Δ190-243 did not cause the sharp decreases in helicity of the deletion in apoAI Δ44-126, supporting the idea of a low degree of helicity of the C-terminal half of the protein in solution.
Table 1.
α-Helix content and Gdn·HCL denaturation of human plasma apoAI and recombinant deletion mutants in the lipid-free state and in discoidal rHDL
| Apolipoprotein* | α-helix content,†
%
|
No. of amino acids in
α-helices‡
|
D1/2,§ M
Gdn·HCl
|
ΔGD°, ¶ kcal/mol
apoAI
|
||||
|---|---|---|---|---|---|---|---|---|
| Lipid-free | rHDL | Lipid-free | rHDL | Lipid-free | rHDL | Lipid-free | rHDL | |
| Plasma apoAI | 57 ± 3 | 80 ± 6 | 139 | 194 | 1.2 ± 0.2 | 3.1 ± 0.5 | 2.5 ± 0.9 | 2.4 ± 0.5 |
| apoAI Δ44-126 | 27 ± 2 | 59 ± 19 | 43 | 96 | 0.8 ± 0.4 | 2.5 ± 0.2 | 1.0 ± 0.1 | 1.7 ± 0.1 |
| apoAI Δ139-170 | 54 ± 9 | 68 ± 0 | 114 | 143 | 0.9 ± 0.2 | 2.8 ± 0.2 | 1.4 ± 0.2 | 1.8 ± 0.2 |
| apoAI Δ190-243 | 56 ± 7 | 63 ± 3 | 106 | 120 | 1.8 | 1.7 | 2.4 | 1.2 |
Values in this table are derived from triplicate measurements from two independent reconstitution experiments except for the apoAI Δ190-243 (n = 1).
Determined from molar ellipticities at 222 nm (±1 SD).
Calculated by multiplying the number of amino acids in the protein by the α-helix content.
Gdn·HCl concentration for the protein to become 50% denatured (±1 SD).
Standard change in free energy of denaturation (±1 SD).
When complexed with POPC and FC, the helical content of all proteins increased. Native apoAI, apoAI Δ190-243, and apoAI Δ139-170 α-helical contents increased about 13–40% upon lipid binding. ApoAI Δ44-126 showed a much larger increase in helicity in the presence of lipids, more than doubling the number of α-helical amino acids. Applying similar logic as for the lipid-free case, this result suggests that the large change in helical content is primarily due to residues normally found in the C-terminal half of apoAI. The fact that neither apoAI Δ139-170 nor apoAI Δ190-243 become as helical as native apoAI upon lipid binding confirms that these regions are important for α-helix formation in response to lipid.
The thermodynamic stability of the α-helices in the various apolipoproteins was monitored by CD in the presence of increasing concentrations of Gdn·HCl (Table 1). The denaturation profiles for all samples were monotonic (Fig. 3), indicating a two-state denaturation process (native state and denatured state) that can be analyzed by the denaturant binding model as proposed by Aune and Tanford (20). In the lipid-free form, native apoAI exhibited the well-reported free energy of unfolding of about 2.5 kcal/mol of apoAI (27). Removal of the extreme C-terminal region (apoAI Δ190-243) had no affect on the stability (2.4 kcal/mol), whereas removal of most of the N-terminal region (apoAI Δ44-126) reduced the stability of the lipid-free form (1 kcal/mol). This indicates that the α-helices in the C-terminal region of apoAI are unstable in solution and the N-terminal region is important for helical stability in the lipid-free state. The sequence of the central region of apoAI (Δ139-170) also appeared to be important for the conformational stability of the protein in solution.
When native apoAI is associated with POPC in rHDL particles, the conformational stability does not change dramatically from that of the lipid-free form (ref. 21; Table 1). However, the deletion mutants apoAI Δ44-126 and apoAI Δ139-170 showed significant increases in α-helical stability, paralleling the increases in α-helix content in response to lipid-binding. By contrast, the helix stability of lipid-bound apoAI Δ190-243 decreased in the presence of lipid. These results confirm a significant role for the extreme C-terminal segment for stabilization of apoAI in the lipid-bound form and suggest that the helices in the N-terminal half of the protein are less important in lipid binding.
Fluorescence Spectroscopy of the Lipid-Free Apolipoproteins and rHDL Particles.
Trp fluorescence is a sensitive indicator of the relative hydrophobicity of the environment of the four Trp residues located in the N-terminal half of apoAI (positions 8, 50, 72, and 108). Apo Δ44-126 differs from the other proteins in this study because three of these residues have been deleted, leaving only a single Trp. Therefore, the fluorescence properties of this mutant cannot be directly compared with the others and are not discussed in detail.
In the case of the lipid-free apolipoproteins, native apoAI and wild-type apoAI exhibited a WMF of 334–335 nm, indicating relatively hydrophobic Trp environments that are likely maintained by protein/protein contacts. The Trp residues of apoAI Δ190-243 (335 nm) were in similar environments to those of native apoAI, but those in apoAI Δ139-170 (337 nm) were red shifted, suggesting a slightly more polar environment. In agreement with the WMF results, native apoAI and apoAI Δ190-243 exhibited similar Trp exposures to solvent as indicated by fluorescence quenching with KI. Apo Δ139-170 again differed by exhibiting homogeneous Trp environments and a lower Ksv. Native apoAI and apoAI Δ190-243 exhibited similar average fluorescent lifetimes of about 3 ns. Consistent with the WMF and quenching results, apoAI Δ139-170 was again different with a longer lifetime of 4.6 ns.
In rHDL complexes, the WMF values for all proteins were reproducibly blue shifted by 2–3 nm with respect to the lipid-free state, indicating a transition to a less polar environment likely resulting from Trp contact with lipids (3). Similarly, the Ksv and fa values indicated a decreased exposure of Trp to solvent. The exception in this case was apoAI Δ190-243, which showed a slight increase in Ksv. Lipid-bound native apoAI and apoAI Δ190-243 exhibited similar fractional accessibilities to the solvent. However, lipid-bound apoAI Δ139-170, as in the lipid-free case, exhibited different quenching properties. The average fluorescent lifetimes were similar for all the lipid-bound proteins at about 4.5 to 4.8 ns. The results show that the deletion of aa 139–170 may affect the N-terminal Trp environments in the lipid-bound form as well as the lipid-free form.
DISCUSSION
ApoAI Structure in the Lipid-Free Form.
The structure of monomeric apoAI. Barbeau et al. (5) have shown that lipid-free apoAI in solution approximates an elongated ellipsoid with a long axis of about 150 Å and a short axis of about 25 Å. It contains significant α-helical structure, but thermodynamic studies indicate that the helices are less stable than those in soluble globular proteins (27), indicating a loosely folded, dynamic conformation. Long-range interactions between the N- and the extreme C-terminal portions are unlikely because: (i) the fluorescence measurements in Table 2 show that the removal of the last 53 aa of the C terminus did not affect Trp environments in the N terminus, (ii) apoAI Δ44-126 is known to exhibit competent binding to lipid, despite the absence of most of the N-terminal half of the protein (15), (iii) studies by Morrison et al. (28) using CNBr fragments of apoAI showed that the N terminus was not needed to maintain the structures in the C-terminal region that were required for cell surface binding. Thus, although deletional mutagenesis may not be appropriate for detailed studies of many rigidly organized proteins, the linear nature of apoAI lends itself well to deletional mutagenesis studies because of the lack of long range protein/protein interactions. Indeed, more subtle, single residue substitutions have had relatively minor effects on overall apoAI structure (26, 29).
Table 2.
Tryptophan fluorescence parameters
| Apolipoprotein* | WMF,
nm
|
Ksv,† M−1
KI
|
fa‡
|
Average
lifetime,§ ns
|
||||
|---|---|---|---|---|---|---|---|---|
| Lipid-free | rHDL | Lipid-free | rHDL | Lipid-free | rHDL | Lipid-free | rHDL | |
| Plasma apoAI | 334 ± 1.2 | 332 ± 1.4 | 7.0 ± 1.4 | 4.9 ± 1.3 | 0.67 ± 0.03 | 0.55 ± 0.08 | 2.9 ± 0.1 | 4.5 ± 0.2 |
| Wild-type apoAI | 335 ± 0.8 | 332 | 6.6 ± 0.3 | 3.9 | 0.67 ± 0.01 | 0.59 | 3.2 | 4.5 |
| apoAI Δ139-170 | 337 ± 1.0 | 334 ± 0.9 | 2.1 ± 0.3 | 1.3 ± 0.4 | 1.04 ± 0.05 | 1.06 ± 0.04 | 4.6 ± 0.2 | 4.6 ± 0.3 |
| apoAI Δ190-243 | 335 ± 1.4 | 333 ± 0.4 | 6.3 ± 0.3 | 7.9 | 0.67 ± 0.01 | 0.56 | 3.0 ± 0.1 | 4.0 ± 0.1 |
Values in this table are derived from two independent reconstitution experiments except for the wild-type apoAI rHDL and the apoAIΔ190-243 rHDL (n = 1). The apoAI Δ44-126 mutant contains only one Trp residue (in position 8 of native apoAI) and cannot be directly compared with the other proteins that contain four Trp residues.
Stern-Volmer constant indicating relative exposure of quenchable fluorescent residues to the quenching agent (KI) (±1 SD).
Fraction of Trp fluorescence that is quenchable by KI; determined from Lehrer (23).
The average lifetime is calculated from the three lifetime fractional components by the formula: (τ1f1) + (τ2f2) + (τ3f3) where τx is the lifetime of a given fractional component and fx is the fractional contribution of that component.
It is clear from this work that the C terminus of lipid-free apoAI exhibited significantly lower α-helicity than intact apoAI (27% for apoAI Δ44-126 versus 57% for native apoAI). In the absence of long-range interactions, it is logical that this low α-helical content arose because the missing N-terminal half of apoAI was responsible for most of the helical structure of intact apoAI. Lipid-free apoAI Δ44-126 had 43 helical aa or about 12 turns of α-helix [at 3.6 aa per turn (2)]. Since native apoAI had 140 helical aa or about 39 helical turns, it can be estimated that the missing N-terminal segment was responsible for approximately 27 helical turns. Similarly, it can calculated that there were about 6 helical turns deleted from apoAI Δ139-170 and about 9 turns deleted from apoAI Δ190-243. These two deletions encompass most of the C-terminal half of apoAI present in apoAI Δ44-126, and the sum of 15 turns within these two deletions agrees generally with the 12 turns predicted from CD measurements on apoAI Δ44-126. Thus, the bulk of the α-helical content of lipid-free apoAI (approximately 24–27 turns) resides in the N-terminal region of apoAI between residues 44 to 126. The C-terminal half of apoAI (residues 126–243) is substantially less helical containing only 12–15 helical turns approximately evenly distributed between residues 139–170 and 190–243. The exact length and organization of the helices in the N-terminal half of apoAI cannot be determined from these studies, but it is likely that they are organized into a bundle to minimize contact of the hydrophobic faces of the α-helices with water.
The proposal of an organized N-terminal region in lipid-free apoAI is consistent with previous studies from this laboratory (3) and with a recent study by Gursky and Atkinson (30). Furthermore, systematic mapping of apoAI epitopes by Marcel and coworkers (31, 32) showed numerous discontinuous epitopes in the N-terminal and central portions of apoAI, consistent with a folded structure in which the N-terminal and central regions make contact. The dependence of the N-terminal Trp environments on the presence of the central region (aa 139–170) supports this hypothesis. No such discontinuous epitopes were found in the C-terminal half of the molecule. Finally, the x-ray crystal structures of the N-terminal part of apoE (11) and apolipophorin III (33) show that these related proteins contain organized helical bundles in solution. It should be noted that the intra helical associations that occur in the apoAI N-terminal domain are less stable than those in the N terminus of apoE [ΔGdo = 10 kcal/mol (11)], but the existence of these domains is likely important for the solubility of the lipid-free form of both proteins in plasma.
The self-association of apoAI.
The observation of the unstable C-terminal half of lipid-free apoAI offers a possible explanation for the propensity of apoAI to self-associate in solution. Intermolecular contact offers the benefits of stabilizing protein/protein interactions that may favor partial formation of C-terminal α-helical segments, providing a driving force for oligomerization. This is consistent with the studies of Osborne and Brewer (34), who demonstrated that the α-helix content of apoAI increases with the extent of oligomerization. The less efficient oligomer formation observed for apoAI Δ190-243 compared with intact apoAI could be due to the removal of potential helical regions from the C terminus. It is interesting to note that the removal of the central region (apoAI Δ139-170) abolished the formation of oligomers higher than dimers. Barbeau et al. (5) have suggested that apoAI monomers oligomerize along their long axis in an overlapping, end-to-end arrangement. If this is the case, then it appears that the region between aa 139 and 170 is important for the propagation of the oligomeric chain.
ApoAI Structural Transitions in Response to Lipid.
Upon lipid binding in rHDL particles, the helical content of apoAI Δ44-126 increased from 43 helical aa or about 12 helical turns in the lipid-free state to 96 helical aa or about 26 turns in rHDL. Assuming an average of 6 turns for a typical lipid-binding helical segment (2), this readily allows for the formation of the 4–5 helical segments that are predicted to form in this region of apoAI upon lipid binding (2) (see Fig. 1). Thus, the C-terminal half of the protein (from aa 126–243) undergoes a substantial increase in helicity upon lipid binding. Interestingly, the lipid-induced increase in α-helical content in the C terminus of apoAI was sufficient to account for the overall lipid induced increase in helical content of native apoAI. As discussed in the case of lipid-free proteins, the region deleted from apoAI Δ44-126 contained about 27 helical turns when the protein was in the lipid-free form. A similar calculation for the same protein in the lipid-bound form yields about 28 helical turns. Thus, although the conformation of the N-terminal half is known to change significantly upon lipid binding (3), there was not a major change in the overall helical content within this region of apoAI. This undoubtedly resulted from a rearrangement of the three-dimensional organization of the N-terminal helices in response to lipid. ApoAI Δ139-170 also exhibited efficient lipid binding characteristics suggesting that, although the removal of this central region affected the conformation of the N-terminal domain of apoAI, the presence of this region was not critical for the lipid binding properties of the C-terminal region.
In summary, we propose that lipid-free apoAI exists in solution as a loosely folded, asymmetric molecule with an organized bundle of α-helices present in the N-terminal half (aa 44–126) of the protein that is primarily responsible for the stability and solubility of the lipid-free form of the protein. The relatively unstable C-terminal half, especially residues 190–243, would be expected to readily participate in interactions which stabilize its potential amphipathic α-helical segments, including self-association and lipid binding. This observation offers a possible explanation for the enhanced efficiency of the lipid-poor forms of apoAI in mobilizing cellular FC and PL. The central region of the molecule, residues 139–170, appears to interact with the N-terminal portions of the molecule in the lipid-free form, possibly stabilizing the α-helical bundle in the N terminus. Finally, upon lipid binding, the C-terminal half (aa 139–243) of the molecule becomes highly α-helical and becomes the major stabilizing domain. The helices involved in protein–protein contacts in the N-terminal half rearrange to make protein-lipid contacts without a major change in overall helical content.
Acknowledgments
We thank Dr. Paul Holvoet (Center for Molecular and Vascular Biology, University of Lueven, Belgium) for his generous gift of the apoAI mutants. We thank Els Brouwers, Tina Sims, and Zhian Zhao for expert technical assistance. This work was supported by National Institutes of Health Grant HL-16059 to A.J., and a postdoctoral fellowship from the American Heart Association, Illinois Affiliate to W.S.D. The spectroscopic experiments were performed at the Laboratory for Fluorescence Dynamics (RR03155, supported by the National Institutes of Health).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: apoAI, apolipoprotein AI; apoE, apolipoprotein E; FC, free (unesterified) cholesterol; Gdn·HCl, guanidine hydrochloride; HDL, high density lipoprotein; rHDL, reconstituted HDL; PAGGE, polyacrylamide gradient gel electrophoresis; PL, phospholipid; POPC, 1-palmitoyl, 2-oleoyl phosphatidylcholine; WMF, wavelength of maximum fluorescence; BS3, bis(sulfosuccinimidyl) suberate.
References
- 1.Miller G J, Miller N E. Lancet. 1975;i:16–19. doi: 10.1016/s0140-6736(75)92376-4. [DOI] [PubMed] [Google Scholar]
- 2.Segrest J P, Jones M K, De Loof H, Brouillette C G, Venkatachalapathi Y V, Anantharamaiah G M. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 3.Jonas A, Wald J H, Toohill K H, Krul E S, Kézdy K E. J Biol Chem. 1990;265:22123–22129. [PubMed] [Google Scholar]
- 4.Jonas A, Kézdy K E, Wald J H. J Biol Chem. 1989;264:4818–4824. [PubMed] [Google Scholar]
- 5.Barbeau D L, Jonas A, Teng T, Scanu A M. Biochemistry. 1979;18:362–369. doi: 10.1021/bi00569a021. [DOI] [PubMed] [Google Scholar]
- 6.Neary R, Bhatnagar D, Durrington P, Ishola M, Arrol S, Mackness M. Arteriosclerosis. 1991;89:35–48. doi: 10.1016/0021-9150(91)90005-n. [DOI] [PubMed] [Google Scholar]
- 7.Liang H Q, Rye K A, Barter P J. J Lipid Res. 1994;35:1187–1199. [PubMed] [Google Scholar]
- 8.Nichols A V, Gong E L, Blanche P J, Forte T M, Shore V G. J Lipid Res. 1987;28:719–732. [PubMed] [Google Scholar]
- 9.Fielding P E, Kawano M, Catapano A L, Zoppo A, Marcovina S, Fielding C J. Biochemistry. 1994;33:6981–6985. doi: 10.1021/bi00188a030. [DOI] [PubMed] [Google Scholar]
- 10.Kawano M, Miida T, Fielding C J, Fielding P E. Biochemistry. 1993;32:5025–5028. doi: 10.1021/bi00070a008. [DOI] [PubMed] [Google Scholar]
- 11.Wilson C, Wardell M R, Weisgraber K H, Mahley R W, Agard D A. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- 12.Chan L, Li W. In: Structure and Function of Apolipoproteins. Rosseneu M, editor. Boca Raton, FL: CRC; 1992. pp. 33–62. [Google Scholar]
- 13.Schmidt H H, Remaley A T, Stonik J A, Ronan R, Wellman A, Thomas F, Zech L A, Brewer H B, Hoeg J M. J Biol Chem. 1995;270:5469–5475. doi: 10.1074/jbc.270.10.5469. [DOI] [PubMed] [Google Scholar]
- 14.Ji Y, Jonas A. J Biol Chem. 1995;270:11290–11297. doi: 10.1074/jbc.270.19.11290. [DOI] [PubMed] [Google Scholar]
- 15.Holvoet P, Zhao Z, Vanloo B, Vos R, Derrider E, Dhoest A, Taveirne J, Brouwers E, Demarsin E, Engelborghs Y, Rosseneu M, Collen D, Brasseur R. Biochemistry. 1995;34:13334–13342. doi: 10.1021/bi00041a009. [DOI] [PubMed] [Google Scholar]
- 16.Sparks D L, Phillips M C, Lund-Katz S. J Biol Chem. 1992;267:25830–25838. [PubMed] [Google Scholar]
- 17.Davidson W S, Gillotte K L, Lund-Katz S, Johnson W J, Rothblat G H, Phillips M C. J Biol Chem. 1994;270:5882–5890. doi: 10.1074/jbc.270.11.5882. [DOI] [PubMed] [Google Scholar]
- 18.Markwell M A, Haas S M, Bieber L L, Tolbert N E. Anal Biochem. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 19.Sokoloff L, Rothblat G H. Proc Soc Exp Biol Med. 1974;146:1166–1172. doi: 10.3181/00379727-146-38267. [DOI] [PubMed] [Google Scholar]
- 20.Aune K C, Tanford C. Biochemistry. 1969;8:4586–4590. doi: 10.1021/bi00839a053. [DOI] [PubMed] [Google Scholar]
- 21.Sparks D L, Lund-Katz S, Phillips M C. J Biol Chem. 1992;267:25839–25847. [PubMed] [Google Scholar]
- 22.Leroy A, Jonas A. Biochim Biophys Acta. 1994;1212:285–294. doi: 10.1016/0005-2760(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 23.Lehrer S S. Biochemistry. 1971;10:3254–3263. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 24.Motulsky H J, Rasnas L A. FASEB J. 1987;1:365–374. [PubMed] [Google Scholar]
- 25.Thompson R B, Gratton E. Anal Biochem. 1988;60:670–674. doi: 10.1021/ac00158a014. [DOI] [PubMed] [Google Scholar]
- 26.Brouillette C G, Anantharamaiah G M. Biochim Biophys Acta. 1995;1256:103–129. doi: 10.1016/0005-2760(95)00018-8. [DOI] [PubMed] [Google Scholar]
- 27.Tall A R, Small D M, Shipley G G, Lees R S. Proc Natl Acad Sci USA. 1975;72:4940–4942. doi: 10.1073/pnas.72.12.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrison J, Fidge N H, Tozuka M. J Biol Chem. 1991;266:18780–18785. [PubMed] [Google Scholar]
- 29.Jonas A, von Eckardstein A, Kezdy K E, Steinmetz A, Assmann G. J Lipid Res. 1991;32:97–106. [PubMed] [Google Scholar]
- 30.Gursky O, Atkinson D. Proc Natl Acad Sci USA. 1996;93:2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcel Y L, Provost P R, Koa H, Raffai E, Ngoc V D, Fruchart J C, Rassart E. J Biol Chem. 1991;266:3644–3653. [PubMed] [Google Scholar]
- 32.Bergeron J, Franck P G, Scales D, Meng Q, Castro G, Marcel Y L. J Biol Chem. 1995;270:27429–27438. doi: 10.1074/jbc.270.46.27429. [DOI] [PubMed] [Google Scholar]
- 33.Breiter D R, Kanost M R, Benning M M, Wesenberg G, Law J H, Wells M A, Rayment I, Holden H M. Biochemistry. 1991;30:603–608. doi: 10.1021/bi00217a002. [DOI] [PubMed] [Google Scholar]
- 34.Osborne J C, Brewer H B. Ann NY Acad Sci. 1980;348:104–121. doi: 10.1111/j.1749-6632.1980.tb21294.x. [DOI] [PubMed] [Google Scholar]