

Abstract
Containment of hepatitis C virus (HCV) and other chronic human viral infections is associated with persistence of virus-specific CD4 T cells, but ex vivo characterization of circulating CD4 T cells has not been achieved. To further define the phenotype and function of these cells, we developed a novel approach for the generation of tetrameric forms of MHC class II/peptide complexes that is based on the cellular peptide-exchange mechanism. HLA-DR molecules were expressed as precursors with a covalently linked CLIP peptide, which could be efficiently exchanged with viral peptides following linker cleavage. In subjects who spontaneously resolved HCV viremia, but not in those with chronic progressive infection, HCV tetramer–labeled cells could be isolated by magnetic bead capture despite very low frequencies (1:1,200 to 1:111,000) among circulating CD4 T cells. These T cells expressed a set of surface receptors (CCR7+CD45RA–CD27+) indicative of a surveillance function for secondary lymphoid structures and had undergone significant in vivo selection since they utilized a restricted Vβ repertoire. These studies demonstrate a relationship between clinical outcome and the presence of circulating CD4 T cells directed against this virus. Moreover, they show that rare populations of memory CD4 T cells can be studied ex vivo in human diseases.
Introduction
The characterization of CD4 T cell populations specific for microbial antigens or self-proteins represents a major challenge in human immunology. Precise definition of the MHC/peptide specificity and functional properties of CD4 T cell populations is required for a mechanistic understanding of human disease processes. Characterization of such T cell populations is particularly relevant to the outcome of chronic viral infections as well as disease mechanisms in chronic inflammatory diseases with proposed autoimmune pathogenesis. Ex vivo characterization of these cell populations is critical, since in vitro culture may significantly alter composition and functional properties of the populations of interest. Current approaches rely on particular effector functions, such as proliferation and cytokine production, and do not permit ex vivo characterization of T cell populations with a defined MHC class II/peptide specificity.
The development of tetrameric forms of MHC class I/peptide complexes has revolutionized the analysis of CD8 T cells, but expanded populations of human CD4 T cells have so far been visualized only in the synovial fluid of two of three patients with chronic Lyme arthritis with an MHC class II tetramer (1, 2). Despite intensive efforts, it has so far not been possible to directly quantify human CD4 T cell populations from peripheral blood with MHC class II tetramers. Both effector and memory CD4 T cells appear to be present at significantly lower frequencies than their CD8 counterparts (3), and it may thus be difficult to distinguish populations of interest from background with conventional tetramer-detection techniques. The robust generation of MHC class II tetramers that are suitable for systematic analysis of human CD4 T cell responses, and the identification of the major CD4 T cell epitopes, have also represented significant challenges.
We have developed a general approach for tetramer-based ex vivo analysis of human pathogen-specific memory CD4 T cells and used it to study the T cell response to hepatitis C virus (HCV). Chronic HCV infection is a major cause of liver cirrhosis and hepatocellular carcinoma worldwide and represents the leading indication for liver transplantation in the US (4, 5). HCV is a single-stranded RNA virus that causes chronic viremia with high-level virus production in the majority of infected subjects. Understanding of the key parameters of the naturally developing immune response to this virus is particularly relevant since spontaneous control of viremia is observed in 20–50% of infected subjects, with the virus becoming undetectable in plasma by RT-PCR. The HCV genome encodes a single polyprotein that is spliced into ten polypeptides, and the relatively small size of its genome makes it amenable to systematic study (4, 6). Spontaneous control of HCV viremia is associated with induction of a vigorous and broadly directed HCV-specific CD4 T cell response that is maintained long-term in peripheral blood after resolution of viremia (7–10). In contrast, individuals with chronic HCV infection exhibit a narrowly directed or undetectable HCV-specific CD4 T cell response (7–12). It is not known whether HCV-specific CD4 T cells are anergic or largely absent in chronically infected subjects lacking functional responses, since current methods do not permit isolation of HCV-specific CD4 T cells independent of a particular function. Isolation of CD4 T cells based on their T cell receptor (TCR) specificity is also important for dissecting mechanisms responsible for different clinical outcomes. We have therefore developed an approach to isolate and characterize HCV-specific CD4 T cell populations ex vivo, despite their low frequency among circulating lymphocytes.
Methods
Subjects.
This study was approved by the Institutional Review Boards of the Massachusetts General Hospital and the Nuffield Department of Medicine; all subjects gave written informed consent. All subjects were anti-HCV–positive as measured by enzyme immunoassay. HCV viral loads were measured by the Roche Amplicor Monitor assay (Roche, Branchburg, New Jersey, USA) (detection limit 300 copies/ml of plasma). Subjects 01-40, 98A, OXS, OXM, OXD and O1E spontaneously resolved HCV viremia, while subject 99D was treated with IFN/ribavirin therapy and subsequently resolved HCV viremia; these subjects had persistent documented undetectable plasma viral loads (<300 copies/ml of plasma). Subject OXK had received unsuccessful combination therapy 6 years previously, but all other subjects with chronic infection were untreated. HLA typing was performed by standard serological and molecular techniques (13). The HLA-DR alleles of the subjects included in this study were as follows: 99D: DRB1*0401; 01-40: DRB1*0401, DRB1*07; 98A: DRB1*0401, DRB1*10; OXS: DRB1*0404, DRB1*07; OXM: DRB1*0401, DRB1*0701; OXD: DRB1*04, DRB1*07; O1E: DRB1*0401, DRB1*11; 99-24: DRB1*0401, DRB1*15; OXB: DRB1*1501, DRB1*0401; OXK: DRB1*0404, DRB1*1101; O2P: DRB1*0401, DRB1*07.
Expression of DR/CLIP precursors.
The constructs for the expression of DR/CLIP complexes were based on those previously described for DR2 (DRA, DRB1*1501), which carried Fos and Jun dimerization domains (14, 15). For the β chain constructs (DRB1*0101, DRB1*0401, DRB1*1501, and DRB5*0101), the covalently linked myelin basic protein (MPP)peptide was replaced with the CLIP peptide (PVSKMRMATPLLMQA, single–amino acid code). The linker carried a thrombin-cleavage site, as reported (16). For the DRα chain construct, a BirA biotinylation site (GLNDIFEAQKIEWHE) was attached via a six–amino acid linker (GGSGGS) to the 3′ end of the Fos dimerization domain (17). The four different DRβ chain constructs were cotransfected with the DRα chain construct into CHO cells, and clones were screened for secretion of DR molecules in an ELISA in which mAb L243 (American Type Culture Collection, Manassas, Virginia, USA) was used for capture and a polyclonal DR antiserum for detection (15). Clones with the highest expression levels were expanded for inoculation into bioreactors. For large-scale protein production, the ACUSYST-miniMAX instrument (Cellex Biosciences Inc., Minneapolis, Minnesota, USA) was used, which allows control of temperature, pH, and media feed rate. Transfectants were grown in hollow fiber bioreactors with an internal surface area of 1.1 m2, and supernatants from the extracellular space were harvested by the instrument at a predetermined flow rate into a collection bottle placed at 4°C. These cultures were maintained for approximately 3 months, and supernatants were frozen at weekly intervals. DR molecules were purified by affinity chromatography with mAb L243 (American Type Culture Collection) as previously described (14).
Processing of DR molecules: biotinylation, thrombin cleavage, and peptide exchange.
Purified DR molecules were biotinylated using a 1:20 molar ratio of BirA to DR in a buffer containing 100 μM biotin, 10 mM ATP, 10 mM magnesium acetate, 50 mM Bicine (Sigma-Aldrich, St. Louis, Missouri, USA), and 1× protease inhibitor cocktail (Sigma-Aldrich) at pH 8.0. The final HLA-DR concentration was adjusted to 2.5 mg/ml with 10 mM Tris, pH 8.0, and the reaction was incubated for 2 hours at 30°C. The protein was then extensively dialyzed against PBS to remove free biotin. Biotinylation was confirmed by electrophoresis on native polyacrylamide gels (6%), and dimers, trimers, and tetramers of DR molecules could be visualized at different molar ratios of streptavidin and DR.
Prior to the peptide-exchange reaction, the linker was cleaved with thrombin for 2 hours to allow release of the CLIP peptide. Twenty units of thrombin (Novagen, Madison, Wisconsin, USA) were used per milligram for DRB1*0101 and DRB1*0401 molecules, and 40 units per milligram for DRB5*0101 and DRB1*1501. Thrombin cleavage was confirmed by SDS-PAGE based on a shift in the molecular weight of the DRβ chain. Thrombin-cleaved complexes are designated as *DR/CLIP. Peptide exchange was performed using dinitrophenol-labeled (DNP-labeled) peptides for affinity purification of defined DR/peptide complexes. The DNP group was attached to the N-terminus of peptides via an aminohexanoic acid linker during synthesis (New England Peptide Inc., Fitchburg, Massachusetts, USA); all peptides were HPLC-purified and analyzed by mass spectrometry.
The sequences of the HCV peptides used for construction of MHC class II tetramers were as follows: HCV 1248: GYKVLVLNPSVAATL; HCV 1579: SGENLPYLVAYQATVCARA; HCV 1770: SGIQYLAGLSTLPGNPAIASL. Control tetramers were generated with peptides from annexin II and gp100 that had been eluted from DR4 molecules of a melanoma cell line (annexin II, residues 208–223: DVPKWISIMTERSVPH; gp100, residues 44–59: WNRQLYPEWTEAQRLD) (18). Peptide-exchange reactions were carried out with 3.3 μM *DR/CLIP and 50 μM of the respective DNP-labeled peptide in a buffer containing 50 mM sodium citrate, pH 5.2, 1% octylglucoside, 100 mM NaCl, and 1× protease inhibitor cocktail (Sigma-Aldrich). The reactions were incubated overnight at 30°C and then concentrated by ultrafiltration. The *DR/CLIP concentration and reaction temperature were chosen based on preliminary experiments designed to minimize aggregation of empty DR molecules created by CLIP dissociation. Aggregation was substantially lower at a *DR/CLIP concentration of 3.3 μM compared with 8.25 μM, and at a reaction temperature of 30°C compared with 37°C.
DR molecules were separated from unbound peptide with a Superose 12 HPLC gel filtration column (Amersham Pharmacia Biotech, Piscataway, New Jersey, USA) using PBS at a flow rate of 0.8 ml/min. The peak representing DR molecules was collected and injected onto an anti-DNP HPLC affinity column. The anti-DNP affinity column was generated by covalent cross-linking of 10 mg of anti–DNP-1 antibody (BIOTREND Chemikalien GmbH, Cologne, Germany) to a 4.6 mm × 50 mm protein G column on POROS 20 XL media (Applied Biosystems, Foster City, California, USA). DR molecules with bound DNP-labeled peptide were eluted from the column using 50 mM 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS), pH 11.5, and eluates were neutralized by addition of 1 M phosphate, pH 6.0. Biotinylated, peptide-loaded DR molecules were concentrated by ultrafiltration (Centricon 2-ml concentrator; Millipore Corp., Bedford, Massachusetts, USA), and the buffer was simultaneously changed to PBS. Biotinylated DR/peptide complexes were frozen in small aliquots at –80°C and multimerized with labeled streptavidin prior to use in staining reactions.
Kinetics of CLIP dissociation and peptide association.
CLIP dissociation and HA peptide association were examined under conditions relevant to preparative loading of *DR/CLIP complexes. To study the kinetics of CLIP dissociation, 8.33 μM of *DR/CLIP was incubated overnight at 37°C with 100 nM Alexa-488–labeled CLIP peptide in 50 mM sodium citrate, pH 5.2, 1% octylglucoside, 100 mM NaCl, and 1× protease inhibitor cocktail (Sigma-Aldrich). A molar excess of the high-affinity influenza HA (residues 306–318) peptide (100 μM) was then added, and the reactions were incubated for different periods of time at 37°C and then frozen until analysis. Complexes of DR and Alexa-488–labeled peptide were separated from unbound peptide using a Bio-Silect SEC 125 gel filtration column (Bio-Rad Laboratories Inc., Hercules, California, USA) run at a flow rate of 1 ml/min with PBS. The Alexa-488–labeled peptide was detected using an HPLC fluorescence detector with excitation and emission wavelengths set at 495 and 519 nm, respectively (Varian ProStar 363; Varian Analytical Instruments, Walnut Creek, California, USA). Peptide association was examined using 3.3 μM *DR/CLIP and 50 μM Alexa-488–labeled influenza HA peptide, under the same conditions as described above. The amount of bound peptide was determined based on the surface area of the peak that represented the DR/peptide complex. Half-life for dissociation was calculated by fitting of data to an equation describing an exponential decay. Half-life for association was calculated by fitting of data to an equation describing exponential rise. The following labeled peptides were used in these experiments: CLIP (residues 87–101) Alexa-488–CGGGPVSKMRMATPLLMQA; influenza HA (residues 306–318) Alexa-488–CGGGPKYVKQNTLKLAT.
Peptide-binding assay.
Competition assays were performed to identify HCV peptides suitable for tetramer production. HCV peptides were used as competitors for binding of a biotinylated influenza HA (residues 306–318) peptide to thrombin-cleaved DR4/CLIP complexes. Competitor peptides were tested at concentrations ranging from 30 nM to 10 μM in the presence of 30 nM labeled HA peptide and 1.7 μM *DR/CLIP. Reactions were incubated for 20 hours at 37°C in 50 mM sodium citrate, pH 5.2, 1% octylglucoside, 100 mM NaCl, 100 μg/ml of BSA, and 1× protease inhibitor cocktail. One hundred nanograms of DR was loaded onto a 96-well plate (Wallac Oy, Turku, Finland) coated with 200 ng/well of mAb L243, and DR-bound peptide was detected with europium-labeled streptavidin. Fluorescence was quantitated using a DELFIA 1234 fluorometer (Wallac Oy).
Establishment of T cell lines specific for the influenza HA peptide.
PBMCs from normal donors with the appropriate DR subtype were plated at 2 × 105 cells per well in 96-well U-bottom plates in the presence of influenza HA (residues 306–318) peptide (0.2–1 μM) and RPMI 1640, 10% human serum, 2 mM L-glutamine, 10 mM HEPES, and 100 μg/ml penicillin/streptomycin. Recombinant IL-2 was added after 3 days to a final concentration of 5 U/ml (Roche Diagnostics, Indianapolis,Indiana, USA), and half of the media was changed every 3 days. Cell lines were maintained by restimulation with autologous irradiated mononuclear cells and HA peptide at 10- to 14-day intervals as described previously (19).
Establishment of HCV-specific CD4 T cell lines.
HCV-specific CD4 T cell lines were generated as previously described (7) by a single round of stimulation with a recombinant HCV antigen. Briefly, 5 × 106 PBMCs were stimulated with 1 μg/ml of recombinant C200 antigen (encoding NS3 and NS4 proteins) in media supplemented with 50 U/ml of recombinant IL-2. After 10–14 days of culture, T cell lines were stained with tetramers and analyzed for Vβ usage.
Intracellular cytokine staining.
Intracellular cytokine staining of PBMCs ex vivo and in short-term stimulated cell lines was performed as previously described (7).
MHC class II tetramer staining of PBMCs.
For tetramer formation, biotinylated DR/peptide complexes were incubated with R-phycoerythrin–labeled streptavidin for at least 1 hour on ice, at a 4:1 molar ratio of DR to streptavidin and a final DR concentration of 0.2 mg/ml. Cells (fresh PBMCs, cryopreserved PBMCs, or cells from a short-term stimulated line) were stained in 100 μl R10 medium (RPMI, 10% FCS, 10 mM HEPES, 2 mM L-glutamine, and 50 U/ml penicillin/streptomycin) with 2 μg of phycoerythrin-conjugated (PE-conjugated) MHC class II tetramer for 2 hours at room temperature. APC-conjugated anti-CD4, peridinin chlorophyll protein–conjugated (PerCP-conjugated) anti-CD14, PerCP-conjugated anti-CD19, and either FITC-conjugated CD27 or FITC-conjugated CD45RA mAb’s were added for the last 20 minutes of incubation. For CCR7 analysis, cells were stained with unconjugated anti–human CCR7 antibody (Becton Dickinson–Immunocytometry Products, San Jose, California, USA) during the last 20 minutes of tetramer incubation, then washed twice and stained with a FITC-conjugated secondary anti-mouse IgM antibody. The cells were incubated for 20 minutes and washed twice, and then anti-CD4, -CD14, and -CD19 were added and the cells incubated for an additional 20 minutes. Cells were washed twice and then incubated with anti-PE MicroBeads (Miltenyi Biotec Inc., Auburn, California, USA) for 20 minutes at 4°C. The cells were washed once, and 90% of the cells were applied to MS separator columns (Miltenyi Biotec Inc.) according to the manufacturer’s instructions. The other 10% of the tetramer-stained cells were reserved for FACS analysis; the total number of cells in this pre-enrichment sample was multiplied by 10 to determine the input number of cells for each sample. Two to three million PBMCs were used for each tetramer stain. The PE-positive cells were then eluted from the columns, stained with Via-Probe (Becton Dickinson–Immunocytometry Products), and analyzed by flow cytometry using CellQuest software (Becton Dickinson–Immunocytometry Products). Cells were gated on CD4+, CD14–, CD19–, and Via-Probe–negative cells. Multiple control tetramers with irrelevant peptides were used in most experiments to demonstrate specificity of binding by the relevant tetramers. The frequency of tetramer-positive cells was determined by division of the number of CD4+/tetramer+ cells after enrichment by the total number of CD4 T cells as calculated by FACS analysis of the pre-enrichment sample. For Vβ analysis, cells were labeled with FITC-conjugated antibodies (Immunotech, Westbrook, Maine, USA) following magnetic enrichment and washed after a 20-minute incubation at room temperature.
Results
MHC class II/CLIP precursors for the creation of tetramers.
We developed a novel approach for the generation of MHC class II tetramers that is based on the cellular peptide-exchange mechanism. In APCs, MHC class II molecules assemble in the ER with invariant chain, and the CLIP segment of invariant chain serves to protect the hydrophobic binding site. Following transport to the endosomal/lysosomal peptide-loading compartment, invariant chain is proteolytically cleaved, and the remaining CLIP peptide is exchanged with other peptides in a reaction catalyzed by HLA-DM (20–24). This cellular peptide-exchange mechanism thus preserves the functionality of MHC class II molecules prior to exposure to peptides in the appropriate cellular compartment and suggests a general strategy for the creation of MHC class II tetramers. We generated DR/CLIP complexes in which the CLIP peptide is covalently linked to the N-terminus of the DRβ chain and converted these precursors to an active, peptide-receptive form by thrombin cleavage of the linker (Figure 1a). Four different HLA-DR molecules were generated in which the nonpolymorphic DRα chain was paired with different DRβ chains (DRB1*0101, DRB1*0401, DRB1*1501, and DRB5*0101). Stable CHO transfectants that secreted these molecules were grown at a high density in bioreactors, and affinity purification from supernatants collected over 1 week of a 2- to 3-month culture yielded substantial quantities of purified DR/CLIP complexes: 45.6 mg (DRB1*0101), 57.7 mg (DRB1*0401), 34.3 mg (DRB1*1501), and 9.4 mg (DRB5*0101). SDS-PAGE demonstrated that these preparations were pure and that the CLIP peptide could be removed by thrombin cleavage (Figure 1b). Since 1 mg of protein is used to generate a tetramer, these DR/CLIP complexes can be used to generate many different DR/peptide complexes for systematic analysis of human CD4 T cell populations.
Figure 1.

Generation of tetramers from MHC class II/CLIP precursors. (a) Recombinant HLA-DR molecules were expressed in which the CLIP peptide was covalently attached to the N-terminus of the DRβ chain. These precursors were converted to the peptide-receptive form by thrombin cleavage of the linker. This strategy allowed a series of tetramers to be generated from a precursor protein. (b) SDS-PAGE (10%) of purified DR/CLIP precursors (7 μg per lane). Four different DR molecules were expressed in which DRα was paired with one of four DRβ chains (lanes 1 and 2: DRB1*0101; lanes 3 and 4: DRB5*0101; lanes 5 and 6: DRB1*1501; lanes 7 and 8: DRB1*0401). Thrombin cleavage of the linker (lanes 2, 4, 6, and 8) removed the CLIP peptide and reduced the molecular weight of the DRβ chain. (c) Kinetics of CLIP dissociation from DR4 (DRA, DRB1*0401). DR4 molecules were incubated with an Alexa-488–labeled CLIP peptide, and dissociation of labeled CLIP was examined at different time points following addition of a molar excess of unlabeled HA (residues 306–318) peptide by separating DR-bound and free peptide on an HPLC gel filtration column. (d) Peptide association is rapid and follows the kinetics of CLIP dissociation. DR4 molecules were incubated for different time intervals with Alexa-488–labeled HA peptide. The kinetics of CLIP dissociation (t1/2 of 5.3 min) closely mirrored those of HA peptide association (t1/2 of 5.4 min) indicating rapid binding of peptide. Addition of DM further accelerated this process (t1/2 of 2.5 min).
Exchange of CLIP with viral peptides.
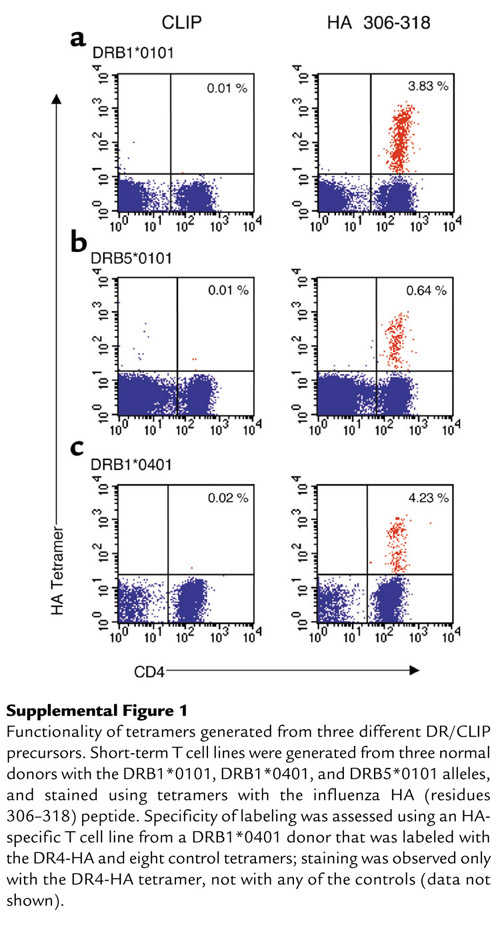
To test the functionality of the DR/CLIP precursors, we compared the kinetics of CLIP dissociation and peptide association using fluorescently labeled peptides under conditions used for the generation of tetramers. CLIP dissociation was assessed by loading of DR molecules (thrombin-cleaved) with an Alexa-488–labeled CLIP peptide and quantification of DR/CLIP–Alexa-488 complexes at different time points following addition of a large molar excess of unlabeled competitor peptide (Figure 1c). The complex with the bound fluorescent peptide was separated from unbound peptide by HPLC gel filtration chromatography and quantitated with an HPLC fluorescence detector. These experiments demonstrated rapid dissociation (t1/2 of 5.3 minutes) of the fluorescent CLIP peptide from DR4 at an acidic pH (5.2) that is characteristic for the endosomal peptide-loading compartment. The kinetics for association of an Alexa-488–labeled influenza HA (residues 306–318) peptide closely mirrored those observed for CLIP dissociation (t1/2 of 5.4 minutes). The rate of the reaction could be further accelerated by addition of HLA-DM (t1/2 of 2.5 minutes) (Figure 1d). CLIP dissociation from DRB5*0101 was slower, with a t1/2 of 3–4 hours, but again the rates of CLIP dissociation and peptide association were similar. The time required for CLIP dissociation is known to be allele dependent and ranged from several minutes to more than 4 hours for three allelic forms of DR molecules solubilized from metabolically labeled cell lines (25). Expression of DR molecules as precursors with a bound CLIP peptide thus greatly increased their biological activity, since the kinetics of peptide binding were substantially faster than reported for empty DR molecules (26). Since the influenza HA (residues 306–318) peptide binds to three of the four DR molecules that we had expressed (27, 28), we generated short-term T cell lines from normal donors with the DRB1*0101, DRB1*0401, or DRB1*1501 haplotype. The corresponding tetramers labeled distinct populations of CD4 T cells in these lines, indicating that tetramers generated by this approach were functional (See Supplemental Figure 1 at http://www.jci.org/cgi/content/full/112/6/831/DC1).
We next applied this tetramer technology to the assessment of a clinically relevant chronic viral disease, infection with HCV. Since the MHC restriction elements are difficult to determine for polyclonal human CD4 T cell populations, we examined DR4 (DRA, DRB1*0401) binding of seven HCV peptides that represent broadly recognized T cell epitopes for which the restricting class II alleles had not been unequivocally defined (7, 29). Three HCV peptides bound to DR4 (residues 1241–1260, 1581–1600, and 1771–1790), and two of these competed for binding of labeled peptide as effectively as did the influenza HA (residues 306–318) peptide that represents one of the highest-affinity DR4 binders (Figure 2a) (27, 28). Based on the DRB1*0401-binding motif, peptides were chosen for synthesis in which the core nine–amino acid segment was flanked by several N- and C-terminal residues, and a DNP-affinity tag was added to the N-terminus of each peptide (Table 1). A two-step purification process was used to isolate complexes loaded with a single peptide. DR/peptide complexes were first separated from free DNP-labeled peptide by HPLC gel filtration chromatography (Figure 2b), and the DR/peptide fraction from the gel filtration column was then injected into an HPLC DNP affinity column (Figure 2c). For the two high-affinity HCV peptides, close to 100% of DR molecules injected into the DNP column carried the DNP-labeled peptide, while a smaller fraction (43%) was loaded for the lower-affinity epitope. This affinity purification step therefore provided a homogenous DR/peptide preparation even for peptides that were not loaded with a high efficiency. This is relevant since the presence of DR molecules without the appropriate peptide would reduce the valency of the resulting staining reagents.
Figure 2.

Purification of HLA-DR molecules loaded with peptides from HCV. (a) Binding of HCV peptides to DR4. Based on a genome-wide analysis of the CD4 T cell response to HCV, seven candidate peptides were evaluated for DR4 binding. Unlabeled HCV peptides were used as unlabeled competitors for a biotinylated influenza HA (residues 306–318) peptide, and the amount of DR-bound biotinylated peptide was measured with europium-labeled streptavidin following capture of DR molecules with the L243 antibody. Two HCV peptides (HCV 1241 and HCV 1771; numbering based on the position of the first residue in the HCV polyprotein) showed a dose-response curve similar to that of the high-affinity influenza HA peptide, while higher concentrations of the HCV 1581 peptide were required for competition with labeled HA peptide. HCV 1531 is shown as an example of a peptide that did not effectively compete for binding of HA peptide to DR4. (b and c) Two-step purification of DR molecules loaded with defined peptides. DR4 molecules were separated from free peptide by HPLC gel filtration (b) and then injected into an HPLC DNP affinity column (c) to isolate DR4 molecules loaded with DNP-labeled HCV peptide. Bound complexes were eluted using 50 mM CAPS, pH 11.5, and immediately neutralized. A280 nm, absorbance measurements at a wavelength of 280 nm.
Table 1.
Peptide sequences

Tetramer-based analysis of CD4 T cells specific for HCV.
Each of the three DR4-HCV tetramers labeled a discrete population of CD4 T cells in short-term T cell lines generated from DRB1*0401-positive subjects with antibodies to HCV by in vitro stimulation of PBMCs with soluble C200 antigen (encoding HCV NS3 and NS4 proteins), while no significant staining was observed with three control tetramers (DR4–annexin II, DR4-gp100, and DR4-CLIP) (Figure 3a and data not shown). A substantial enrichment of labeled cells was obtained when anti-PE magnetic microbeads were used to capture cells stained with tetramers composed of DR4–HCV peptide complexes and streptavidin-PE (Figure 3b). The CD4+/tetramer+ cells displayed effector function, as shown by parallel intracellular cytokine staining for IFN-γ and tetramer staining in an HCV NS3/NS4–stimulated line from subject 98A. A pool of the three HCV tetramers (DR4–HCV 1248, DR4–HCV 1579, and DR4–HCV 1770) stained approximately 5% of CD4 T cells from this line, consistent with 5% CD4+IFN-γ+ cells upon stimulation with a pool of the three HCV peptides corresponding to the tetramer epitopes (Figure 3c).
Figure 3.

Staining of short-term HCV-specific CD4 T cell lines with MHC class II tetramers. (a) A short-term T cell line was generated from PBMCs by a single round of stimulation with 1 μg/ml of recombinant C200 antigen encoding the HCV NS3 and NS4 proteins. CD4 T cells were stained with PE-labeled MHC class II tetramers, including a control tetramer, DR4–annexin II, as well as three HCV tetramers, DR4–HCV 1248, DR4–HCV 1579, and DR4–HCV 1770. Cells were analyzed by flow cytometry prior to enrichment for PE-tetramer–positive cells; the percentage of CD4+/tetramer+ cells is indicated in the upper right quadrant. (b) Magnetic enrichment of tetramer-positive cells by positive selection of anti-PE–labeled cells. Data are shown for stimulated CD4 T cells from subject 01-40; similar data were obtained with short-term stimulated lines from subjects 99D and 98A. (c) Cells from an HCV NS3/NS4–stimulated line from subject 98A were stained with a pool of three HCV tetramers, DR4–HCV 1248, DR4–HCV 1579, and DR4–HCV 1770. Data are shown gated on CD4 T cells, without enrichment for tetramer-positive cells. Intracellular cytokine staining was performed by stimulation of cells from this same line with a pool of HCV peptides corresponding to the tetramer epitopes. Data are shown gated on CD4 T cells, and the percentage of IFN-γ+CD4+ cells is indicated in the upper right quadrant.
Tetramer-positive cells could not be unambiguously detected in fresh PBMCs, indicating that enrichment of labeled cells prior to FACS analysis might be essential. Fresh PBMCs from subject 01-40, who spontaneously resolved HCV viremia, were stained with three control tetramers and three HCV tetramers, and tetramer-positive cells were then magnetically enriched with anti-PE microbeads. Following magnetic bead enrichment of cells labeled with control tetramers, very few or no CD4+/tetramer+ cells were isolated (zero to two cells in the experiment shown in Figure 4a), whereas discrete populations were identified for each of the three HCV tetramers with frequencies of 1 in 12,200 CD4 cells (0.008%) for DR4–HCV 1248, 1 in 66,000 (0.0015%) for DR4–HCV 1579, and 1 in 42,000 (0.0024%) for DR4–HCV 1770 (Figure 4a). To ensure that the HCV tetramer–selected cells represented specific populations of CD4 T cells, a similar analysis was performed on PBMCs from a DRB1*0401-positive, HCV-seronegative donor (Figure 4b). All three DR4-HCV tetramers were tested as a pool, and no CD4+/tetramer+ cells were isolated in this seronegative control subject. A discrete population of cells was only observed with an influenza DR4-HA tetramer in this subject, and seven CD4+/tetramer+ cells were isolated after enrichment (1 in 110,000; 0.0009%). Also, no staining with DR4-HCV tetramers was observed with PBMCs from three MHC-mismatched (DRB1*0401-negative), HCV-infected control subjects (data not shown).
Figure 4.

Ex vivo MHC class II tetramer staining of HCV-specific CD4 T cells. (a) PBMC from subject 01-40 with spontaneously resolved HCV viremia were stained with three control tetramers and three HCV tetramers. Cells were positively selected with anti-PE microbeads and analyzed by flow cytometry; the number of CD4+/tetramer+ cells is indicated in the upper right quadrant. An example of FACS analysis before enrichment for tetramer+ cells is shown for control tetramer DR4-CLIP and HCV tetramer DR4–HCV 1248. The frequencies of tetramer-positive cells in the CD4 T cell pool were determined by splitting the sample 9:1 following labeling with anti-PE beads; 90% of the cells were magnetically enriched, while 10% of the cells were analyzed without enrichment to determine the number of CD4 T cells. The frequencies of the tetramer+ cells of total CD4 cells are as follows: DR4–HCV 1248: 8.2 per 100,000 (0.008%); DR4–HCV 1579: 1.5 per 100,000 (0.0015%); DR4–HCV 1770: 2.4 per 100,000 (0.0024%). Representative data are shown from two independent experiments with fresh PBMC from subject 01-40. (b) PBMC from a DRB1*0401 HCV-seronegative control were stained with the following MHC class II tetramers: 3 control tetramers (DR4-CLIP, DR4-gp100, and DR4–annexin II), a pool of three HCV tetramers, and an HA (residues 306–318) tetramer. The number of CD4+/tetramer+ cells following enrichment with anti-PE microbeads is indicated in the upper right quadrant. The frequency of DR4–influenza HA–specific T cells in this subject was 0.9 per 100,000 (0.0009%).
HCV-specific CD4 T cells are not detected in patients with chronic viremia.
In contrast to patients who spontaneously resolve infection with HCV, a CD4 T cell response is difficult to detect in chronically infected subjects using assays based on particular effector functions, raising the question of whether HCV-specific CD4 T cells are anergic or absent (7, 9). PBMCs from four patients with chronic viremia were labeled with a pool of three DR4-HCV tetramers, as well as a pool of control tetramers (Figure 5 and Table 2). HCV tetramer–positive cells were not detected in any of these patients, indicating that such T cells are absent or present at very low frequencies in patients with chronic viremia. Consistent with these findings, T cell proliferation assays performed with PBMCs from three of these subjects were negative. PBMCs from DRB1*0401-positive subject 99-24 with chronic HCV were also subjected to additional functional assays, and no response to these HCV epitopes was detected based on intracellular cytokine staining for IFN-γ production or the ability to generate specific lines by culture with peptide and IL-2 (data not shown). In contrast, HCV tetramer–positive T cells were detected in six of seven subjects with resolved viremia (Figure 5 and Table 2). Eight of the 11 subjects carried the DRB1*0401 subtype; subject OXD was positive for DRB1*04, but subtyping for the DRB1*0401 allele was not performed. Two subjects (OXS and OXK) carried the closely related DRB1*0404 subtype. One of these patients (OXS) had spontaneously resolved viremia, and HCV-specific CD4 T cells could be visualized with the DRB1*0401 tetramers; while the other patient (OXK) had chronic viremia. To assess interassay variability, we performed two staining reactions with PBMCs from subject 01-40 with a pool of HCV tetramers on the same day; the total frequencies in these experiments were 437 and 447 per million CD4 T cells (less than 10% variability).
Figure 5.

Comparison of the T cell response to HCV in subjects with resolved viremia and chronic persistent infection. PBMCs from subject OXM (a spontaneous resolver) and OXB (chronic viremia) were labeled with a pool of the three HCV tetramers (left panels) or three control tetramers (DR4-CLIP, DR4-gp100, and DR4–annexin II). The number of CD4+/tetramer+ cells following enrichment with anti-PE microbeads is indicated in the upper right quadrant. HCV tetramer–positive CD4 T cells could not be detected in the four patients with chronic viremia (Table 2).
Table 2.
Analysis of MHC class II tetramer staining in different patient groups

The frequency of HCV tetramer–positive cells declined over time following resolution of viremia. In subject 99D, relatively large numbers of HCV tetramer–positive cells were detected among cells frozen just after resolution of viremia (850 per million CD4 T cells; Table 2), but tetramer-positive cells were almost undetectable in a sample taken approximately 3 years later, since they could only be visualized following in vitro stimulation. In subject 01-40, the frequency of HCV tetramer–positive T cells was highest at time points close to the resolution of viremia, and the frequency dropped from 447 per million to 121 and 130 per million CD4 T cells in two separate experiments performed 3–4 months later (cells from one of these later time points were used for Figure 4a). T cells from subject 98A were analyzed more than 4.5 years after resolution of viremia, and the frequency of tetramer-positive T cells was considerably lower (13 per million CD4 T cells) than in subjects 99D and 01-40 after resolution of viremia. In subject O1E with resolved HCV viremia, in whom HCV tetramer–positive cells were not detected, functional assays (T cell proliferation, intracellular cytokine staining, generation of T cell lines) were also negative, and it is not known how much time had elapsed between these measurements and exposure to the virus. The time interval is also not known for subjects OXS, OXM, and OXD, who had antibodies directed against HCV but no viral RNA in the plasma, indicating spontaneous resolution of viremia.
Ex vivo phenotype of HCV-specific CD4 T cells.
These data indicated that it is possible to study virus-specific CD4 T cells ex vivo, and we therefore used this approach to examine expression of surface receptors important for memory T cell function. We examined the expression of the surface antigens CCR7, CD45RA, and CD27 on tetramer-positive cells enriched from three DRB1*0401 subjects with resolved HCV viremia. Isolated tetramer-positive cells were CCR7+CD45RA–CD27+, consistent with a surveillance function for secondary lymphoid structures (Figure 6a) (30). The expression of the lymph node homing marker CD62L was analyzed on fresh PBMCs from subject 01-40 in conjunction with the DR4–HCV 1579 and DR4–HCV 1770 tetramers, and 67% and 70% of CD4+/tetramer+ cells, respectively, were CD62L+ (Figure 6b).
Figure 6.

Ex vivo phenotypic analysis of HCV-specific memory CD4 T cells using MHC class II tetramers. (a) PBMCs from three subjects with resolved HCV viremia were stained with a pool of three DR4-HCV tetramers as well as antibodies to CCR7, CD45RA, or CD27. Tetramer-positive cells were positively selected with anti-PE microbeads and analyzed by flow cytometry. Plots are gated on CD4+/tetramer+ cells. (b) PBMCs from subject 01-40 were labeled with an antibody to CD62L in conjunction with the DR4–HCV 1579 and DR4–HCV 1770 tetramers.
The HCV 1248 peptide has been described as an immunodominant CD4 T cell epitope in HCV infection (29), offering the possibility to determine the breadth of TCR usage by memory CD4 cells in controlled HCV infection. We analyzed the TCR Vβ repertoire of CD4 T cells specific for the DR4–HCV 1248 complex in subjects 01-40 and 98A. Fresh PBMCs or short-term lines were enriched with the DR4–HCV 1248 tetramer and stained individually with a panel of 16 Vβ antibodies. In fresh PBMCs from subject 01-40, 30% and 23% of DR4–HCV 1248+ cells were Vβ5.1+ and Vβ2+, respectively, indicating strong selection in vivo. A small population of tetramer-positive cells (1.6%) was Vβ12+ ex vivo in subject 01-40; no other Vβ antibodies tested in this panel stained DR4–HCV 1248+ cells, indicating that additional TCR Vβ segments were used by these HCV 1248–specific CD4 T cells that were not detected here. Similar results were obtained with a short-term line from the same subject (Figure 7, a and b). A restricted TCR repertoire for the DR4–HCV 1248 complex was also observed in a short-term line from subject 98A, since greater than 80% of CD4+/DR4–HCV 1248+ cells were Vβ12+ (Figure 7c). The TCR Vβ repertoires against the DR4–HCV 1248 complex may be different in these two subjects because in vivo selection may result in the expansion of clones with Vα-Jα and Vβ-Jβ rearrangements whose TCRs have a relatively high avidity for this MHC class II/peptide complex. Since the TCR repertoire is likely to be distinct between individuals, different clones may be selected during the infection.
Figure 7.

TCR Vβ repertoire of HCV class II tetramer–positive cells. (a) Fresh PBMCs from subject 01-40 were stained with DR4–HCV 1248 tetramer, positively selected by labeling with anti-PE microbeads, and then split into separate tubes for staining with a panel of 16 Vβ antibodies. (b and c) Similarly, short-term stimulated HCV-specific CD4 T cell lines from subjects 01-40 (b) and 98A (c) were stained with DR4–HCV 1248 and subsequently stained with the 16 Vβ antibodies. The Vβ antibodies that positively stained DR4–HCV 1248+ cells are shown for each subject, as well as an example of a negative control Vβ antibody that did not stain the tetramer-positive cells. In subject 98A, the frequency of T cells in bulk PBMCs expressing Vβ5.1 and Vβ12 was 6.6% and 2.2% of CD4 T cells, respectively.
Discussion
We show here that low-frequency human virus-specific memory CD4 T cells can be visualized in PBMCs using a novel combination of MHC class II tetramer labeling and magnetic bead capture techniques. Discrete populations of CD4 T cells could be identified with all three HCV tetramers, even though the frequencies of these cells were extremely low, about 1–2 logs lower than those of memory CD8 T cells in resolved HCV infection (31). The enrichment technique provides high sensitivity by allowing the analysis of large cell numbers and greatly reducing background. The cells visible after enrichment are strong tetramer binders and potentially “high-avidity” T cells; use of this technique for “low-avidity” T cells may require generation of higher-order multimers of MHC class II/peptide complexes. A high TCR avidity may be relevant to immune control of this infectious agent, and the limited TCR Vβ repertoire of T cells specific for a defined HLA-DR/HCV complex indicates that these T cells are the product of a vigorous in vivo selection process. The frequency of HCV tetramer–positive memory T cells declined following resolution of viremia, but memory populations nevertheless persisted in vivo for extended periods of time, since they could be detected in subject 98A more than 4.5 years after resolution of viremia. These results are consistent with studies in a murine model of lymphocytic choriomeningitis virus (LCMV) infection in which CD8 T cell memory was stably maintained for life, while the levels of specific CD4 memory T cells gradually declined (3).
HCV-specific CD4 T cell populations displayed a distinctive CCR7+CD45RA–CD27+ central-memory phenotype. The CCR7 chemokine receptor controls homing to secondary lymphoid organs, and the CCR7 ligand SLC is displayed on high endothelial venules (32). In CCR7-deficient mice, lymph node localization of circulating T cells and activated DCs from the skin is impaired, indicating that this chemokine receptor plays a critical role in coordinating recruitment of circulating T cells and tissue-localized DCs to lymph nodes (33). Expression of CCR7 and CD62L by the majority of circulating HCV-specific memory CD4 T cells thus indicates a surveillance function for secondary lymphoid structures. There was no evidence for ongoing hepatic inflammation in the subjects with resolved viremia, since liver enzyme markers in the serum were within the normal range, and it is thus unlikely that CCR7– T cells specific for HCV were sequestered in the liver.
Since spontaneous control of viremia is observed in a subgroup of HCV-infected subjects, analysis of the immune response to HCV offers an opportunity to study key determinants of immune control. Loss of the HCV-specific CD4 T cell response can result in recurrence of viremia (8), further supporting the conclusion that this cell population is critical. While HCV-specific CD4 T cell responses are more vigorous in subjects who spontaneously resolve the infection than in individuals with chronic viremia (9–11), HCV-specific CD8 T cells are present at higher frequency in patients with chronic viremia (34). However, these CD8 T cells display impaired effector function (34), which may be related to the deficiencies in the CD4 T cell response. HCV tetramer–positive CD4 T cells were only detected ex vivo in subjects with resolved HCV viremia, not in patients with chronic uncontrolled infection. These results indicate that lack of HCV-specific CD4 T cell responses in individuals with chronic uncontrolled infection is not due to substantial anergic populations, but rather that these cells may either be lost or present at extremely low frequencies. Future studies in individuals with acute HCV infection will be essential, as increasing evidence suggests that antiviral CD4 T cell responses are at the highest frequency during this stage (9–11, 29), although the precise role and fate of these cells over time remain unclear. The ability to isolate such T cells based on their TCR specificity may thus be valuable in longitudinal studies initiated during acute infection.
The CLIP peptide binds with intermediate to low affinity to all human and murine MHC class II molecules (35), and the strategy presented here is therefore likely to be generally applicable to the creation of MHC class II tetramers. Since the peptide-binding site is highly hydrophobic, empty MHC class II molecules have a tendency to aggregate, and a substantial fraction of molecules can be inactive. These properties are dependent on the isotype and allele and are reflected by substantially slower peptide-binding kinetics compared to DR/CLIP precursors (26, 36, 37). Tetramers have been generated by incubation of empty MHC class II molecules with peptides of interest and have been shown to stain T cell lines grown in vitro with viral antigen/peptide in the presence of recombinant IL-2, but they have not yet been used to directly identify virus-specific T cell populations from peripheral blood without prior in vitro expansion (38–40). Tetramers also have been generated by covalent attachment of individual antigenic peptides to the MHC class II β chain. Since a new transfectant/recombinant virus has to be generated for every peptide of interest, this approach is suitable when the major T cell epitope(s) has already been defined, such as for well-characterized murine model antigens (3, 41). In contrast, a single MHC class II/CLIP precursor can be used to generate tetramers with many different peptides, permitting systematic analysis of CD4 T cell responses in human diseases in which the relevant T cell epitopes are frequently not yet known with certainty. The approaches described here for the creation of MHC class II tetramers and the analysis of rare CD4 T cell populations in peripheral blood are therefore widely applicable to the investigation of CD4 T cells in human diseases.
Supplementary Material
Acknowledgments
We thank Zhi Li and Karin Overzet for expert technical assistance in the development of the MHC class II tetramers, Graham Pawelec for providing the gp100 and annexin II peptides, and Dennis Zaller for the DM transfectant. This work was supported by grants from the NIH to K.W. Wucherpfennig (AI45757), B.D. Walker (AI31563), R.T. Chung (DK57857), and C.L. Day (T332 AI07387-12); from the Doris Duke Charitable Foundation to B.D. Walker; and from the Deutsche Forschungsgemeinschaft to G.M. Lauer (DFG LA 1241/1-1). P. Klenerman, M. Lucas, and G. Harcourt are supported by the Wellcome Trust and the European Union (grants QLK2-CT-2002-01329 and QLK2-CT-1999-00356).
Footnotes
Cheryl L. Day, Nilufer P. Seth, and Michaela Lucas contributed equally to this work.
Conflict of interest:The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: hepatitis C virus (HCV); T cell receptor (TCR); dinitrophenol (DNP); 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS); hemagglutinin (HA); phycoerythrin (PE).
References
- 1.Altman JD, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 2.Meyer AL, et al. Direct enumeration of Borrelia-reactive CD4 T cells ex vivo by using MHC class II tetramers. Proc. Natl. Acad. Sci. U. S. A. 2000;97:11433–11438. doi: 10.1073/pnas.190335897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 2001;7:913–919. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 4.Lauer GM, Walker BD. Hepatitis C virus infection. N. Engl. J. Med. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 5.Alter MJ, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N. Engl. J. Med. 1999;341:556–562. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- 6.Rehermann B, Chisari FV. Cell mediated immune response to the hepatitis C virus. Curr. Top. Microbiol. Immunol. 2000;242:299–325. doi: 10.1007/978-3-642-59605-6_14. [DOI] [PubMed] [Google Scholar]
- 7.Day CL, et al. Broad specificity of virus-specific CD4+ T-helper-cell responses in resolved hepatitis C virus infection. J. Virol. 2002;76:12584–12595. doi: 10.1128/JVI.76.24.12584-12595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlach JT, et al. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology. 1999;117:933–941. doi: 10.1016/s0016-5085(99)70353-7. [DOI] [PubMed] [Google Scholar]
- 9.Missale G, et al. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J. Clin. Invest. 1996;98:706–714. doi: 10.1172/JCI118842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thimme R, et al. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001;194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosen HR, et al. Frequencies of HCV-specific effector CD4+ T cells by flow cytometry: correlation with clinical disease stages. Hepatology. 2002;35:190–198. doi: 10.1053/jhep.2002.30293. [DOI] [PubMed] [Google Scholar]
- 12.Lamonaca V, et al. Conserved hepatitis C virus sequences are highly immunogenic for CD4(+) T cells: implications for vaccine development. Hepatology. 1999;30:1088–1098. doi: 10.1002/hep.510300435. [DOI] [PubMed] [Google Scholar]
- 13.Bunce M, et al. Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP) Tissue Antigens. 1995;46:355–367. doi: 10.1111/j.1399-0039.1995.tb03127.x. [DOI] [PubMed] [Google Scholar]
- 14.Gauthier L, et al. Expression and crystallization of the complex of HLA-DR2 (DRA, DRB1*1501) and an immunodominant peptide of human myelin basic protein. Proc. Natl. Acad. Sci. U. S. A. 1998;95:11828–11833. doi: 10.1073/pnas.95.20.11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalandadze A, Galleno M, Foncerrada L, Strominger JL, Wucherpfennig KW. Expression of recombinant HLA-DR2 molecules. Replacement of the hydrophobic transmembrane region by a leucine zipper dimerization motif allows the assembly and secretion of soluble DR alpha beta heterodimers. J. Biol. Chem. 1996;271:20156–20162. doi: 10.1074/jbc.271.33.20156. [DOI] [PubMed] [Google Scholar]
- 16.Kozono H, White J, Clements J, Marrack P, Kappler J. Production of soluble MHC class II proteins with covalently bound single peptides. Nature. 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- 17.Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halder T, et al. Isolation of novel HLA-DR restricted potential tumor-associated antigens from the melanoma cell line FM3. Cancer Res. 1997;57:3238–3244. [PubMed] [Google Scholar]
- 19.Wucherpfennig KW, et al. Clonal expansion and persistence of human T cells specific for an immunodominant myelin basic protein peptide. J. Immunol. 1994;152:5581–5592. [PubMed] [Google Scholar]
- 20.Sherman MA, Weber DA, Jensen PE. DM enhances peptide binding to class II MHC by release of invariant chain-derived peptide. Immunity. 1995;3:197–205. doi: 10.1016/1074-7613(95)90089-6. [DOI] [PubMed] [Google Scholar]
- 21.Sloan VS, et al. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature. 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 22.Roche PA, Cresswell P. Invariant chain association with HLA-DR molecules inhibits immunogenic peptide binding. Nature. 1990;345:615–618. doi: 10.1038/345615a0. [DOI] [PubMed] [Google Scholar]
- 23.Riberdy JM, Newcomb JR, Surman MJ, Barbosa JA, Cresswell P. HLA-DR molecules from an antigen-processing mutant cell line are associated with invariant chain peptides. Nature. 1992;360:474–477. doi: 10.1038/360474a0. [DOI] [PubMed] [Google Scholar]
- 24.Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 25.Avva RR, Cresswell P. In vivo and in vitro formation and dissociation of HLA-DR complexes with invariant chain-derived peptides. Immunity. 1994;1:763–774. doi: 10.1016/s1074-7613(94)80018-9. [DOI] [PubMed] [Google Scholar]
- 26.Stern LJ, Wiley DC. The human class II MHC protein HLA-DR1 assembles as empty alpha beta heterodimers in the absence of antigenic peptide. Cell. 1992;68:465–477. doi: 10.1016/0092-8674(92)90184-e. [DOI] [PubMed] [Google Scholar]
- 27.Hammer J, et al. Promiscuous and allele-specific anchors in HLA-DR-binding peptides. Cell. 1993;74:197–203. doi: 10.1016/0092-8674(93)90306-b. [DOI] [PubMed] [Google Scholar]
- 28.O’Sullivan D, et al. Characterization of the specificity of peptide binding to four DR haplotypes. J. Immunol. 1990;145:1799–1808. [PubMed] [Google Scholar]
- 29.Diepolder HM, et al. Immunodominant CD4+ T-cell epitope within nonstructural protein 3 in acute hepatitis C virus infection. J. Virol. 1997;71:6011–6019. doi: 10.1128/jvi.71.8.6011-6019.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 31.He XS, et al. Quantitative analysis of hepatitis C virus-specific CD8(+) T cells in peripheral blood and liver using peptide-MHC tetramers. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5692–5697. doi: 10.1073/pnas.96.10.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell JJ, et al. 6-C-kine (SLC), a lymphocyte adhesion-triggering chemokine expressed by high endothelium, is an agonist for the MIP-3beta receptor CCR7. J. Cell Biol. 1998;141:1053–1059. doi: 10.1083/jcb.141.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forster R, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 34.Wedemeyer H, et al. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 2002;169:3447–3458. doi: 10.4049/jimmunol.169.6.3447. [DOI] [PubMed] [Google Scholar]
- 35.Malcherek G, Gnau V, Jung G, Rammensee HG, Melms A. Supermotifs enable natural invariant chain-derived peptides to interact with many major histocompatibility complex-class II molecules. J. Exp. Med. 1995;181:527–536. doi: 10.1084/jem.181.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zarutskie JA, et al. The kinetic basis of peptide exchange catalysis by HLA-DM. Proc. Natl. Acad. Sci. U. S. A. 2001;98:12450–12455. doi: 10.1073/pnas.211439398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frayser M, Sato AK, Xu L, Stern LJ. Empty and peptide-loaded class II major histocompatibility complex proteins produced by expression in Escherichia coli and folding in vitro. Protein Expr. Purif. 1999;15:105–114. doi: 10.1006/prep.1998.0987. [DOI] [PubMed] [Google Scholar]
- 38.Kwok WW, et al. HLA-DQ tetramers identify epitope-specific T cells in peripheral blood of herpes simplex virus type 2-infected individuals: direct detection of immunodominant antigen-responsive cells. J. Immunol. 2000;164:4244–4249. doi: 10.4049/jimmunol.164.8.4244. [DOI] [PubMed] [Google Scholar]
- 39.Novak EJ, Liu AW, Nepom GT, Kwok WW. MHC class II tetramers identify peptide-specific human CD4+ T cells proliferating in response to influenza A antigen. J. Clin. Invest. 1999;104:R63–R67. doi: 10.1172/JCI8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cameron TO, Cochran JR, Yassine-Diab B, Sekaly RP, Stern LJ. Detection of antigen-specific CD4+ T cells by HLA-DR1 oligomers is dependent on the T cell activation state. J. Immunol. 2001;166:741–745. doi: 10.4049/jimmunol.166.2.741. [DOI] [PubMed] [Google Scholar]
- 41.Gutgemann I, Fahrer AM, Altman JD, Davis MM, Chien YH. Induction of rapid T cell activation and tolerance by systemic presentation of an orally administered antigen. Immunity. 1998;8:667–673. doi: 10.1016/s1074-7613(00)80571-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}