

Abstract
Transcription factor TFIID is a multiprotein complex composed of the TATA box-binding protein (TBP) and multiple TBP-associated factors (TAFs). TFIID plays an essential role in mediating transcriptional activation by gene-specific activators. Numerous transcriptional activators have been characterized from mammalian cells; however, molecular analysis of the components of mammalian TFIID has been incomplete. Here we describe isolation of cDNAs encoding two TAF subunits of the human transcription factor TFIID. The first cDNA is predicted to encode the C-terminal 947 residues of the 130-kDa human TAF subunit, hTAFII130. The second cDNA encodes the C-terminal 801 residues of the 100-kDa subunit, hTAFII100. Recombinant TAFs expressed in human cells by transient transfections are capable of associating with the endogenous TAFs and TBP to form a TFIID complex in vivo. Protein binding experiments demonstrate that hTAFII130, like its Drosophila homolog dTAFII110, interacts with the glutamine-rich activation domains of the human transcription factor Sp1. Furthermore, hTAFII130 shows reduced binding to the Sp1 mutants with impaired ability to activate transcription, suggesting a role for hTAFII130 as a direct coactivator target for Sp1.
Transcriptional regulation of the genes transcribed by eukaryotic RNA polymerase II is a complex process that requires concerted action of promoter-specific transcription factors and the general transcription machinery (reviewed in refs. 1, 2, 3). In response to extracellular and intracellular signals, promoter-specific transcription factors bind to specific target DNA sequences and affect transcription. Recent research has focused on the mechanisms by which the DNA-binding transcriptional activators modulate transcription initiation. A search for proteins that might serve as their targets has revealed a class of transcription factors termed coactivators that function as intermediary factors between DNA-bound transcription factors and the components of the basal machinery. One such target is TFIID, a multiprotein complex that consists of the TATA box-binding protein (TBP) and at least eight TBP-associated factors, or TAFs. TAFs have been shown to directly bind to selected activators and are thought to mediate activator-dependent stimulation of transcription (4, 5, 6, 7).
The Drosophila dTAFII110, one of the first TAFIIs to be cloned, interacts directly with the human transcription factor Sp1 (5). Subsequent in vitro transcription studies using a partial TFIID complex reconstituted with dTBP, dTAFII250, dTAFII150, and dTAFII110 demonstrated that dTAFII110 functions as a coactivator to mediate Sp1-dependent activation of transcription (4). Most importantly, it was shown that the interaction between dTAFII110 and Sp1 is mediated by the glutamine-rich activation domain of Sp1 and that mutations in this domain that decrease the ability of Sp1 to activate transcription also decrease its ability to interact with dTAFII110 (8), lending further support to the notion that dTAFII110 serves as the coactivator for Sp1.
To compare the activities of human and Drosophila transcription factors, we set out to isolate the cDNA clones encoding a subset of human TAF proteins. Here we report the isolation and characterization of cDNAs encoding human TAFII130 and human TAFII100.
MATERIALS AND METHODS
Protein Preparation and Microsequencing.
Nuclear extracts were prepared from 200–250 liters of α3 cells, a HeLa cell line stably transformed with hemagglutinin antigen (HA)-tagged human TBP (9), and TFIID complex immunopurified with α-HA and α-TBP antibodies (10). TBP and TAFs were eluted with HA peptide, resolved on a 6–12% gradient SDS/polyacrylamide gel, and blotted onto a poly(vinylidene difluoride) membrane. Specific bands corresponding to TAFs were visualized by staining with Ponceau S, excised, and digested with trypsin (11). Peptide sequences were obtained from the tryptic digests eluted from the membrane.
Preparation of Probes for cDNA Library Screening.
Degenerate oligonucleotides were synthesized based on the microsequencing data and used in a PCR reaction to amplify the corresponding cDNA fragment from a library of cDNAs. In the case of hTAFII130, two sets of degenerate PCR primers were designed corresponding to each end of a long peptide sequence (pep4, see Fig. 2A). An amplified product (40 nucleotides) was obtained in a PCR reaction using cDNA prepared from HeLa poly(A)+ RNA. In the case of hTAFII100, two peptides (pep5 and pep6, see Fig. 3) were found to be nearly identical in sequence to those found in dTAFII80. Degenerate oligonucleotides designed from these peptides (which were 13 amino acids apart in dTAFII80) were used to amplify a cDNA fragment that spanned between the two peptides. The cDNA fragments thus isolated were used as probes to screen multiple human cDNA libraries. Positive clones for both TAFs were obtained from screening λgt10 and λZAPII HeLa cell cDNA libraries (12) and the NTera2D1 line of human teratocarcinoma cDNA library (13).
Figure 2.

(A) Predicted C-terminal 947 amino acid sequence of hTAFII130. Peptides obtained from microsequencing of immunopurified hTAFII130 protein are underlined. (B) Schematic comparison of hTAFII130 with Drosophila dTAFII110. Region I (residues 449–528): 68% sequence similarity (46% identity) over 80 residues. Region II (residues 689–947): 72% similarity (55% identity) over 259 residues as determined by the bestfit sequence analysis program (Genetics Computer Group, Madison, WI). Glutamine-rich regions (presence of 20–30% glutamines in hTAFII130) are shaded and denoted by Q. hTAFII130N/C subdomain used in protein binding assays is indicated by a bracket. Dotted box represents the predicted missing N-terminal region. (C) Amino acid sequence alignment of conserved residues within regions I and II of dTAFII110 and hTAFII130.
Figure 3.

Predicted C-terminal 801 amino acid sequence of hTAFII100. Peptide sequences obtained from microsequencing the endogenous hTAFII100 are indicated above the amino acid sequence. The dotted line below residues 557–611 represents 55 amino acids missing from a spliced variant of hTAFII100 isolated from the NTera cDNA library. The six WD repeats are indicated by the dotted arrows.
In Vivo TFIID Complex Assembly Assay for Recombinant TAFs.
The procedure was carried out as described (14). hTAFII100 and hTAFII130 cDNAs were tagged with an HA epitope and transfected by calcium-phosphate coprecipitation into human embryonic kidney 293 cells under the control of a cytomegalovirus promoter. Mininuclear extracts were prepared from transfected cells (15) and TFIID immunopurified with a polyclonal α-TBP antibody. Proteins separated by SDS/PAGE were immunoblotted with the α-HA monoclonal antibody (12CA5) to detect for the presence of the recombinant protein.
In Vitro Protein Binding Studies.
Sp1 DNA affinity columns were prepared by coupling biotinylated oligonucleotides containing multiple Sp1 binding sites. Sp1 prepared from recombinant vaccinia virus-infected HeLa cells (4) was bound to the DNA affinity resin and incubated with in vitro-translated radiolabeled hTAFII130. HA-tagged hTAFII130N/C was expressed from a pET expression vector in Escherichia coli by subcloning downstream from the HA sequence an NdeI–ClaI fragment derived from the central portion of hTAFII130 (NdeI site introduced at residue 270 by PCR; ClaI present at residue 700). HA-hTAFII130N/C was purified and bound to an α-HA antibody resin. A control resin was prepared with an E. coli extract harboring the pET-HA expression vector alone. The resins were incubated with a crude HeLa cell nuclear extract (16) and proteins in the bound fraction were analyzed by SDS/PAGE and immunoblotting with α-Sp1 antibody to detect for binding of the endogenous Sp1.
Yeast Two-Hybrid Methods.
The NdeI–ClaI fragment of hTAFII130 was subcloned in-frame into the yeast pAS1 vector (17) to generate G4-hTAFII130N/C. Plasmids expressing fusions of Sp1 to the acidic activation domain of GAL4 (8) were cotransformed with G4-hTAFII130N/C into yeast strains (Y153 and Y526) and assayed for β-galactosidase activity (5). For studies using the mutant Sp1 constructs, pGAD plasmids carrying deletions or a linker substitution mutation (M37) in Sp1 domain Bc (8) were cotransformed with a pEG202 plasmid (18) expressing hTAFII130N/C into the yeast strain W303 (18). Quantitative β-galactosidase assays (19) were performed in triplicate.
RESULTS
Cloning of the cDNAs Encoding Human TAFII130 and TAFII100.
To obtain the cDNAs corresponding to the subunits of the human TFIID complex we immuopurified TFIID from HeLa α3 cells and separated the components by SDS/PAGE (Fig. 1). Multiple tryptic peptides were obtained from the protein bands corresponding to hTAFII130 and hTAFII100. Microsequencing of the purified peptides allowed us to design degenerate oligonucleotides for PCR amplification of the corresponding cDNA sequences. Four independent cDNA clones were isolated for hTAFII130 from the λZAPII HeLa cDNA library. The longest hTAFII130 cDNA (clone 3a2) was 4.2 kb in length with 2.8 kb of open reading frame encoding 947 amino acids (Fig. 2) and 1.4 kb of 3′ untranslated sequence. The 5′ end of the remaining three cDNAs were found to be ≈1 kb downstream of the 5′ end of clone 3a2. Based on the Northern blot analysis and comparison of the gel mobility of the in vitro-translated recombinant product and the native protein, clone 3a2 appears to be missing the N-terminal ≈100 amino acid residues of hTAFII130 (data not shown). Multiple attempts to obtain the 5′ end of the cDNA have failed because of the extraordinarily high GC content of the gene in this region (80% G+C in the first 1 kb of cDNA). However, we are confident that the unusual cDNA sequence in clone 3a2 is genuine since we have isolated genomic hTAFII130 clones that include this region (data not shown).
Figure 1.
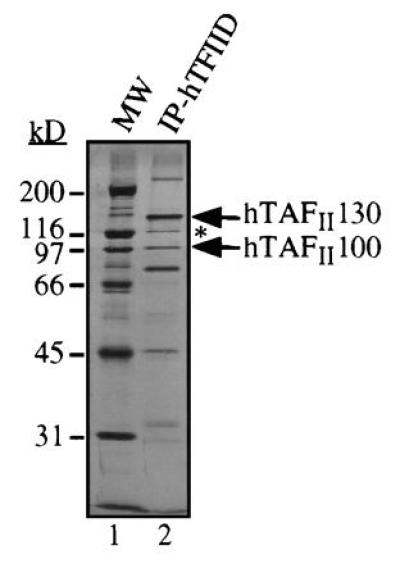
TFIID complex immunopurified from HeLa cells. hTAFIIs and hTBP were separated by SDS/PAGE and stained with silver. Positions of hTAFII130 and hTAFII100 are indicated. A 110-kDa hTAFII protein immunologically related to hTAFII130 is indicated by an asterisk (∗). We have obtained one peptide from this protein whose sequence is found in the predicted amino acid sequence of hTAFII130, suggesting that the 110-kDa hTAFII is a derivative or a breakdown product of hTAFII130.
Multiple independent cDNAs were also isolated for hTAFII100 from the λgt10 HeLa cell library and the NTera cDNA library. The longest cDNA obtained from the NTera library was 2.4 kb in length, and included an open reading frame encoding 801 amino acids (Fig. 3) with a very short 3′ untranslated sequence of 19 nucleotides. In the recently reported cDNA sequence of hTAFII100, the authors suggest that a methionine equivalent to residue 2 in our predicted amino acid sequence (Fig. 3) is the first codon of the hTAFII100 protein (20). However, neither their reported cDNA sequence nor ours contain an in-frame stop codon upstream of the methionine in question. Since the N-terminal amino acid sequence obtained from sequencing the endogenous hTAFII100 protein is different from the N-terminal sequence predicted by the cloned hTAFII100 (S. Zhou and R. Tjian, personal communication), this methionine (position 2 in Fig. 3) may not be the N-terminal methionine of hTAFII100. We have also made multiple attempts to extend the current hTAFII100 cDNA to include the complete 5′ end. Since the translated product of the cDNA is similar in size to the endogenous protein, the missing cDNA sequence is probably very short.
The identity of the cDNA clones was confirmed by the presence of multiple peptide sequences obtained from microsequencing (Figs. 2A and 3). In addition, the cDNAs were translated in vitro and the protein products found to react with monoclonal antibodies raised against native human TAFIIs (data not shown). Comparison of the sequences of hTAFII130 and the previously reported Drosophila TAFII110 (5) is shown (Fig. 2 B and C). Two regions (designated I and II) share a high degree of sequence similarity, suggesting that hTAFII130 is a true homolog of dTAFII110. Region I (residues 449–528) is an 80 amino acid stretch within the central domain of hTAFII130 that shares 46% identity (68% similarity) with dTAFII110. Interestingly, a data base search found a similar region present within the putative transcription factor ETO/MTG8 that is associated with a chromosomal translocation in acute myelogenous leukemia (21, 22). Nervy, a Drosophila homolog of ETO, has also been cloned and shares extensive sequence similarity (31% identity over 71 residues) to the corresponding region I of dTAFII110 (23). These findings suggest that region I represents a domain that has been conserved through evolution in multiple proteins involved in transcriptional regulation.
Region II (residues 689–947) located near the C terminus of hTAFII130 also shows a high degree of sequence similarity with dTAFII110 (55% identity, 72% similarity over 259 amino acid residues). This region of dTAFII110 was shown to interact with dTAFII30α (24). Besides regions I and II, the remaining hTAFII130 and dTAFII110 show little similarities except for multiple glutamine-rich domains present in both TAFIIs. Finally, the N-terminal 200 amino acids of hTAFII130 are rich in glycine, proline, and alanine residues. This region is absent from the N terminus of dTAFII110, suggesting a function unique to hTAFII130.
The predicted sequence for hTAFII100 (Fig. 3) is very similar to the sequence of Drosophila TAFII80 (25, 26) and yeast TAFII90 (27, 28), again indicating that they are true homologs. The C-terminal 730 amino acids of hTAFII100 share 44% identity (65% similarity) with dTAFII80 and 35% identity (57% similarity) with yTAFII90. All three TAFs contain at least six WD repeats (Fig. 3) that are found in proteins of diverse biological function. The recently solved crystal structure of the β-subunit of the G protein transducin suggests that proteins containing WD repeats form β-propellers (29, 30). Interestingly, we have isolated at least one spliced form of hTAFII100 from the NTera cDNA library that is missing one and a half WD repeats (Fig. 3); however, its significance has not been determined.
Recombinant hTAFII100 and hTAFII130 Become Stably Incorporated into the TFIID Complex in Transfected 293 Cells.
To determine whether the near full-length cDNAs are capable of inclusion in a TFIID complex in vivo, the cDNAs encoding hTAFII100 and hTAFII130 tagged with an HA epitope at the N terminus, and HA-tagged full-length hTAFII70 as a control (14), were individually expressed in human 293 cells by transient transfection. Nuclear extracts were prepared from transfected cells and the TFIID complex immunopurified using an anti-TBP antibody. An immunoblot of the immunopurified TFIID complex probed with α-HA antibody is shown (Fig. 4). The HA-ΔNhTAFII100 polypeptide showed a similar mobility to HA-hTAFII70 since the ΔNhTAFII100 construct used in this experiment contained the C-terminal 704 residues of hTAFII100 (lanes 2 and 3). Each HA-tagged polypeptide was recovered in the immunopurified TFIID fraction, demonstrating that the recombinant hTAFIIs were capable of being incorporated into a TFIID complex with the enodgenous TBP and TAFs in vivo.
Figure 4.
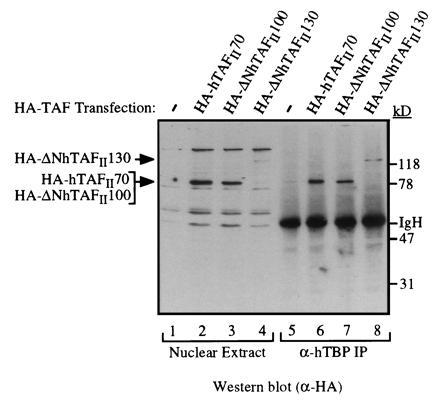
Recombinant HA-tagged hTAFIIs can be stably incorporated into the TFIID complex in vivo. Nuclear extracts were prepared from 293 cells transfected with a control plasmid (lanes 1 and 5) or expression plasmids encoding HA-hTAFII70 (lanes 2 and 6), HA-ΔNhTAFII100 containing C-terminal 704 residues (lanes 3 and 7), HA-ΔNhTAFII130 (C-terminal 947 residues, lanes 4 and 8) for 48 hours and TFIID was immunopurified using a polyclonal α-hTBP antibody. Presence of each recombinant hTAFII was detected by Western blotting using the α-HA antibody. Presence of endogenous hTAFIIs in immunopurified TFIID was detected by probing the same blot with a mixture of α-hTAFII monoclonal antibodies (data not shown).
hTAFII130 Binds to Sp1 In vitro. TAFs are thought to serve as transcriptional coactivators by interacting with site-specific transcription activators as well as basal factors. It has been demonstrated that dTAFII110 binds Sp1 and functions as a coactivator for Sp1 (4, 5). Since Sp1 is a human transcription factor, it is more relevant to determine whether human TAFII130 can interact with Sp1, and this was addressed using a variety of protein–protein interaction assays. Recombinant Sp1 purified from HeLa cells was immobilized on a DNA affinity resin bearing tandem Sp1 binding sites (Fig. 5A).
Figure 5.
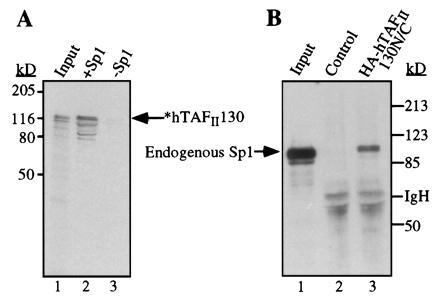
Interactions between Sp1 and hTAFII130 detected in vitro. (A) Radiolabeled hTAFII130 was incubated with purified Sp1 bound to a DNA-affinity resin (lane 2) or a control resin prepared without Sp1 protein (lane 3). Input lane represents approximately 10% of each reaction. (B) Endogenous Sp1 was detected by Western blotting after incubating a crude HeLa cell nuclear extract (1.5 mg) with a resin containing immobilized HA-hTAFII130N/C, expressed and purified from E. coli (lane 3), and a control resin (lane 2).
Incubation with in vitro-translated radiolabeled hTAFII130 resulted in retention of hTAFII130 by the Sp1 resin but not by the control resin (lanes 2 and 3).
To determine whether this association was mediated by the Sp1 activation domains, glutathione S-transferase pulldown experiments were performed using glutathione S-transferase fusion proteins expressed and purified from mammalian cells (data not shown). The results demonstrate specific binding of hTAFII130 to the Sp1 B domain rather than to a control fusion. In a reciprocal experiment, the central domain of hTAFII130N/C (containing residues 270–700) tagged with HA was expressed and purified from E. coli and immobilized on an α-HA antibody affinity column. Incubation of this matrix with in vitro-translated full-length Sp1 resulted in retention of Sp1 by HA-hTAFII130N/C but not by the control resin prepared with an extract from E. coli harboring the expression vector with an HA-tag (data not shown). The immobilized HA-hTAFII130N/C was also mixed with a crude nuclear extract prepared from HeLa cells and found to retain endogenous Sp1, which was detected by Western blot analysis followed by incubation with a polyclonal α-Sp1 antibody (Fig. 5B, lanes 2 and 3). Together, these in vitro experiments argue for a direct protein–protein interaction between the activation domain B of Sp1 and the central domain of hTAFII130.
hTAFII130-Sp1 Interaction Domain Is Localized to the Central Glutamine-Rich Region of hTAFII130 and Correlates with Transactivation by Sp1.
In addition to the protein binding assays performed in vitro, the region of hTAFII130 involved in interaction with Sp1 was also defined in vivo using the yeast two-hybrid system (Fig. 6A and data not shown). The Sp1 interaction domain was localized to a central region of hTAFII130 that includes multiple glutamine-rich regions (20–30% glutamines). To examine the correlation between the ability of Sp1 to interact with hTAFII130 and its ability to activate transcription, we tested subfragments of the Sp1 activation domain (8) using yeast two-hybrid experiments. For each Sp1 mutant, the results obtained using human TAFII130 were consistent with the previous results for the interaction of Sp1 mutants and Drosophila TAFII110 (Fig. 6 B and C and data not shown). Mutations in Sp1 B that reduce transcription also interfere with the binding to hTAFII130. Mutations in the C-terminal half of Sp1 B domain have been shown to significantly decrease transactivation by Sp1 (8, 31). Therefore, we conclude that the ability of hTAFII130 to bind to Sp1 correlates with the ability of Sp1 to activate transcription. As with dTAFII110 (32), hTAFII130 also interacted with the N-terminal domain of CREB that contains the Q2 activation domain but not with a derivative missing one-half of the Q2 domain (data not shown). In contrast, the C-terminal proline-rich activation domain of CTF (33) did not interact with hTAFII130 (data not shown). These results suggest that hTAFII130 interacts with the glutamine-rich activation domains present in Sp1 and CREB but not with the proline-rich activation domain of CTF.
Figure 6.
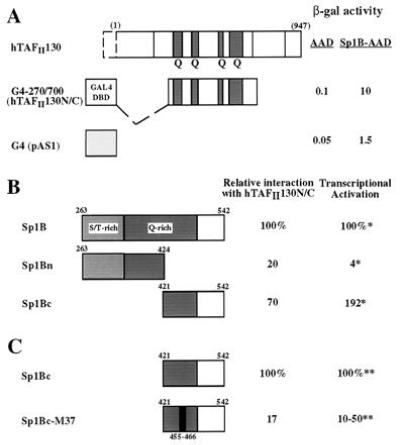
(A) Sp1-hTAFII130 interaction detected by yeast two-hybrid assay. hTAFII130N/C fused to the GAL4 DNA binding domain (residues 1–147, lightly shaded) is shown schematically. β-Galactosidase activity measured from lysates of yeast cotransformed with AAD fusions are indicated on the right. (B) hTAFII130 interacts preferentially with the C-terminal subdomain of Sp1 B that activates transcription in HeLa cells. Yeast plasmids containing the full-length Sp1 B domain (amino acids 263–542), or the subdivisions Bn (263-424), or Bc (421-542) fused to the acidic activation domain (8) were cotransformed with lexADBD-hTAFII130N/C fusion. The resulting β-galactosidase activity measured relative to the activity of the full-length Sp1 B, is indicated on the right. ∗, Previously reported relative values of transcriptional activation by equivalent fusion constructs expressed and assayed in HeLa cells (8). (C) A linker substitution mutation (indicated by the black bar) in the C-terminal subdomain of Sp1 B reduces both interaction with hTAFII130 and transcriptional activation in human cells. The yeast two-hybrid experiments were conducted as described for B. ∗∗, Transcriptional activation values measured in 293 cells by transient transfection were taken from (31) and S. Smale (personal communication).
DISCUSSION
Recent progress in uncovering the mechanisms of transcriptional regulation from eukaryotic promoters transcribed by RNA polymerase II has led to the notion that TFIID functions as an intermediary between site-specific transcriptional activators and components of the basal transcription machinery. Direct interactions between specific activators and TAF subunit(s) in the TFIID complex have been demonstrated, suggesting that TAFs may make multiple contacts with different transcriptional regulators thereby integrating signals transmitted to the gene promoter from multiple sources (4, 5, 6, 7). To investigate the role of each TAF subunit in mediating transcriptional activation, we have isolated and characterized cDNA clones encoding two human TAF proteins: hTAFII130 and hTAFII100. Our goal is to identify the mammalian activators that contact these human TAFs and to characterize TAF-activator interactions relevant to transcriptional regulation. This paper provides several lines of evidence that suggest that hTAFII130 is the target of the glutamine-rich activation domain of Sp1.
When expressed in mammalian cells the recombinant hTAFII130 and hTAFII100 proteins were capable of stably associating with the endogenous TFIID complex, demonstrating that the isolated cDNAs encode bona fide hTAFIIs. The predicted C terminus of human TAFII130 shares a high degree of sequence similarity with the C terminus of Drosophila TAFII110, arguing that hTAFII130 is the mammalian counterpart of dTAFII110. Sequence comparisons between two homologs have revealed conserved and non-conserved regions in hTAFII130. The conserved C-terminal domain might be involved in interaction with other TAFs and basal factors. For example, TAFII250, TFIIA and dTAFII30α have previously been shown to interact with dTAFII110 (24, 34, 35).
Outside of the conserved C-terminal region, hTAFII130 shows less sequence similarity to dTAFII110; however, both proteins interact with the activation domain of Sp1, suggesting that they share structural similarities. The central domain of hTAFII130 interacts with the B activation domain of Sp1 both in in vitro protein-binding assays and in the yeast two-hybrid assay. Most importantly, we observed a tight correlation between Sp1’s ability to activate transcription in mammalian cells and its ability to interact with hTAFII130, consistent with a role for hTAFII130 in mediating Sp1-dependent transcriptional activation. These results are in agreement with previously reported interactions between the activation domain of Sp1 and dTAFII110 (8).
In contrast, the most N-terminal portion of hTAFII130 appears to be unique to this protein and might engage in some as yet unidentified function such as binding to promoter DNA. The Drosophila TAFII150 has been shown to bind specifically to the initiator and downstream sequences of the adenovirus major late promoter and play a role in promoter selectivity (36). Indeed, TFIID immunopurified from HeLa cells shows extensive interaction with promoter DNA (9), consistent with additional DNA contacts involving components of TFIID. Interestingly, the mammalian homolog of dTAFII150 is not tightly associated with TFIID (37); therefore, other TAFs such as hTAFII130 in the tightly associated TFIID complex might be responsible for interaction with promoter DNA, thereby contributing to promoter selectivity and transcriptional regulation. Indeed, a recent study showed that a 135-kDa polypeptide present in immunopurified TFIID can be crosslinked to promoter DNA (38).
We have found that both Sp1 B and the N-terminal activation domain of CREB seem to interact through the central domain of hTAFII130 (residues 270–700) in the yeast two-hybrid assay. This contrasts with the interaction between dTAFII110 and the DNA binding/transactivation domain of the progesterone receptor, that requires more C-terminal domain of dTAFII110 (39). Therefore, multiple activators may contact the same TAF protein via different structural domains. It has been demonstrated for the Drosophila transcriptional regulators Bicoid and Hunchback that two activators that target different TAFs in the TFIID complex function synergistically when bound to the same promoter, presumably by enhancing recruitment of TFIID and/or by increasing the stability of the preinitiation complex (40). These findings are significant in the context of naturally occurring promoters where site-specific regulators bind to multiple sites adjacent to or overlapping with other factor binding sites. Therefore, it will be important to determine the nature of protein–protein interactions between hTAFII130 and those activators that contact hTAFII130 including Sp1 and CREB.
Our results, together with those of others, suggest that Sp1 may use independent domains to make separate contacts with distinct TAFs to activate transcription. In addition to the interaction between the B activation domain and hTAFII130 reported here, hTAFII55 has been shown to contact a C-terminal portion of Sp1 consisting of the DNA binding domain and activation domain D (6). Interestingly, Smale and colleagues performed transient transfection studies with reporter constructs driven by synthetic core promoter elements and found that stimulation of initiator-containing core promoters by Sp1 occurred with the glutmaine-rich activation domains of Sp1 (31). By contrast, TATA box-mediated transcription required full-length Sp1. Therefore, they proposed that interaction with hTAFII130 might function through the initiator element whereas interaction with hTAFII55 might be additionally required for Sp1-dependent stimulation of transcription from the TATA box-containing promoters.
Recent findings suggest that hTAFII130 may be a member of a family of related proteins. A cDNA clone has been isolated that encodes a B-cell specific form of hTAFII130 that is present in substoichiometric amounts (41). It appears that no true homolog of hTAFII130 exists in yeast, raising the possibility that hTAFII130 is a TAF unique to higher eukaryotes. This is consistent with the reported observations that glutamine-rich activators such as Sp1 lack the ability to activate transcription when introduced into yeast while acidic activators activate transcription from a variety of host cells including yeast (42). hTAFII130 and related proteins may have evolved to accommodate an increasingly large repertoire of transcriptional activators required to carry out complex programs of gene expression during cell growth and differentiation in higher eukaryotes.
Acknowledgments
This work was initiated in the laboratory of R. Tjian. We are grateful to members of the Tjian lab for their generous gifts and support for the project. We thank R. Dikstein for sharing the unpublished data; S. Zhou and J. Goodrich for peptide microsequencing; K. Goodrich and J. Goodrich for DNA sequencing; S. Ruppert for an exceptional cDNA library; and L. Comai for the cDNAs. We thank Q. Zhou and A. Berk for the generous gift of HeLa α3 cells. We thank E. Rojo, L. Sun, K. Vogel, J. Sok, and A. Shih for technical contributions to the project. We are especially grateful to G. Gill for her generous gifts of the yeast plasmids and advice, and R. Tjian for his advice and support. We thank A. Wilson, M. Garabedian, and G. Gill for the critical reading of the manuscript. This work was supported by American Cancer Society Institutional Grant IRG-14-35 and a grant from the National Institutes of Health (R01-GM51314). N.T. was supported in part by The Irma T. Hirschl Trust, and D.S. was supported by National Institutes of Health Training Grant 5T32 AI07180. We thank the National Science Foundation for its support of the computing resources through Grant BIR-9318128.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Roeder R G. Trends Biochem Sci. 1991;16:402–408. doi: 10.1016/0968-0004(91)90164-q. [DOI] [PubMed] [Google Scholar]
- 2.Tjian R, Maniatis T. Cell. 1994;77:5–8. doi: 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 3.Zawel L, Reinberg D. Annu Rev Biochem. 1995;64:533–561. doi: 10.1146/annurev.bi.64.070195.002533. [DOI] [PubMed] [Google Scholar]
- 4.Chen J L, Attardi L D, Verrijzer C P, Yokomori K, Tjian R. Cell. 1994;79:93–105. doi: 10.1016/0092-8674(94)90403-0. [DOI] [PubMed] [Google Scholar]
- 5.Hoey T, Weinzierl R O, Gill G, Chen J L, Dynlacht B D, Tjian R. Cell. 1993;72:247–260. doi: 10.1016/0092-8674(93)90664-c. [DOI] [PubMed] [Google Scholar]
- 6.Chiang C-M, Roeder R G. Science. 1995;267:531–536. doi: 10.1126/science.7824954. [DOI] [PubMed] [Google Scholar]
- 7.Jacq X, Brou C, Lutz Y, Davidson I, Chambon P, Tora L. Cell. 1994;79:107–117. doi: 10.1016/0092-8674(94)90404-9. [DOI] [PubMed] [Google Scholar]
- 8.Gill G, Pascal E, Tseng Z H, Tjian R. Proc Natl Acad Sci USA. 1994;91:192–196. doi: 10.1073/pnas.91.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Q, Lieberman P M, Boyer T G, Berk A J. Genes Dev. 1992;6:1964–1974. doi: 10.1101/gad.6.10.1964. [DOI] [PubMed] [Google Scholar]
- 10.Tanese N, Pugh B F, Tjian R. Genes Dev. 1991;5:2212–2224. doi: 10.1101/gad.5.12a.2212. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez J, DeMott M, Atherton D, Mische S M. Anal Biochem. 1992;201:255–264. doi: 10.1016/0003-2697(92)90336-6. [DOI] [PubMed] [Google Scholar]
- 12.Ruppert S, Wang E H, Tjian R. Nature (London) 1993;362:175–179. doi: 10.1038/362175a0. [DOI] [PubMed] [Google Scholar]
- 13.Skowronski J, Fanning T G, Singer M F. Mol Cell Biol. 1988;8:1385–1397. doi: 10.1128/mcb.8.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinzierl R O, Ruppert S, Dynlacht B D, Tanese N, Tjian R. EMBO J. 1993;12:5303–5309. doi: 10.1002/j.1460-2075.1993.tb06226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee K A, Bindereif A, Green M R. Gene Anal Technol. 1988;5:22–31. doi: 10.1016/0735-0651(88)90023-4. [DOI] [PubMed] [Google Scholar]
- 16.Dignam J D, Lebovitz R M, Roeder R G. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durfee T, Becherer K, Chen P-L, Yeh S-H, Yang Y, Kilburn A E, Lee W-H, Elledge S J. Genes Dev. 1993;7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- 18.Gyuris J, Golemis E A, Chertkov H, Brent R. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- 19.Garabedian M J. Methods Companion Methods Enzymol. 1993;5:138–146. [Google Scholar]
- 20.Dubrovskaya V, Lavigne A-C, Davidson I, Acker J, Staub A, Tora L. EMBO J. 1996;15:3702–3712. [PMC free article] [PubMed] [Google Scholar]
- 21.Erickson P F, Robinson M, Owens G, Drabkin H A. Cancer Res. 1994;54:1782–1786. [PubMed] [Google Scholar]
- 22.Miyoshi H, Kozu T, Shimizu K, Enomoto K, Maseki N, Kaneko Y, Kamamda N, Ohki M. EMBO J. 1993;12:2715–2721. doi: 10.1002/j.1460-2075.1993.tb05933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feinstein P G, Kornfeld K, Hogness D S, Mann R S. Genetics. 1995;140:573–586. doi: 10.1093/genetics/140.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yokomori K, Chen J L, Admon A, Zhou S, Tjian R. Genes Dev. 1993;7:2587–2597. doi: 10.1101/gad.7.12b.2587. [DOI] [PubMed] [Google Scholar]
- 25.Dynlacht B D, Weinzierl R O, Admon A, Tjian R. Nature (London) 1993;363:176–179. doi: 10.1038/363176a0. [DOI] [PubMed] [Google Scholar]
- 26.Kokubo T, Gong D-W, Yamashita S, Takada R, Roeder R G, Horikoshi M, Nakatani Y. Mol Cell Biol. 1993;13:7859–7863. doi: 10.1128/mcb.13.12.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poon D, Bai Y, Campbell A M, Bjorklund S, Kim Y-J, Zhou S, Kornberg R D, Weil P A. Proc Natl Acad Sci USA. 1995;92:8224–8228. doi: 10.1073/pnas.92.18.8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reese J C, Apone L, Walker S S, Griffin L A, Green M R. Nature (London) 1994;371:523–527. doi: 10.1038/371523a0. [DOI] [PubMed] [Google Scholar]
- 29.Sondek J, Bohm A, Lambright D G, Hamm H, Sigler P B. Nature (London) 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- 30.Wall M A, Coleman D E, Lee E, Iniguez-Lluhi J A, Posner B A, Gilman A G, Sprang S R. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 31.Emami K H, Navarre W W, Smale S T. Mol Cell Biol. 1995;15:5906–5916. doi: 10.1128/mcb.15.11.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferreri K, Gill G, Montminy M. Proc Natl Acad Sci USA. 1994;91:1210–1213. doi: 10.1073/pnas.91.4.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mermod N, O’Neill E A, Kelley T J, Tjian R. Cell. 1989;58:741–753. doi: 10.1016/0092-8674(89)90108-6. [DOI] [PubMed] [Google Scholar]
- 34.Weinzierl R, Dynlacht B D, Tjian R. Nature (London) 1993;362:511–517. doi: 10.1038/362511a0. [DOI] [PubMed] [Google Scholar]
- 35.Yokomori K, Admon A, Goodrich J A, Chen J L, Tjian R. Genes Dev. 1993;7:2235–2245. doi: 10.1101/gad.7.11.2235. [DOI] [PubMed] [Google Scholar]
- 36.Verrijzer C P, Chen J L, Yokomori K, Tjian R. Cell. 1995;81:1115–1125. doi: 10.1016/s0092-8674(05)80016-9. [DOI] [PubMed] [Google Scholar]
- 37.Kaufmann J, Verrijzer C P, Shao J, Smale S T. Genes Dev. 1996;10:873–886. doi: 10.1101/gad.10.7.873. [DOI] [PubMed] [Google Scholar]
- 38.Oelgeschlager T, Chiang C-M, Roeder R G. Nature (London) 1996;382:735–738. doi: 10.1038/382735a0. [DOI] [PubMed] [Google Scholar]
- 39.Schwerk C, Klotzbucher M, Sachs M, Ulber V, Klein-Hitpass L. J Biol Chem. 1995;270:21331–21338. doi: 10.1074/jbc.270.36.21331. [DOI] [PubMed] [Google Scholar]
- 40.Sauer F, Hansen S K, Tjian R. Science. 1995;270:1783–1788. doi: 10.1126/science.270.5243.1783. [DOI] [PubMed] [Google Scholar]
- 41.Dikstein R, Zhou S, Tjian R. Cell. 1996;87:137–146. doi: 10.1016/s0092-8674(00)81330-6. [DOI] [PubMed] [Google Scholar]
- 42.Ponticelli A S, Pardee T S, Struhl K. Mol Cell Biol. 1995;15:983–988. doi: 10.1128/mcb.15.2.983. [DOI] [PMC free article] [PubMed] [Google Scholar]