

Abstract
The jellyfish Aequorea victoria possesses in the margin of its umbrella a green fluorescent protein (GFP, 27 kDa) that serves as the ultimate light emitter in the bioluminescence reaction of the animal. The protein is made up of 238 amino acid residues in a single polypeptide chain and produces a greenish fluorescence (λmax = 508 nm) when irradiated with long ultraviolet light. The fluorescence is due to the presence of a chromophore consisting of an imidazolone ring, formed by a post-translational modification of the tripeptide -Ser65-Tyr66-Gly67-. GFP has been used extensively as a reporter protein for monitoring gene expression in eukaryotic and prokaryotic cells, but relatively little is known about the chemical mechanism by which fluorescence is produced. To obtain a better understanding of this problem, we studied a peptide fragment of GFP bearing the chromophore and a synthetic model compound of the chromophore. The results indicate that the GFP chromophore consists of an imidazolone ring structure and that the light emitter is the singlet excited state of the phenolate anion of the chromophore. Further, the light emission is highly dependent on the microenvironment around the chromophore and that inhibition of isomerization of the exo-methylene double bond of the chromophore accounts for its efficient light emission.
Keywords: imidazolone, excited state, phenolate anion, low-temperature fluorescence, photoisomerization
When stimulated electrically or mechanically, the bioluminescent jellyfish Aequorea victoria produces numerous spots of bluish-green light along the margin of its umbrella (1). The light is due to the presence of two closely associated proteins: aequorin (21.4 kDa) (2, 3, 4) and a green fluorescent protein (GFP, 27 kDa) (5, 6, 7). Aequorin is a monomeric Ca2+-binding protein with three Ca2+-binding sites (EF-hand structures). It is made up of apoaequorin (apoprotein), coelenterazine (imidazole compound, 423 Da), and molecular oxygen. On binding Ca2+, an intramolecular reaction takes place in which apoaequorin is converted to a luciferase (oxygenase), which then catalyzes the oxidation of coelenterazine to coelenteramide by the bound oxygen to yield light (λmax = 470 nm), CO2 and a blue fluorescent protein (BFP). BFP consists of coelenteramide bound to apoaequorin and the excited state of coelenteramide is the light emitter in the reaction (8, 9). In the presence of GFP, however, there is a radiationless energy transfer by resonance from the excited state of the coelenteramide to a chromophore in GFP, resulting in a blue-green light emission (λmax = 508 nm), which is identical to that observed in the animal (5, 6, 10, 11).
The cDNA for GFP has been cloned and the amino acid sequence has been deduced from the nucleotide sequence (11, 12, 13). GFP is made up of 238 amino acid residues in a single polypeptide chain and emits a greenish fluorescence when irradiated with long ultraviolet light. The light is due to the presence of a chromophore with an imidazolone ring structure, formed by the post-translational cyclization of the tripeptide -Ser65-Tyr66-Gly67- in the protein chain (14, 15). Extensive digestion of GFP with papain has yielded a hexapeptide (from Phe64 to Gln69), which contains the chromophore, but the peptide has been found to be nonfluorescent. When native GFP is denatured in acid or guanidine hydrochloride, the protein loses its fluorescence and a new absorption spectrum (λmax = 380 nm) is obtained (16), similar to that of the hexapeptide in acid (14, 15). When the absorption and fluorescence spectra of native and renatured native GFP are compared at neutral pH, the spectra are found to be identical, indicating that the denaturation of GFP is reversible and that the fluorescence is dependent on the protein environment (16). However, a model compound of the chromophore has been found to be nonfluorescent in a variety of solvents (17).
At the present time, GFP fluorescence is still not completely understood. In this paper, we describe the results of a study on the properties of a large chromophore-bearing fragment, derived from GFP by lysyl endopeptidase digestion, and on a synthetic model compound of the chromophore. The findings indicate that (i) the chromophore has an imidazolone ring structure, (ii) fluorescence is highly dependent on the chromophore environment, (iii) the light emitter is a phenolate anion of the chromophore, and (iv) inhibition of isomerization of the exo-methylene double bond of 4-hydroxyphenylmethylidene group attached to the C-4 carbon of the imidazolone ring is responsible for the highly efficient green light emission of GFP.
MATERIALS AND METHODS
Materials.
Acetamidine hydrochloride, ethyl bromoacetate, 4-hydroxybenzaldehyde, and tert-butyldimethylchlorosilane were purchased from Tokyo Kasei (Tokyo) and α-cyano-4-hydroxycinnamic acid was obtained from Aldrich. Lysyl endopeptidase (Achromobacter protease I, sequencing grade), proteinase K, dithiothreitol, guanidine hydrochloride, imidazole, acetonitrile (HPLC grade), trifluoroacetic acid (amino acid analysis grade), C2HCl3 and tetramethylsilane were purchased from Wako Pure Chemicals (Osaka). Chelating Sepharose-Fast Flow gel was obtained from Pharmacia. All other chemicals used were of reagent grade quality.
Synthesis of Model Compound 1.
Compound 1, ethyl 4-(4-hydroxyphenyl)methylidene-2-methyl-5-oxo-1-imidazolacetate, was synthesized according to Scheme SI shown below (18, 19, 20). Acetamidine hydrochloride (1.01 g) was
Scheme I.
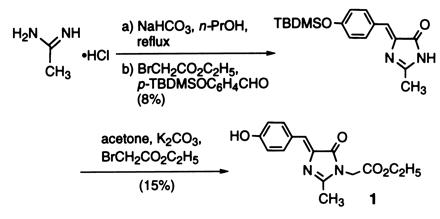
added to anhydrous 1-propanol (25 ml) containing NaHCO3 (1.78 g) and refluxed with heating under N2 for 15 min. A solution of ethyl bromoacetate (1.30 ml) and 4-tert-butyldimethylsiloxybenzaldehyde (p-TBDMSOC6H4CHO, 2.82 g) in anhydrous 1-propanol (25 ml) was added and the mixture refluxed with heating for 4.5 hr. p-TBDMSOC6H4CHO was prepared from 4-hydroxybenzaldehyde by O-silyation with tert-butyldimethylchlorosilane (21). After refluxing and cooling, the orange-colored mixture was concentrated in vacuo and the residue was dissolved in 60 ml of H2O. After extraction with toluene (4 × 60 ml), the organic layer was dried over Na2SO4, concentrated in vacuo, and purified by silica gel chromatography using ethyl acetate/CHCl3 (1:3, vol/vol) as the developing solvent to give 4-(4-tert-butyldimethylsiloxyphenyl)methylidene-2-methylimidazol-5-one, compound 2 (269 mg, 8% yield) in the form of yellow crystals: mp, 129–132°C; infra-red (IR) (KBr) νmax 3350–2750 (br), 1720, 1645, 1595, 1510, 1410, 1270, 1170 cm−1; 1H NMR (270 MHz, C2HCl3) δ 8.30 (br s, 1H, NH), 8.04 (d, J = 8.9 Hz, 2H), 7.04 (s, 1H), 6.88 (d, J = 8.4 Hz, 2H), 2.36 (s, 3H), 0.22 (s, 6H); MS [electron inpact (EI), 70 eV] m/z (relative intensity) 316 (M+, 68) 260 (34), 259 (100), 42 (21).
To a solution of compound 2 (70.6 mg, 0.223 mmol) in anhydrous acetone (1.2 ml) containing K2CO3 (37.3 mg, 0.270 mmol), ethyl bromoacetate (29 ml, 0.26 mmol) was added and the mixture was stirred at room temperature for 6 hr under N2. After concentrating the mixture in vacuo, the residue was dissolved in H2O (15 ml) and extracted with CHCl3 (4 × 15 ml). The organic layer was dried over Na2SO4, concentrated in vacuo and purified by silica gel chromatography using ethyl acetate/CHCl3 (1:3, vol/vol) as the developing solvent, followed by preparative thin-layer chromatography using the same solvent. Recrystallization from ethyl ether gave compound 1 (9.9 mg, 15% yield) in the form of yellow crystals: mp 164–165°C; IR (KBr) νmax 3700–2900 (br), 1735, 1715, 1640, 1600, 1445, 1370, 1275, 1220, 1170, 1145, 1020 cm−1; 1H NMR (500 MHz, C2HCl3) δ 8.08 (d, J = 8.8 Hz, 2H), 7.l0 (s, 1H), 6.89 (d, J = 8.8 Hz, 2H), 5.35 (s, 1H, OH), 4.38 (s, 2H), 4.24 (q, J = 7.2 Hz, 2H), 2.33 (s, 3H), 1.30 (t, J = 7.0 Hz, 3H); MS (EI, 70 eV) m/z (relative intensity) 288 (M+, 100), 146 (25), 100 (37), 55 (47), 54 (23); high-resolution mass spectrum (EI, 70 eV) m/z 288.1109, M+ calculated for C15H16N2O4 288.1111.
Spectroscopic Measurements.
Ultraviolet-visible absorption spectra were measured with a Hitachi (Tokyo) model 320 spectrophotometer. Fluorescence emission spectra were measured with a Hitachi model F4010 spectrofluorimeter (excitation band pass, 5 nm; emission band pass, 5 nm; scan speed, 60 nm/min). Fluorescence spectra were corrected according to manufacturer’s instructions. Fluorescence spectra of compound 1 and chromophore-containing lysyl endopeptidase fragment (in digest of GFP) were measured at 77 K in an all quartz tube (5 mm diameter) immersed in liquid N2 in an all quartz Dewar. For obtaining 1H NMR spectra, a Varian Unity (500 MHz) spectrometer and a JEOL GX270 (270 MHz) FT-NMR spectrometer, both calibrated with tetramethylsilane as an internal standard, were used. IR spectra were recorded on a Jasco (Easton, MD) IR-810 spectrometer. Low- and high-resolution EI mass spectra were obtained using a Hitachi M-80B mass spectrometer.
Preparation of Recombinant GFP.
The recombinant GFP used was His-GFP, the histidine-tagged fusion protein of GFP, which was overexpressed in Escherichia coli and purified by nickel-chelate affinity chromatography, as described (11, 13). The eluted GFP fraction was desalted and concentrated with an Amicon Centricon 30 microconcentrator. The purity of the GFP was estimated to be >95% by SDS/PAGE (12% gels), using heat and a reducing agent (22).
Purification of Chromophore-Containing Peptide Fragment.
One milligram of GFP was dissolved in 50 μl of denaturation buffer made up of deoxygenated 6 M guanidine hydrochloride/1 mM dithiothreitol in 0.4 M ammonium bicarbonate (pH 8.0) and heated at 88°C for 30 sec, after which the mixture was cooled in an ice bath. The denatured protein was suspended in 140 μl of H2O containing 10 μg of lysyl endopeptidase [enzyme/GFP = 1:100 (wt/wt)] and incubated at 37°C for 16 hr. A portion of the GFP digest was removed to measure absorption and fluorescence emission spectra. For further digestion with proteinase K, 10 μg of the enzyme was added and the incubation was continued for another 16 hr at 37°C. The digest was then diluted with 5 vol of 0.1 M acetic acid and subjected to reversed-phase HPLC using a 0.39 × 15 cm μBondasphere 5C4-300A column (Waters) and a linear gradient of 15–55% acetonitrile in 0.1% trifluoroacetic acid over a period of 80 min at a flow rate of 0.5 ml/min. The fractions eluting from the column were monitored at 220 and 380 nm. The lysyl endopeptidase fragment containing the chromophore eluted as a major peak at 49% acetonitrile, while the lysyl endopeptidase-proteinase K fragment eluted at 22% acetonitrile (data not shown). These fractions were pooled, lyophilized, and stored at −20°C until used.
Protein Analysis.
Protein concentration was determined by the dye-binding method of Bradford (23), using a Bio-Rad kit and bovine serum albumin as a standard (Pierce). From the GFP concentration, the molar extinction coefficient (ɛ) was calculated to be 19,400 M−1·cm−1 at 398 nm. N-terminal amino acid sequence analysis was performed using an Applied Biosystems model 470A gas-phase protein sequencer connected to an on-line model 120A phenylthiohydantoin analytical system (Applied Biosystems).
Mass Spectroscopy of Peptide Fragment.
Electrospray ionization (ESI) mass spectra were obtained using a JEOL HX110/HX110A tandem mass spectrometer, equipped with an electrospray ion source (Analytica, Branford, CT). The accelerating voltage was 7 kV and the ionization voltage was approximately 2.5 kV. A solution (50 pmol/ml) of the peptide fragment dissolved in acetonitrile/water, 65:35 (vol/vol), containing 1% acetic acid, was introduced into the electrospray ion source at a rate of 1.0 ml/min using a Harvard Apparatus model 22 syringe pump. A Brüker (Bremen, Germany) reflex time-of-flight (TOF) mass spectrometer was used to obtain matrix-assisted laser desorption/ionization (MALDI) mass spectra for determining molecular weight in a linear TOF mode and for carrying out post-source decay (PSD) fragment ion mass analysis. The nitrogen laser was set to deliver 337-nm wavelength pulses (5 ns) to the sample. α-Cyano-4-hydroxycinnamic acid was used as the matrix. On a sample target, 0.5 ml of saturated matrix solution in acetonitrile/0.1% aqueous trifluoroacetic acid, 50:50 (vol/vol), was mixed with 0.5 ml of the peptide fragment solution at 50 pmol/ml in acetonitrile/water, 65:35 (vol/vol), containing acetic acid (1%, vol/vol). Cocrystals of the sample–matrix target were obtained by drying the mixture at room temperature.
RESULTS AND DISCUSSION
Fig. 1A shows the amino acid sequence of the N-terminal region of GFP, including the amino acid residues involved in the formation of the fluorescent chromophore (11, 13), while Fig. 1B shows the proposed dehydration–dehydrogenation mechanism of chromophore formation (14, 15). According to this mechanism, the tripeptide -Ser65-Tyr66-Gly67- is cyclized in a post-translational modification with the elimination of one water molecule and two hydrogen atoms to give an imidazolone ring. The use of the expression vector, pHis-AGP, yields the protein His-GFP, consisting of Gly-Gly-Ser-His-His-His-His-His-His-Gly-Met-Ala-Ser-Met-Thr-Gly-Gly-Gln-Gln-Met-Gly-Arg-Asp-Leu-Tyr-Asp-Asp-Asp-Asp-Lys-Asp-Arg-Trp-Ile-Pro-Lys- fused to the N terminus of GFP (11, 13). Amino acid sequencing showed the protein to lack a methione at the N terminus. The fusion protein, however, has fluorescence properties identical to those of native GFP (11, 13). The expressed GFP was purified by nickel-chelate affinity chromatography and digested with lysyl endopeptidase, and the digest was subjected to reversed-phase HPLC. By monitoring the HPLC column at 380 nm and 220 nm, a colored fraction was isolated containing two peptide fragments. Amino acid sequencing showed the main peptide fragment present to have the sequence Phe-Ile-(Xaa)-Thr-Thr-Gly-Lys-Leu-Pro-Val-Pro-Trp-Pro-Thr-Leu-Val-Thr-Thr-Phe-(Xaa)-(Xaa)-(Xaa)-(Xaa)-(Xaa)-(Xaa)-, where (Xaa) denotes an undetectable amino acid residue. The second peptide was present as a minor contaminant, which had the beginning sequence Gly-Glu-Glu-Leu-Phe- and was identified as the peptide from Gly4 to Lys26. Thus, the chromophore-containing peptide isolated was Phe46 to Lys79, with a modification at Ser65 (13).
Figure 1.
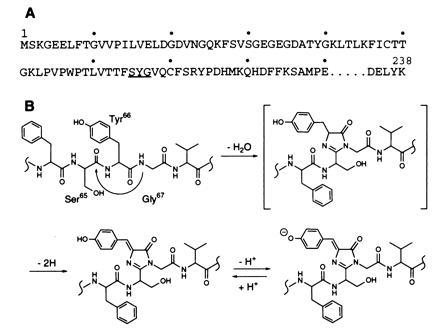
Scheme for the formation of the chromophore in Aequorea GFP. (A) Partial amino acid sequence of the N-terminal region of GFP showing the amino acid residues involved (underlined) in chromophore formation. (B) Dehydration–dehydrogenation mechanism for the formation of the chromophore.
The colored HPLC fraction containing the chromophore peptide gave ESI mass spectrum with peaks of the multiply charged ions at m/z 1301.0 and 976.0, corresponding to [M+3H]3+ and [M+4H]4+, respectively (data not shown). From this result, the molecular weight of the peptide in the fraction was determined to be m/z 3900.0, whereas the calculated mass for the peptide fragment from Phe46 to Lys79 is 3921.6 (average mass). The difference of 21.6 Da was attributed to the loss of one water molecule (18 Da) and four hydrogen atoms (4 Da), of which 20 Da was assumed to be due to the loss of one water molecule (18 Da) and two hydrogens (2 Da) during chromophore formation (Fig. 1B). This fragment is also seen to contain two cysteine residues, namely, Cys48 and Cys70 (Fig. 1A). If an intramolecular disulfide bond were to form in solution between the two, two more hydrogens would be lost and the total decrease would be 22.0 Da. The observed decrease of 21.6 Da is sufficiently close to this theoretical value to suggest that the chromophore-containing peptide originated from Phe46 to Lys79 by a modification of -Ser65-Tyr66-Gly67- (Fig. 1B) in the primary structure. The ESI mass spectrum also showed a clearly distinguishable peak with a mass of 2429.0 (data not shown), which was assumed to be due to the minor contaminating peptide Gly4 to Lys26, with a calculated average mass of 2428.7. The close agreement between the observed and calculated values lends credence to the accuracy of the mass measurements.
To establish the chemical structure of the chromophore, the purified GFP was digested with lysyl endopeptidase/proteinase K and a small peptide was isolated by reversed-phase HPLC. In the linear TOF mode MALDI mass spectrum (Fig. 2 Inset), this peptide gave a [M+H]+ ion at m/z 781.3, accompanied by a dehydrated [M+H-H2O]+ ion at m/z 763.8. The observed mass was 20.6 Da smaller than the calculated mass for the heptapeptide Thr63-Phe64-Ser65-Tyr66-Gly67-Val68-Gln69, which has an average mass of 801.9. The difference of 20.6 Da was attributed to the loss of one water molecule (18 Da) and two hydrogens (2 Da) during chromophore formation (Fig. 1B). PSD fragment ions generated by dissociation of the peptide bonds from the precursor ion m/z 781.3 were observed at m/z 515.0, 473.7, 368.8, 245.9, 220.9, and 146.9 (Fig. 2). These results are consistent with the degradation pattern and mass assignments shown for the peptide in Fig. 2 and strongly support the dehydration–dehydrogenation mechanism (Fig. 1B) (14, 15).
Figure 2.
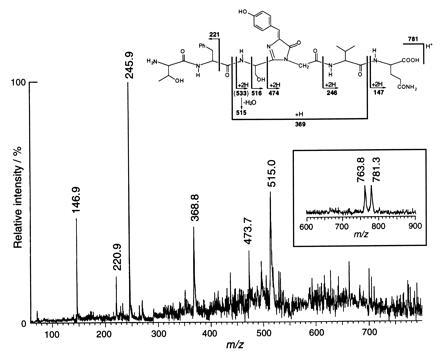
Linear mode MALDI–TOF mass spectrum (Inset), MALDI–PSD fragment ion mass spectrum of the isolated lysyl endopeptidase fragment of GFP and proposed fragmentation products of the chromophore with their assigned masses. Details of the mass spectroscopic procedures are described in the text.
The spectroscopic evidence also shows that the chemical structure of the GFP chromophore is similar to that of model compound 1. The GFP fusion protein dissolved in 0.1 M ammonium bicarbonate (pH 8.0) had an absorption maximum at 398 nm, with a smaller maximum at 476 nm. Under acidic conditions (0.1 M HCl), the GFP showed a single maximum at 382 nm, which shifted to 447 nm under alkaline conditions (0.1 M NaOH) (data not shown). This shift was reversible and the isosbestic point was at 405 nm (data not shown), which is similar to the shift previously reported for native GFP (24). The lysyl endopeptidase fragment (in digest of GFP) also showed the same spectral behavior, indicating that the covalent structure of the chromophore was not altered by the digestion and that the chromophore structure is similar to that in GFP. That the chromophore structure in the lysyl endopeptidase fragment and compound 1 resemble each other is further seen by the solvent-polarity dependency of the absorption spectra of compound 1 under neutral, acidic, and basic conditions. The absorption spectra of compound 1 in dimethyl sulfoxide (DMSO) (Fig. 3), 2-propanol, ethanol, and H2O showed absorption maxima at 373 nm (ɛ = 29,000), 371 nm (ɛ = 30,000), 372 nm (ɛ = 42,000), and 368 nm (ɛ = 31,000), respectively. In the same solvents containing 1 M HCl, the absorption spectra of compound 1 also showed a small solvent dependency with maxima between 373 and 386 nm. In DMSO (Fig. 3), 2-propanol, ethanol, and H2O containing 1 M NaOH, compound 1 in the form of a phenolate anion species showed strong absorption maxima at 462 nm (ɛ = 41,000), 443 nm (ɛ = 41,000), 435 nm (ɛ = 55,000), and 425 nm (ɛ = 41,000), respectively. The absorption spectra shifted to a shorter wavelength with increasing solvent polarity, indicating that in a polar solvent the ground state of the phenolate anion of compound 1 is stabilized to a greater extent than in the singlet excited state.
Figure 3.
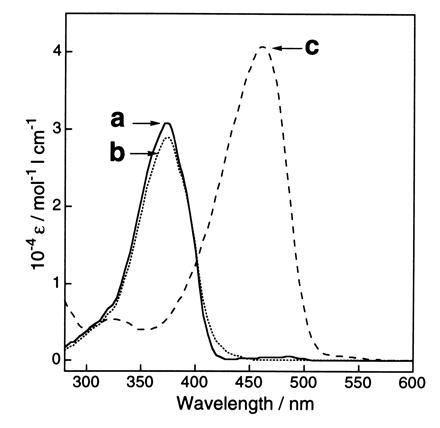
UV–visible absorption spectra of compound 1 in DMSO. Traces: a, compound 1 in DMSO (neutral); b, compound 1 in DMSO containing 1 M HCl aqueous, 5% (vol/vol) (acidic); c, compound 1 in DMSO containing 1 M NaOH aqueous, 5% (vol/vol) (basic). Concentration of compound 1 = 5.0 × 10−5 M.
The similarity of the GFP chromophore structure with compound 1 is further seen in the absorption spectra of compound 1 dissolved in 2-propanol/1 M NaOH, 2.5% (vol/vol), and 2-propanol/1 M HCl, 2.5% (vol/vol) (Fig. 4B), which match closely those of the lysyl endopeptidase fragment (in GFP digest) dissolved in 0.1 M NaOH and 0.1 M HCl (Fig. 4A). Aside from the absorbancy contributed by the aromatic amino acid residues (in the digest) of GFP, the absorption maxima and shapes of the curves for the peptide and compound 1 at basic and acid pH values correspond very well, with maxima at 447 nm and 382 nm for the peptide and at 443 nm and 376 nm for compound 1, which are also close to those (447 nm and 382 nm) of native GFP under the same conditions. The close agreement in the observed curves suggests that the chemical structures of the chromophore and compound 1 are similar, if not identical, and that the microenvironment around the chromophore in GFP is similar to that of compound 1 in 2-propanol, that is, nonpolar. Under basic conditions, compound 1 has the resonance structure of a phenolate anion shown in Fig. 4B. Under the same basic conditions, the absorption spectra of the lysyl endopeptidase fragment (in the GFP digest) and compound 1, as noted above, both have a maximum that is not too far from the maximum (476 nm) of the native GFP spectrum, which mirrors the fluorescence spectrum.
Figure 4.
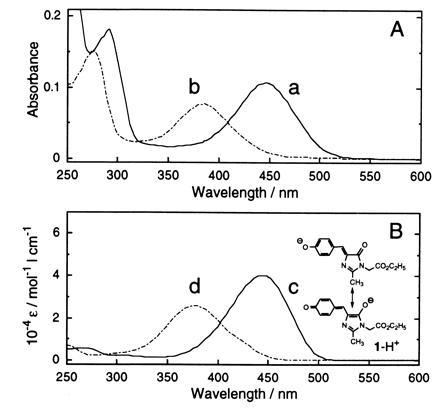
UV–visible absorption spectra of lysyl endopeptidase digest of GFP (A) and compound 1 (B). Traces: a, GFP digest in 0.1 M NaOH; b, GFP digest in 0.1 M HCl; c, compound 1 in 2-propanol containing 0.1 M NaOH aqueous solution, 2.5%, (vol/vol); d, compound 1 in 2-propanol containing 0.1 M HCl aqueous solution, 2.5% (vol/vol). Estimated original concentration of GFP = 4.8 × 10−6 M; concentration of compound 1 = 5.0 × 10−5 M.
On protease digestion or heat denaturation, the fluorescence of GFP was completely lost. The isolated lysyl endopeptidase fragment and lysyl endopeptidase/proteinase K fragment did not show any fluorescence in any of the organic solvent/aqueous mixtures examined. Similarly, solutions of compound 1 in DMSO, ethanol, and H2O gave only a weak fluorescence with quantum yields of less than 0.0001 and the addition of HCl or NaOH did not cause a significant increase in fluorescence intensity. However, when ethanol solutions of the lysyl endopeptidase fragment (in digest of GFP) and compound 1 were frozen as ethanol glass in liquid N2 (77 K), the fragment and compound 1 became highly fluorescent. At this temperature, the fluorescence emission maxima of compound 1 in ethanol and in ethanol containing 1 M aqueous HCl, 1% (vol/vol), were 435 nm and 437 nm, respectively, whereas in ethanol containing 1 M aqueous NaOH, 1% (vol/vol), the fluorescence of compound 1 was a bluish-green with a maximum at around 490 nm (Fig. 5). Similarly, the lysyl endopeptidase fragment (in GFP digest) in ethanol containing 1 M aqueous NaOH, 1% (vol/vol), was strongly fluorescent at 77 K, with the fluorescence emission spectrum (λmax = ≈475 nm) coinciding almost exactly with that of compound 1 (Fig. 5). The agreement in spectra provides support for the belief that the chemical structures of the GFP chromophore and compound 1 are the same, that is, it consists of a 4-(4-hydroxyphenyl)methylideneimidazol-5-one ring. Under basic conditions, the low-temperature fluorescence maxima of the lysyl endopeptidase fragment and compound 1 are also close to that of native GFP at room temperature, suggesting that the excited state phenolate anion of the 4-(4-hydroxyphenyl)methylideneimidazol-5-one ring is the greenish light emitter. However, the sharp and structured shape of the fluorescence spectrum of native GFP could not be reproduced under the low-temperature conditions. The reason why compound 1 is nonfluorescent at room temperature may be explained by competition with the cis–trans photoisomerization of a 4-methylideneimidazol-5-one derivative, which gives a mixture of cis–trans stereoisomers of an exo-methylene double bond (20). In a liquid medium, the cis–trans photoisomerization of the exo-methylene double bond may proceed as the main deactivation process from the singlet excited state, whereas in rigid glass at 77 K cis–trans isomerization is inhibited and fluorescence emission becomes the primary process (25, 26).
Figure 5.
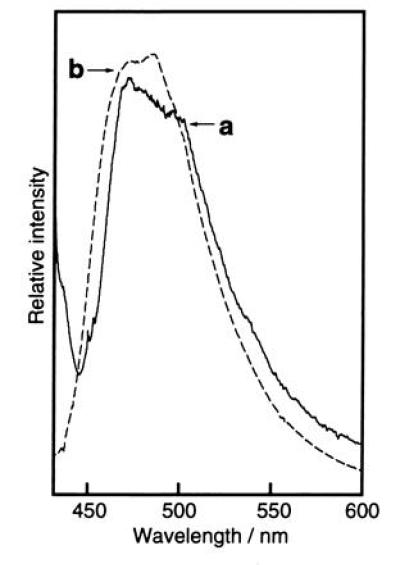
Fluorescence emission spectra of lysyl endopeptidase digest of GFP and compound 1 in ethanol glass at 77 K. Traces: a, digest of GFP in ethanol containing 0.1 M NaOH aqueous solution, 1% (vol/vol); b, compound 1 dissolved in ethanol containing 0.1 M NaOH aqueous solution, [% (vol/vol)] Estimated original concentration of GFP = 5.5 × 10−7 M; concentration of compound 1 = 5.0 × 10−7 M.
The clear overlapping of the absorption spectra (Fig. 4) and fluorescence emission spectra (Fig. 5) of compound 1 with those of the lysyl endopeptidase fragment indicates that the GFP chromophore consists of the 4-(4-hydroxyphenyl)methylideneimidazol-5-one ring. In particular, the evidence obtained under basic conditions supports the excited state phenolate anion of this ring as the greenish light emitter of GFP. When GFP is in the electronically excited state, either by exposure to ultraviolet irradiation at 390 nm or by resonance energy transfer from the excited state of the Aequorea blue fluorescent protein, the green light emission at 508 nm originates from the singlet excited state of the phenolate anion. A recent study (27) on the excited-state dynamics of GFP also appears to indicate the existence of a proton transfer process in the generation of a singlet excited-state species of the GFP chromophore, which emits the green light. This species may correspond to the phenolate anion in our scheme. The nonfluorescence and fluorescence of the lysyl endopeptidase fragment at room temperature and 77 K, respectively, may also be explained by a competition between the isomerization of the exo-methylene double bond and fluorescence emission, as in the case of compound 1. Thus, the characteristic greenish fluorescence of GFP at room temperature may be considered to be due to a restriction of the molecular motion of the chromophore in the peptide environment, while the loss of fluorescence on denaturation may be considered the result of isomerization of the exo-methylene double bond in the singlet excited state.
Acknowledgments
We thank Dr. Yoshihiro Ohmiya for carrying out N-terminal amino acid analysis for methionine in His-GFP. This work was supported in part by a Grant-in-Aid for Scientific Research in Priority Area from the Ministry of Education, Science and Culture of Japan [Grants 06240221 and 06239106 (M.O.)] and by National Science Foundation Research Grant MCB-9104684 (F.I.T.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: GFP, green fluorescent protein; EI, electron impact; ESI, electrospray ionization; MALDI, matrix-assisted laser desorption/ionization; PSD, post-source decay; TOF, time-of-flight; IR, infra-red; DMSO, dimethyl sulfoxide.
References
- 1.Harvey E N. Biol Bull (Woods Hole, MA) 1921;41:280–287. [Google Scholar]
- 2.Johnson F H, Shimomura O. Methods Enzymol. 1978;57:271–291. [Google Scholar]
- 3.Inouye S, Noguchi M, Sakaki Y, Takagi Y, Miyata T, Iwanaga S, Miyata T, Tsuji F I. Proc Natl Acad Sci USA. 1985;82:3154–3158. doi: 10.1073/pnas.82.10.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charbonneau H, Walsh K A, McCann R O, Prendergast F G, Cormier M J, Vanaman T C. Biochemistry. 1985;24:6762–6771. doi: 10.1021/bi00345a006. [DOI] [PubMed] [Google Scholar]
- 5.Johnson F H, Shimomura O, Saiga Y, Gershman L C, Reynolds G T, Waters J R. J Cell Comp Physiol. 1962;60:85–103. [Google Scholar]
- 6.Morise H, Shimomura O, Johnson F H, Winant J. Biochemistry. 1974;13:2656–2662. doi: 10.1021/bi00709a028. [DOI] [PubMed] [Google Scholar]
- 7.Prendergast F G, Mann K G. Biochemistry. 1978;17:3448–3453. doi: 10.1021/bi00610a004. [DOI] [PubMed] [Google Scholar]
- 8.Shimomura, O. & Johnson, F. H. (1973) Tetrahedron Lett. 2963–2966.
- 9.Hirano, T., Mizoguchi, I., Yamaguchi, M., Chen, F.-Q., Ohashi, M., Ohmiya, Y. & Tsuji, F. I. (1994) J. Chem. Soc. Chem. Commun. 165–167.
- 10.Morin J G, Hastings J W. J Cell Physiol. 1971;77:313–318. doi: 10.1002/jcp.1040770305. [DOI] [PubMed] [Google Scholar]
- 11.Inouye S, Tsuji F I. FEBS Lett. 1994;341:277–280. doi: 10.1016/0014-5793(94)80472-9. [DOI] [PubMed] [Google Scholar]
- 12.Prasher D C, Eckenrode V K, Ward W W, Prendergast F G, Cormier M J. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- 13.Inouye S, Tsuji F I. FEBS Lett. 1994;351:211–214. doi: 10.1016/0014-5793(94)00859-0. [DOI] [PubMed] [Google Scholar]
- 14.Shimomura O. FEBS Lett. 1979;104:220–222. [Google Scholar]
- 15.Cody C W, Prasher D C, Westler W M, Prendergast F G, Ward W W. Biochemistry. 1993;32:1212–1218. doi: 10.1021/bi00056a003. [DOI] [PubMed] [Google Scholar]
- 16.Ward W W, Bokman S H. Biochemistry. 1982;21:4535–4540. doi: 10.1021/bi00262a003. [DOI] [PubMed] [Google Scholar]
- 17.McCapra, F., Razavi, Z. & Neary, A. P. (1988) J. Chem. Soc. Chem. Commun. 790–791.
- 18.Devasia, G. M. (1976) Tetrahedron Lett., 571–572.
- 19.Devasia G M, Shafi P M. Indian J Chem. 1981;20B:657–660. [Google Scholar]
- 20.Dalla Croce, P. & La Rosa, C. (1985) J. Chem. Res. Synop., 360–361.
- 21.Corey E J, Venkateswarlu A. J Am Chem Soc. 1972;94:6190–6191. [Google Scholar]
- 22.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Ward W W, Cody C W, Hart R C, Cormier M J. Photochem Photobiol. 1980;31:611–615. [Google Scholar]
- 25.Sharafy S, Muszkat K A. J Am Chem Soc. 1971;93:4119–4125. [Google Scholar]
- 26.Turro N J. Modern Molecular Photochemistry. Menlo Park, CA: Benjamin/Cummings; 1978. pp. 110–114. [Google Scholar]
- 27.Chattoraj M, King B A, Bublitz G U, Boxer S G. Proc Natl Acad Sci USA. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]