

Abstract
The present study was undertaken to determine if the neuroprotective effect of 17β-estradiol (E2) when administrated after ischemia is dose-dependent and if the therapeutic window for estrogen can be prolonged. Ischemic injury was induced by permanent middle cerebral artery occlusion (p-MCAO). Administration of E2 at 30 minutes after ischemia resulted in a reduction in lesion volume. A higher dose of E2 extended the therapeutic window to 6 hour after cerebral ischemia in 33% of the rats. These findings suggest that postischemic treatment with estrogen affords protection against ischemic damage and that it acts within a clinically useful therapeutic window.
Keywords: Estrogen, Cerebral-ischemia, Neuroprotection
Introduction
We initially reported that estrogens cause a dose-dependent protection of SK-N-SH cells under conditions of serum- deprivation [3, 26]. Since these initial observations of in vitro neuroprotection with estrogen treatment, there have been several hundred reports describing the neuroprotective effects of estrogens [4, 11]. This neuroprotection is seen in a variety of neuronal cell types in response to over 14 different neurotoxic insults in vitro, including serum-deprivation [14, 26], β-amyloid [2, 10] and excitatory amino acid treatment [5, 10].
We have now extensively assessed the neuroprotective effects of estrogens in an animal model of cerebral ischemia. Following our first reports of neuroprotection with estrogens in an animal model of ischemia [25], we and other have demonstrated that estrogens protects the brain from ischemic damage induced by transient cerebral ischemia [15, 21], permanent cerebral ischemia [6, 30], subarachnoid hemorrhage [30], and global ischemia [13]. The protective effects of estrogens are seen with 17β-estradiol (E2), as well as non-feminizing estrogens, such as 17α-estradiol [25], ENT-estradiol [12] and 2-adamantyl-estrone [16], suggesting that at pharmacological doses, estrogen receptors are not required for neuroprotection in stroke. Dubal et al. [9] reported that ERαKO, but not ERβKO, mice were resistant to the neuroprotective effects of E2 administered chronically at low physiological concentrations and concluded that ERα is a necessary mediator of estrogen neuroprotection. Later, however, Sampie et al. [21] observed that ERβKO mice have unusually small infarcts, likely due to the extremely high levels of circulating E2, and McCullough et al [18] demonstrated estrogen neuroprotection in ERαKO mice using pharmacological doses of estrogens.
While much is known about the ability of pretreatment with estrogens to protect brain tissue from ischemia, few studies have assessed the effects of estrogens when administered after the onset of ischemia. We reported that the protection afforded by E2 in ovariectomized rats when high doses are administered by an intravenous injects can be observed between 30 min and 3 hours following the onset of cerebral ischemia [25, 32]. McCullough et al. [17] administered Premarin®, a conjugated equine estrogen preparation, to male rats after transient MCA occlusion and observed protection at 10 min, but not 90 min after the onset of occlusion. In the preliminary dose-finding study [31], with E2 doses of 0, 100, 500, 1000, 2000 and 5,000 μg/kg body weight when administered at 6 hours after the onset of the occlusion, we observed that there was a dose-dependent reduction in lesion volume from 100 to 500 μg/kg body weight, a persistence of the effect at 1,000 μg/kg body weight and a relative loss of protection at the higher two doses. As such, in the present study, we selected the optimal dose for delayed neuroprotection, 1000 μg/kg body weight, using a subcutaneous preparation for rapid, sustained delivery of estrogens.
Materials and Methods
Experimental animals and E2 preparation
Female Charles Rivers Sprague-Dawley rats (12 weeks old, Wilmington, MA) were acclimatized to animal facilities three days prior to surgery. Bilateral ovariectomy was performed 2 weeks before permanent middle cerebral artery occlusion (MCAO). All animal procedures were approved by the University of Florida and the University of North Texas Health Science Center Animal Care and Use Committees. E2 was first dissolved in absolute ethanol. Then, sesame oil with 2% benzyl alcohol was added to the solution to yield the final concentration of 100 μg/ml, 500 μg/ml, or 1 mg/ml. The ethanol was evaporated off.
E2 serum concentrations
A group of animals (n=7) were ovariectomized to eliminate endogenous ovarian steroids. After two weeks, E2 was injected subcutaneously (s.c.) at the dose of 100 μg/kg. Blood samples were taken via a permanent atrial cannula prior to administration of E2 (time 0), and at 5, 15, 30 and 60 min, and 4, 8, 24 hours after E2 administration. Blood samples were centrifuged for 20 minutes and plasma was separated and stored at −20 °C until assayed. Plasma concentrations of E2 were determined with an ultra-sensitive estradiol radioimmunoassay kit (Diagnostic Systems Lab, Webster, TX.).
Cerebral ischemia
Two weeks after ovariectomy, animals were anesthetized by intraperitioneal injection of ketamine (60 mg/kg) and xylazine (10 mg/kg). During the procedures, rectal temperature was monitored and maintained between 36.5 ºC and 37 ºC with heating lamps and warming pads. With the aid of an operating microscope, the left common carotid artery and internal carotid artery were exposed through a midline cervical skin incision. A 4-0 monofilament suture with a rounded-tip was introduced into the internal carotid artery via the external carotid artery lumen and advanced until resistance was encountered. The distance between the common carotid artery bifurcation and the resistant point was 2.2 cm. A 6-0 silk ligature was placed around the external carotid artery to prevent bleeding and movement of suture position. The common carotid artery and pterygopalatine artery temporary ligatures were then released, and the skin incision was closed.
Measurement of Lesion Volume
Animals were decapitated 48 hours after MCAO, and the brain was harvested and placed in a metallic brain matrix for tissue slicing. Seven 2 mm coronal slices were made at 3, 5, 7, 9, 11, 13, and 15 mm posterior to the olfactory bulb. Each slice was incubated for 30 minutes at 37 °C in a 2% solution of 2,3,5-triphenyltetrazolium chloride (Sigma, St. Louis, MO) followed by fixation in 10% formalin. Stained slices were photographed by a digital camera (Sony, Tokyo, Japan). The ischemic lesion area in both side of each slice was traced and subsequently calculated for ischemic lesion volume (Image-Pro Plus, Media Cybernetics, Silver Spring, MD). The total ischemic lesion volume was calculated as the sum of the volume of the ischemic lesion across the 7 slices.
Protocol 1
This study was conducted to determine if the neuroprotective effects of E2 were dose-dependent when administrated 30 minutes after MCAO. A total of 21 rats were divided into 3 groups. The animals were treated either with a single subcutaneous injection of E2 at the dose of 100 μg/kg (OVX+ E2 100 μg/kg, n=5) or 500 μg/kg (OVX+E2 500 μg/kg, n=7). For controls, ovariectomized females (OVX, n=9) were subcutaneous injected with equivalent volumes of the vehicle. The animals were decapitated 2 days after MCAO and the ischemic lesion volumes were determined.
Protocol 2
Our previous study demonstrated that E2 exerts neuroprotective effects when intravenously administrated up to 3 hours after an ischemic insult at a concentration of 100 μg/kg (43). Herein, we tested whether higher doses of E2 could prolong this therapeutic window. E2 was administered subcutaneously with the dose of 1 mg/kg at 30 minutes (OVX+E2 30 minutes group, n=6) or 6 hours (OVX+E2 6 hours group, n=12) after MCAO. Control animals received equal volume of vehicle at 30 minutes (OVX 30 minutes group, n=7) or 6 hours (OVX 6 hours group, n=8) after MCAO. The ischemic lesion volume was measured as described in protocol 1.
Statistic analysis
Statistical analysis was performed using Prism software (GraphPad Software, Inc, San Diego, CA.). All data are presented as means ± SEM. Ischemic lesion volumes were compared by one-way ANOVA followed by Tukey tests. A probability of <0.05 was considered significant.
Results
Effect of E2 on mortality
The permanent MCAO caused inadvertent death of some animals due to excessively large lesion volume. Deaths prior to the planned 48 hr sampling of animals occurred in 5 OVX and 4 E2-treated rats. These rats fully recovered from surgery and anesthesia, then died within 48 hours after occlusion. Interestingly, in this subset of animals, OVX rats displayed more severe motor deficits and died earlier (<12 h after the occlusion), while, the E2-treated rats had milder motor deficit and survived up to 24–36 h after occlusion. Due to the poor staining of the ischemic lesion, all the premature died animals were excluded from the lesion volume analysis.
Plasma E2 concentration after subcutaneous administration
Plasma estradiol increased rapidly after subcutaneous injection of E2 (100 μg/kg), reaching over 2000 pg/ml within 5 min after administration. The plasma concentration of E2 peaked around 1 hour post-injection, and decreased thereafter (Fig. 1). At 24 hours after administration, plasma E2 was still elevated with a concentration of 167.9 ± 71.8 pg/ml.
Figure 1.
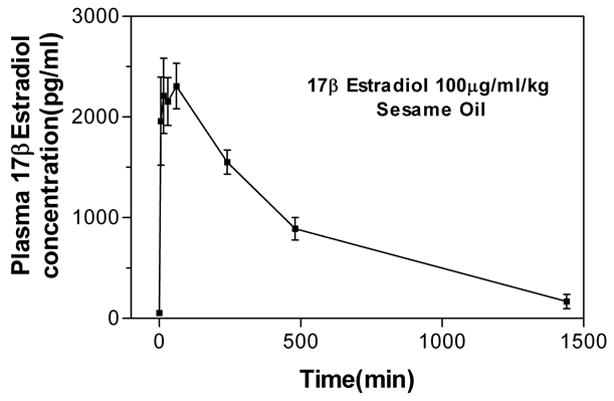
Plasma estradiol levels after E2 administration. E2 was subcutaneous administered at the dose of 100 μg/kg in 7 ovariectomized rats. Blood samples were taken prior (time 0), and 5, 15, 30 minutes, and 1, 4, 8, 24 hours after E2 administration. Plasma estradiol concentration is given in pg/ml: 1nM = 272 pg/ml.
Dose-dependent neuroprotective effect of E2 when administered at 30 minutes after cerebral ischemia
Permanent occlusion of the MCA resulted in a mean total lesion volume of 352 ± 28 mm3, with the lesion localized primarily in the parietal cortex (223 ± 21 mm3) and basal ganglia (130 ± 12.0mm3) (Fig. 2 & 4). Subcutaneous E2 administration at 30 minutes after permanent MCA occlusion significantly decreased the total lesion volume to 224 ± 21 mm3 and 130 ± 31 mm3 with the dose of 100 μg/kg or 500 μg/kg, respectively. This reduction corresponds to 36 ± 6% and 63 ± 9% of the control, respectively (Fig. 2A). The estrogen-mediated protection was observed in both cortex and subcortex regions (Fig. 2B & 2C). In the ischemic cortex, subcutaneous E2 injection at the dose of 100 μg/kg or 500 μg/kg E2 resulted in a mean cortical lesion volume of 137 ± 21 mm3 (a 39 ± 9% reduction) or 66 ± 26 mm3 (a 71 ± 12% reduction), respectively. In the subcortex, E2 treatment decreased lesion volumes to 84 ± 6 mm3 (a 35 ± 5% reduction) at the dose of 100 μg/kg, and 73 ± 9mm3 (a 44 ± 7 % reduction) at the dose of 500 μg/kg.
Figure 2.
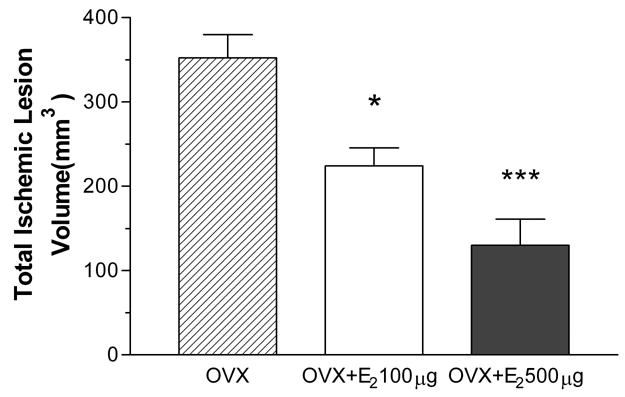
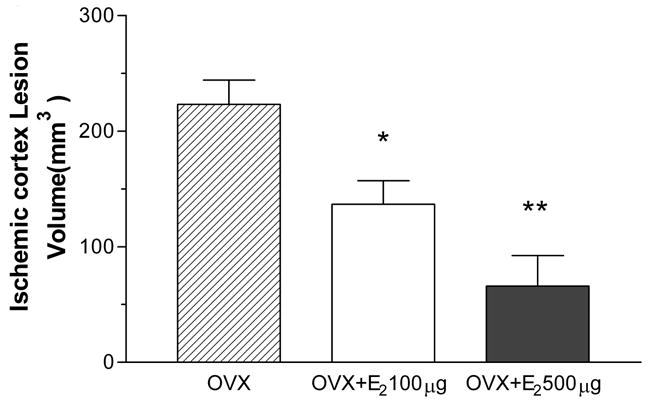

Effect of 30 minutes posttreatment with 100 or 500 μg/kg of E2 on total lesion volume (A), cortical lesion volume (B), and subcortical lesion volume (C) induced by p-MCAO. E2 significantly decreases total lesion volume by 36.4% ± 6.0% and 63.1% ± 8.9% in 100 μg/kg (n=5) or 500 μg/kg (n=7) group vs. OVX group (n=9), respectively. E2 significantly decreases cortical lesion volume by 38.8% ± 9.2% or 70.5% ± 11.8% compared to OVX rats in 100 μg/kg (n=5) or 500 μg/kg (n=7) group vs. control group (n=9), respectively. E2 significantly decreases subcortical lesion volume by 35.3% ± 4.7% or 43.8% ± 7.2% compared to OVX rats in 100 μg/kg (n=5) or 500 μg/kg (n=7) group vs. control group (n=9), respectively. *P<0.05 vs. control. **P<0.01 vs. control. ***P<0.001 vs. control
Figure 4.

Photograph displaying representative brain sections obtained from ovariectomized rats treated with E2 at 30 minutes or 6 hours after MCA occlusion. Rat were treated with vehicle (OVX), or E2 at the dose of 100 μg/kg (E2 100), 500 μg/kg (E2 500), or 1000 μg/kg (E2 1000).
Dose dependent therapeutic window of E2 protection
Ischemic lesion volumes were significantly reduced from 352 ± 28 mm3 in controls to 212 ± 24 mm3 (a reduction of 40 ± 7%) in E2 treatments when E2 was administered with the dose of 1 mg/kg at 30 minutes after ischemic insult. When administered at 6 hours after occlusion, protective effect of estrogen was demonstrated in 33% of animals tested (4 of 12 rats) with the dose of 1 mg/kg. In this subgroup, the ischemic lesion volume was reduced from 318 ± 29 mm3 in control group to 136.4 ± 28.6mm3(57 ± 9% reduction) in E2 treated group. However, protective effect of E2 was not indicated in 67%, or 8 out of 12 rats, in the E2 treated group when E2 was administered with the dose of 1 mg/kg at 6 hours after the onset of MCAO.
Discussion
The present study demonstrates 3 important features for the potential use of estrogens for the treatment of ischemic stroke. First, the protective effect of estrogen is dose-dependent when administered after the onset of cerebral ischemia. Second, the therapeutic window of E2 appears to be at least 6 hours in a subgroup of animals. Finally, subcutaneous administration of an oil formulation of E2 produces a rapid and sustained increase in circulating estrogens at the concentrations that are neuroprotective for at least 24 hours. Collectively, these data indicate that estrogen formulations and dosing regimens can profoundly protect the brain from ischemic damage.
The oil formulation tested in the present study produced a rapid increase in circulating E2 concentrations, with high level seen by the first sampling time at 5 min. and levels were maintained above those needed to protect brain tissue [22, 32] for at least 24 hours. In as much as no toxicity was seen with this formulation and mode of dosing E2, it appears to be a safe therapeutic. Indeed, even in animals that died as a result of large ischemic lesion produced, the E2 treatment appears to improve symptoms and survival time.
Our observation of dose-dependent protection with E2 administered after the onset of MCAO is important since the safety of E2 have not been established for acute high dose administration. The peak concentrations of E2 achieved and sustained for 2 hours with subcutaneous administration of 100 μg/ml with this formulation were 2000 pg/ml (7 nM). We assume that the 1 mg/ml dose of E2 would produce 10 times higher peak concentrations (or 70 nM). In a variety of in vitro studies these peak concentrations of E2 are potently neuroprotective and do not exhibit toxicity [2, 3, 10].
Our observation of a therapeutic window for E2 administration in an oil formulation of 6 hours for a third of animals tested indicates that estrogens can protect brain tissue through mechanisms that are initiated long after the onset of the ischemic event. A variety of mechanisms for estrogen neuroprotection have been proposed, among which are activation of anti-apoptotic proteins [1, 7, 14, 28] and preservation of mitochondrial integrity [4, 27]. Our results suggest that estrogen treatment delayed by as much as 6 hours can affect these neuroprotective processes in a way the affords substantial benefit for a subpopulation of subjects. Although it is uncertain whether the therapeutic window of estrogen could be extended beyond 6 hours, this 6-hour time frame is clinically relevant in that it is logistically difficult to institute therapy in many patients with acute stroke at earlier times.
It is uncertain why some subjects responded, while others didn’t, to the E2 treatment when it was administrated at 6 hours after the onset of cerebral ischemia. This model and treatment paradigm is significant for its potential clinical relevance. Our study indicated that this model could be used to determine the mechanism contribute to the different response of each subject to the therapeutic intervention, which frequently happens in clinical circumstance.
We observed cortical as well as subcortical protection with delayed E2 treatment. In pretreatment paradigms, consistent protection of the cortex has been reported [22, 23]. On the other hand, subcortical protection of E2 has been reported by some studies [20, 29], while others do not [6, 8]. The apparent resistance of the subcortex to E2 protection could be due to the lack of estrogen receptors (ER) expression at the basal ganglion [24]. This could contribute to the relative resistance of the basal ganglion to E2 treatment through the lack of ER-mediated neuroprotection in this region of the brain, as has been proposed. However, the basal ganglion is an exclusive territory of the middle cerebral artery and as such does not receive blood flow from collateral arteries, in contrast to the extensive collateral circulation of the cortex [22]. As such, MCAO results in a more severe ischemia in the subcortex than the cortex [8, 19]. Our observation that high doses of E2, even when administered after the onset of the ischemia can protect the subcortex supports the hypothesis that this brain region is estrogen protectable.
In conclusion, we have shown that a subcutaneous oil preparation for E2 delivery that produces rapid and sustained elevations in circulating E2 causes a dose-dependent protection against focal cerebral ischemia. This protection is seen for as long as 6 hours after the onset of ischemia. Collectively, these results suggest that subcutaneous estrogen treatment with oil formulations during ongoing strokes may improve neurological outcome.
Figure 3.

Effect of 30 minutes or 6 hours posttreatment with 1 mg/kg of E2 on ischemic total lesion volume induced by p-MCAO. When administered at 30 minutes after occlusion, E2 (n=6) significant decreases the total infarct volume by 40.0% ± 6.9% vs. OVX group (n=7). When administered at 6 hours after occlusion, protective effect of estradiol was demonstrated in 4 of 12 rats (E2# group), with a 57.0 ± 9.0% reduction of the total infarct volume vs. OVX group (n=8), while, no protective effect was identified in the other 8 rats (E2 group). *P<0.05 vs. control.
Acknowledgments
This work is supported in part by grants NIA AG 10485 and AG 22550
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21:7543–50. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Behl C, Widmann M, Trapp T, Holsboer F. 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1995;216:473–82. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- 3.Bishop J, Simpkins JW. Estradiol treatment increases viability of glioma and neuroblastoma cells in vitro. Mol Cell Neurosci. 1994;5:303–8. doi: 10.1006/mcne.1994.1036. [DOI] [PubMed] [Google Scholar]
- 4.Brinton RD. Cellular and molecular mechanisms of estrogen regulation of memory function and neuroprotection against Alzheimer’s disease: recent insights and remaining challenges. Learn Mem. 2001;8:121–33. doi: 10.1101/lm.39601. [DOI] [PubMed] [Google Scholar]
- 5.Brinton RD, Tran J, Proffitt P, Montoya M. 17 beta-Estradiol enhances the outgrowth and survival of neocortical neurons in culture. Neurochem Res. 1997;22:1339–51. doi: 10.1023/a:1022015005508. [DOI] [PubMed] [Google Scholar]
- 6.Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18:1253–8. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 7.Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci. 1999;19:6385–93. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubal DB, Wise PM. Neuroprotective effects of estradiol in middle-aged female rats. Endocrinology. 2001;142:43–8. doi: 10.1210/endo.142.1.7911. [DOI] [PubMed] [Google Scholar]
- 9.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–7. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–44. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 11.Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18:347–58. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- 12.Green PS, Yang SH, Nilsson KR, Kumar AS, Covey DF, Simpkins JW. The nonfeminizing enantiomer of 17beta-estradiol exerts protective effects in neuronal cultures and a rat model of cerebral ischemia. Endocrinology. 2001;142:400–6. doi: 10.1210/endo.142.1.7888. [DOI] [PubMed] [Google Scholar]
- 13.He Z, He YJ, Day AL, Simpkins JW. Proestrus levels of estradiol during transient global cerebral ischemia improves the histological outcome of the hippocampal CA1 region: perfusion-dependent and-independent mechanisms. J Neurol Sci. 2002;193:79–87. doi: 10.1016/s0022-510x(01)00648-7. [DOI] [PubMed] [Google Scholar]
- 14.Honda K, Shimohama S, Sawada H, Kihara T, Nakamizo T, Shibasaki H, Akaike A. Nongenomic antiapoptotic signal transduction by estrogen in cultured cortical neurons. J Neurosci Res. 2001;64:466–75. doi: 10.1002/jnr.1098. [DOI] [PubMed] [Google Scholar]
- 15.Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20:631–52. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Liu R, Yang SH, Perez E, Yi KD, Wu SS, Eberst K, Prokai L, Prokai-Tatrai K, Cai ZY, Covey DF, Day AL, Simpkins JW. Neuroprotective effects of a novel non-receptor-binding estrogen analogue: in vitro and in vivo analysis. Stroke. 2002;33:2485–91. doi: 10.1161/01.str.0000030317.43597.c8. [DOI] [PubMed] [Google Scholar]
- 17.McCullough LD, Alkayed NJ, Traystman RJ, Williams MJ, Hurn PD. Postischemic estrogen reduces hypoperfusion and secondary ischemia after experimental stroke. Stroke. 2001;32:796–802. doi: 10.1161/01.str.32.3.796. [DOI] [PubMed] [Google Scholar]
- 18.McCullough LD, Alkayed NJ, Blizzard KK, Hurn PD. Estrogen protects ischemic brain of ERa and ERß knock out mice. The Physiologist. 2001;44:269. [Google Scholar]
- 19.Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27:1641–6. doi: 10.1161/01.str.27.9.1641. discussion 1647. [DOI] [PubMed] [Google Scholar]
- 20.Rusa R, Alkayed NJ, Crain BJ, Traystman RJ, Kimes AS, London ED, Klaus JA, Hurn PD. 17beta-estradiol reduces stroke injury in estrogen-deficient female animals. Stroke. 1999;30:1665–70. doi: 10.1161/01.str.30.8.1665. [DOI] [PubMed] [Google Scholar]
- 21.Sampei K, Goto S, Alkayed NJ, Crain BJ, Korach KS, Traystman RJ, Demas GE, Nelson RJ, Hurn PD. Stroke in estrogen receptor-alpha-deficient mice. Stroke. 2000;31:738–43. doi: 10.1161/01.str.31.3.738. discussion 744. [DOI] [PubMed] [Google Scholar]
- 22.Shi J, Bui JD, Yang SH, He Z, Lucas TH, Buckley DL, Blackband SJ, King MA, Day AL, Simpkins JW. Estrogens decrease reperfusion-associated cortical ischemic damage: an MRI analysis in a transient focal ischemia model. Stroke. 2001;32:987–92. doi: 10.1161/01.str.32.4.987. [DOI] [PubMed] [Google Scholar]
- 23.Shi J, Zhang YQ, Simpkins JW. Effects of 17beta-estradiol on glucose transporter 1 expression and endothelial cell survival following focal ischemia in the rats. Exp Brain Res. 1997;117:200–6. doi: 10.1007/s002210050216. [DOI] [PubMed] [Google Scholar]
- 24.Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–25. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 25.Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, Bodor N, Day AL. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–30. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- 26.Simpkins JW, Singh M, Bishop J. The potential role for estrogen replacement therapy in the treatment of the cognitive decline and neurodegeneration associated with Alzheimer’s disease. Neurobiol Aging. 1994;15(Suppl 2):S195–7. doi: 10.1016/0197-4580(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 27.Simpkins JW, Wang J, Wang X, Perez E, Prokai L, Dykens JA. Mitochondria play a central role in estrogen-induced neuroprotection. Curr Drug Targets CNS Neurol Disord. 2005;4:69–83. doi: 10.2174/1568007053005073. [DOI] [PubMed] [Google Scholar]
- 28.Singh M. Ovarian hormones elicit phosphorylation of Akt and extracellular-signal regulated kinase in explants of the cerebral cortex. Endocrine. 2001;14:407–15. doi: 10.1385/ENDO:14:3:407. [DOI] [PubMed] [Google Scholar]
- 29.Toung TJ, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke. 1998;29:1666–70. doi: 10.1161/01.str.29.8.1666. [DOI] [PubMed] [Google Scholar]
- 30.Yang SH, He Z, Wu SS, He YJ, Cutright J, Millard WJ, Day AL, Simpkins JW. 17-beta estradiol can reduce secondary ischemic damage and mortality of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2001;21:174–81. doi: 10.1097/00004647-200102000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Yang SH, Liu R, Wu SS, Simpkins JW. The Use of Estrogen and Related Compounds in the Treatment of Damage from Cerebral Ischemia,, Steroids and the Nervous System. Annuals of the New York Academy of Sciences. 2004;1007:101–107. doi: 10.1196/annals.1286.010. [DOI] [PubMed] [Google Scholar]
- 32.Yang SH, Shi J, Day AL, Simpkins JW. Estradiol exerts neuroprotective effects when administered after ischemic insult. Stroke. 2000;31:745–9. doi: 10.1161/01.str.31.3.745. discussion 749–50. [DOI] [PubMed] [Google Scholar]