

Abstract
Classical Hodgkin lymphomas (cHLs) contain small numbers of neoplastic Reed–Sternberg (RS) cells within an extensive inflammatory infiltrate that includes abundant T helper (Th)-2 and T regulatory (Treg) cells. The skewed nature of the T cell infiltrate and the lack of an effective host antitumor immune response suggest that RS cells use potent mechanisms to evade immune attack. In a screen for T cell-inhibitory molecules in cHL, we found that RS cells selectively overexpressed the immunoregulatory glycan-binding protein, galectin-1 (Gal1), through an AP1-dependent enhancer. In cocultures of activated T cells and Hodgkin cell lines, RNAi-mediated blockade of RS cell Gal1 increased T cell viability and restored the Th1/Th2 balance. In contrast, Gal1 treatment of activated T cells favored the secretion of Th2 cytokines and the expansion of CD4+CD25high FOXP3+ Treg cells. These data directly implicate RS cell Gal1 in the development and maintenance of an immunosuppressive Th2/Treg-skewed microenvironment in cHL and provide the molecular basis for selective Gal1 expression in RS cells. Thus, Gal1 represents a potential therapeutic target for restoring immune surveillance in cHL.
Keywords: immunomodulation, microenvironment, Th2, Treg, c-Jun
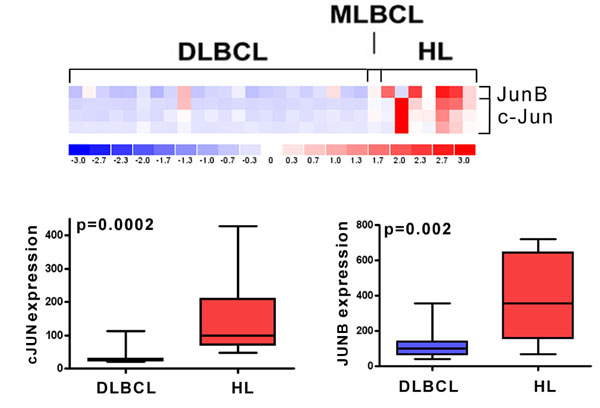
Classical Hodgkin lymphoma (cHL) is a B cell malignancy diagnosed in ≈20,000 new patients in North America and Europe each year; >90% of these patients are young adults. Classical HLs include small numbers of malignant Reed–Sternberg (RS) cells within an extensive inflammatory infiltrate (1). The tumor cells derive from preapoptotic germinal center B cells that have undergone crippling mutations of their rearranged Ig genes (1, 2). Classical HL RS cells lack B cell receptor-mediated signals and rely on alternative survival and proliferation pathways activated by transcription factors such as NF-κB and activator protein 1 (AP1) (3–5). In cHL, the tumor cells exhibit constitutive AP1 activation, express high levels of the AP1 components, c-Jun and JunB, and depend on AP1-mediated proliferation signals (3).
Although primary cHLs have a brisk inflammatory infiltrate, there is little evidence of an effective host antitumor immune response. The reactive T cell population includes predominantly T helper 2 (Th2)-type and CD4+CD25highFOXP3+ regulatory T cells (Treg cells) that directly suppress immune responses and protect cHL RS cells from immune attack (1, 6–8); Th1, natural killer, and cytotoxic T cells are markedly underrepresented. In addition, primary cHLs are characterized by a unique cytokine and chemokine profile, including IL-4, IL-5, IL-10, and IL-13 (1, 9). In fact, IL-13 is a critical growth factor for cHL RS cells (1, 9). However, the molecular signals and endogenous factors responsible for creating and maintaining the Th2-skewed immunosuppressive microenvironment in cHL remain to be defined.
In a screen for T cell-inhibitory molecules in cHL, we found that cHL RS cells overexpressed the carbohydrate-binding lectin, galectin-1 (Gal1). Galectins have recently emerged as novel regulators of immune cell homeostasis and tumor-immune escape (10–13). Gal1, an evolutionarily conserved member of this family (14), preferentially recognizes multiple Gal β1,4 GlcNAc (N-acetyllactosamine) units that may be presented on the branches of N- or O-linked glycans on cell surface glycoproteins such as CD45, CD43, and CD7 (15). Through binding and cross-linking of specific glycoconjugates, Gal1 has the potential to inhibit T cell effector functions and regulate the inflammatory response (16–20). In several murine models of chronic inflammatory diseases, recombinant Gal1 suppressed Th1-dependent responses and increased T cell susceptibility to activation-induced cell death (17–20).
In a recently described solid tumor (murine melanoma) model, Gal1 was also found to play a pivotal role in promoting escape from T cell-dependent immunity and conferring immune privilege to tumor cells (12). In this model, Gal1 blockade markedly enhanced syngeneic tumor rejection and tumor-specific T cell-mediated immune responses (12). In another recently described solid tumor (head and neck squamous cell carcinomas), Gal1 overexpression was inversely correlated with the number of infiltrating T cells and was an independent prognostic factor for shorter overall survival (13). Because cHL is a unique lymphoid malignancy characterized by an extensive but ineffective host inflammatory/immune response, we evaluated the causes and consequences of Gal1 overexpression in this disease.
Results
Gal1 Is Overexpressed in cHL RS Cells.
To identify novel cHL-specific T cell-inhibitory molecules, we compared the gene expression profiles of a series of cHL and diffuse large B cell lymphoma (DLBCL) and mediastinal large B cell lymphoma (MLBCL) cell lines. Gal1 transcripts were 4- to 29-fold more abundant in cHL cell lines than in the LBCL lines (P = 0.002, FDR = 0.014, Fig. 1 A and B). Gal1 protein expression was also uniformly high in cHL cell lines and low or undetectable in DLBCL and MLBCL lines by Western blotting (Fig. 1C). Immunohistochemical staining of primary tumor sections revealed abundant Gal1 expression in cHL RS cells, whereas LBCLs were uniformly negative (Fig. 1D). In a series of primary lymphoid tumors, 10 of 10 cHLs were Gal1+, whereas 10 of 10 primary DLBCLs and 5 of 5 primary MLBCLs lacked Gal1 expression (data not shown). Taken together, these data indicate that Gal1 is selectively up-regulated in the RS cells of cHLs.
Fig. 1.

Gal1 is overexpressed in cHL cell lines and primary tumors. Relative Gal1 mRNA abundance (A and B) and protein expression (C) in a panel of LBCL and cHL cell lines are shown. (A) Gal1 expression profiles of DLBCL, MLBCL, and cHL cell lines. Color scale at the bottom indicates relative expression ± SEM. Red connotes high-level expression; blue indicates low-level expression. (B) Median expression of Gal1 (boxes) in LBCL vs. cHL cell lines ± 25–75 percentile (bars) and ± range (whiskers). (C) Respective cHL cell lines (KMHZ, HDLM2, SupHD1, L1236, L540, L428, and HD-MY-Z), the MLBCL cell line (Karpas 1106), and DLBCL cell lines (all others). (D) Gal1 immunohistochemistry (IHC). Gal1 IHC of a representative primary cHL (Upper) and DLBCL (Lower) are shown. [Original magnifications: ×40 (Upper) and ×400 (Lower).]
RS Cell Gal1 Expression Is Regulated by an AP1-Dependent Enhancer.
To elucidate the mechanisms responsible for Gal1 overexpression in cHL RS cells, we first analyzed the Gal1 locus on chromosome 22. We identified a candidate GC-rich regulatory element with a conserved putative AP1-binding site ≈1.5 kb downstream from the Gal1 transcription start site. Because the AP1 components, c-Jun and JunB, are overexpressed in cHL and are critical for the pathogenesis of the disease (3), we asked whether AP1 mediates Gal1 expression in cHL. We first generated luciferase vectors driven by the previously described Gal1 promoter (−403 to +67) (21) and the putative Gal1 enhancer element (or mutated controls), and we assessed associated luciferase activity in a cHL cell line (HD-MY-Z) known to have constitutive activation of AP1 (Fig. 2A) (3). Constructs including the GC-rich regulatory element (bp 459–1746 or 1346–1746) up-regulated luciferase expression ≈8- to 10-fold, whereas constructs lacking the candidate sequence (459–777) or containing a deletion in the AP1-binding site (1346–1746del) exhibited significantly lower luciferase activity (Fig. 2A). Similar results were obtained with a set of constructs in which the regulatory element was cloned upstream from the Gal1 promoter, demonstrating that the identified sequence (bp 1346–1746) is a bona fide Gal1 enhancer (data not shown).
Fig. 2.

Gal1 transcription is regulated by an AP1-dependent enhancer. (A) Analysis of the AP1-dependent Gal1 enhancer. The previously described Gal1 promoter (21) and putative enhancer element including or lacking the predicted AP1-binding site (represented by a black bar) were cloned into a luciferase reporter vector, transiently transfected into cHL HD-MY-Z cells, and assayed for luciferase activities. Representative luciferase activities from three independent experiments are normalized to Renilla luciferase activity and are presented as bars ± SD. (B) Selective activity of the Gal1 enhancer. Classical HL, DLBCL, and fibroblast lines were transfected with either the Gal1 promoter-only vector (pGL3-Gal1−403 +67-Luc) or the promoter–enhancer construct (pGL3-Gal1403 +67-Luc-e1346–1746) and assessed as in A for their respective luciferase activities. (C) AP-1 dependence of the Gal1 enhancer in electrophoretic mobility shift assays. Nuclear extracts from DLBCL cell lines (DHL4, DHL7, and Toledo) or cHL cell lines (HD-MY-Z, L428, and SupHD1) were incubated with WT or mutant 32P-labeled, double-stranded DNA probe corresponding to AP1-binding site in Gal1 enhancer. Specific, unlabeled competitor and antibodies against c-Jun or β-actin (control) were included in certain assays as indicated. The gel-shift band corresponding to the probe–protein complex is indicated with an arrow, and supershift bands corresponding to the probe–protein–antibody complex are noted with asterisks. (D) c-Jun dependence of the Gal1 enhancer. HD-MY-Z cells were cotransfected with the Gal1 promotor-only vector or the Gal1 promotor–enhancer construct with either the dominant-negative c-Jun (c-Jun-DN) construct (c-Jun-DN) or empty vector. Luciferease activities were determined as in A. (E) AP1 inhibition decreases Gal1 transcript abundance. HD-MY-Z cells were transfected with either c-Jun-DN or empty vector. Thereafter, relative Gal1 mRNA abundance was assessed by real-time QPCR.
Given the AP1 dependence of the Gal1 enhancer and the constitutive AP1 activity in cHL (3), we next asked whether the Gal1 enhancer was selectively active in this disease. For these experiments, we transfected cHL, DLBCL, and fibroblast cell lines with either the Gal1 promoter-only vector (pGL3-Gal1−403+67-Luc) or the promoter–enhancer construct (pGL3-Gal1403–67-Luc- e1346–1746) and compared the respective luciferase activities (Fig. 2B). The Gal1 promoter–enhancer construct specifically up-regulated luciferase expression in cHL cells but not DLBCL cells or fibroblasts (Fig. 2B).
After demonstrating the specificity and activity of the Gal1 AP1 enhancer element in a cHL cell line (Fig. 2 A and B), we directly evaluated the requirement for AP1 transcription factors in electrophoretic mobility shift assays (Fig. 2C). Nuclear extracts from three cHL and three DLBCL cell lines were incubated with radiolabeled wild-type (WT) or mutant probes corresponding to AP1 element in the Gal1 enhancer. Gal1 WT, but not mutant probe, directly bound to nuclear proteins extracted from cHL, but not DLBCL, cell lines (Fig. 2C). The complexes formed with Gal1 WT probe were displaced by unlabeled WT competitor, further confirming the binding specificity (Fig. 2C). In supershift assays, the Gal1–AP1 complex was retarded by c-Jun antibody (Fig. 2C). Furthermore, the simultaneous overexpression of a dominant-negative c-Jun construct (c-Jun-DN) significantly reduced Gal1-driven luciferase activity in cHL cells (Fig. 2D). In addition, when AP1 was at least partially inhibited by the overexpression of c-Jun-DN, there was a significant decrease in Gal1 transcript abundance in cHL cells (Fig. 2E). Taken together, these studies indicate that cHL RS cells selectively overexpress Gal1, at least in part, by an AP1-driven enhancer.
Endogenous cHL Gal1 Expression Contributes to the Immunosuppressive and Th2-Skewed Microenvironment in cHL.
After delineating the mechanism for cHL-specific overexpression of Gal1, we assessed the functional consequences of cHL RS cell Gal1 expression on the associated inflammatory/immune infiltrate. We first generated stable HD-MY-Z transfectants expressing Gal1-specific short hairpin RNA (Gal1 shRNA) or a scrambled control shRNA (SCR, Fig. 3A). Activated T cell blasts were then added to Gal1 shRNA or SCR HD-MY-Z cells grown as adherent monolayers, and the cHL line and T cells were cocultured. Thereafter, total T cell and Th cell viabilities were assessed by triple-color annexin-V, CD3, and CD4 flow cytometry. There were significantly fewer viable total (CD3+) and Th (CD4+) T cells in control (SCR) HD-MY-Z cocultures than in cocultures of HD-MY-Z cells with Gal1 knockdown (Fig. 3B). These studies directly demonstrate that endogenous cHL RS cell Gal1 decreases the viability of infiltrating activated T cells.
Fig. 3.

Gal1 confers immune privilege to cHL RS cells by favoring the expansion of Th2 cells and Treg cells. (A) RNAi-mediated blockade of Gal1 expression in the cHL HD-MY-Z cell line. HD-MY-Z cells were transduced with pSIREN-RetroQ vector encoding Gal1-specific shRNA (Gal1 shRNA, G) or scrambled control shRNA (SCR shRNA, S) and analyzed thereafter for Gal1 protein expression. (B) Viability of total (CD3+) and CD4+ T cells cocultured with Gal1 shRNA cHL or control SCR shRNA cHL cells. After coculture, T cell viability was assessed by using triple-color annexin-V, CD3, and CD4 flow cytometry. (C) Relative abundance of the Th1- and Th2-specific transcription factors, T-bet and GATA-3, in CD4+ cells from the Gal1 shRNA and SCR shRNA (control) cHL/T cell cocultures. (D) Th2 cytokine production by Gal1-treated T cells. Activated T cells were either untreated or treated with rGal1 in the presence or absence of TDG. Th2 cytokine (IL-4, IL-5, IL-10, and IL-13) production was then assessed by cytometric bead arrays. (E) Treg cell abundance in Gal1-treated T cells. Activated T cells were cultured in the presence of rGal1 or rGal1+TDG or left untreated. The percentage of CD4+CD25+FOXP3+ T cells was then assessed by triple-color flow cytometry. The histograms (Left) are representative of three separate experiments averaged to obtain the percent of CD4+CD25highFOXP3+ cells in the bar graph (Right).
Given the skewed nature of inflammatory infiltrate in cHL, we next asked whether endogenous cHL RS cell Gal1 may contribute to this Th1/Th2 imbalance. To address this question, we isolated CD4+ Th cells from the cHL/T cell cocultures and analyzed the relative expression of the Th1- and Th2-specific transcription factors, T-bet and GATA-3, by real-time quantitative PCR (QPCR). CD4+ Th cells cocultured with control (SCR) HD-MY-Z cells exhibited significantly lower expression of T-bet and higher expression of GATA-3 than CD4+ T cells from Gal1 shRNA HD-MY-Z cocultures (Fig. 3C). Taken together, these results indicate that endogenous cHL RS cell Gal1 selectively decreases the viability of associated Th1 cells, resulting in a skewed Th2-type infiltrate.
Gal1 Promotes Immune Privilege by Favoring the Secretion of Th2 Cytokines and the Expansion of CD4+CD25highFOXP3+ Treg Cells.
Given the profound immunosuppressive activity of Gal1 in Th1/Th17-mediated autoimmune settings (17–19), we next asked whether this glycan-binding protein was directly implicated in the skewed Th2 cytokine profile associated with primary cHL. For this purpose, we treated activated T cells with recombinant Gal1 (rGal1) in the presence or absence of the Gal1 inhibitor, thiodigalactoside (TDG) (17). As expected, rGal1 induced apoptosis of total activated T cells [supporting information (SI) Fig. 4]. Concurrent treatment with TDG completely blocked rGal1-induced apoptosis, confirming the specificity of the Gal1 effect (SI Fig. 4). We next quantified Th2 cytokines in supernatants from activated T cells that were untreated or treated with rGal1 in the presence or absence of TDG. Supernatants from rGal1-treated T cells contained significantly higher amounts of the Hodgkin-associated Th2 cytokines, IL-4, IL-5, IL-10, and IL-13, and TDG specifically blocked this effect (Fig. 3D). These data further support the hypothesis that RS cell Gal1 expression promotes Th2-type cytokine production in primary cHLs.
In addition to Th2 cells, the inflammatory infiltrate in primary cHL includes abundant T regulatory cells (CD4+CD25highFOXP3+) that directly blunt the host antitumor immune response (6–8, 22). Therefore, we next assessed the role of Gal1 in the selective expansion of Treg cells by the above-mentioned assay. Activated T cells were treated with rGal1 in the presence or absence of TDG and analyzed thereafter for Treg cells (CD4+CD25highFOXP3+) by triple-color immunofluorescence as described (23). The CD4+CD25highFOXP3+ population was significantly increased in rGal1-treated cells, and TDG completely blocked this effect (Fig. 3E). Taken together, these results demonstrate that Gal1 fosters the skewed and immunosuppressive microenvironment in cHL by enhancing the production of Th2 cytokines (IL-4, IL-5, IL-10, and IL-13) and increasing the relative abundance of Treg cells.
Discussion
Herein, we describe the overexpression of Gal1 by cHL RS cells and identify the mechanism as a phylogenetically conserved AP1-responsive enhancer. In functional in vitro assays, we directly show that cHL RS cell Gal1 decreased the viability of activated T cells and skewed the balance toward a Th2 immune response. Consistent with this observation, Gal1 markedly increased the secretion of Th2 cytokines, including IL-4, IL-5, IL-10, and IL-13. In addition, Gal1 fostered the expansion and/or retention of CD4+CD25highFOXP3+ Treg cells. Taken together, these data directly implicate RS cell Gal1 in the development and maintenance of the unique Th2/Treg-skewed immunosuppressive microenvironment in primary cHL.
Although cHL RS cells exhibit near uniform Gal1 expression, DLBCL and MLBCL are largely Gal1-negative, prompting speculation that Gal1 may distinguish cHL from certain “gray-zone” lymphomas that share characteristics of DLBCL and cHL (24). A common feature of these gray-zone lymphomas is an increased host inflammatory response, highlighting the interaction between the tumor cells and their host microenvironment (24). Gal1 overexpression is a defining feature of cHL that is not shared with its closely related counterpart, primary MLBCL (25), providing insights into the relative efficacy of host immune responses in these tumors.
The differential expression of Gal1 in these lymphomas is likely caused by the cHL-specific overexpression of the AP1 transcription factor components, c-Jun and JunB, and the constitutive activation of the AP1 pathway (3). Gal1 expression is regulated, at least in part, by a cHL-specific, AP1-driven enhancer. AP1 also functions in synergy with NF-κB to control the proliferation and limit the apoptosis of cHL RS cells (3). Therefore, in addition to its prosurvival functions in cHL RS cells, AP1 also regulates the interplay between RS cells and the tumor microenvironment through a Gal1-mediated pathway.
RS Gal1 is likely to be a critical factor shaping the immuno- and histopathologic features of primary cHL. Endogenous RS cell Gal1 specifically induced the apoptosis of activated T lymphocytes, suggesting that a similar mechanism operates in primary cHLs and that these tumors represent sites of immune privilege. Of note, Gandhi et al. (26) also recently described increased Gal1 expression in cHL.
Our short-term in vitro assays likely underestimate the long-term in vivo effects of Gal1 in cHL because the lectin is also deposited in the extracellular matrix and stroma where it kills susceptible T cells (27). In our in vitro assays, Gal1-expressing RS cells selectively decreased the viability of infiltrating Th1 cells, resulting in a Th2-predominant infiltrate. Consistent with the observed reduction in Th1 cells and enrichment in Th2 cells, Gal1 significantly increased the levels of the Th2 cytokines, IL-4, IL-5, IL-10, and IL-13. The Gal1-associated cytokine profile suggests that this glycan-binding protein may have additional functions beyond modulating T cell responses in primary cHLs. Because IL-13 is a critical RS cell growth factor (9), Gal1 may indirectly stimulate tumor growth by fostering the production of this Th2 cytokine. Through its effect on another Th2 cytokine, IL-5, Gal1 may also promote the characteristic eosinophilic infiltrate in primary cHL (28).
Our observations regarding Gal1 function in cHL are consistent with reports regarding the role of the lectin in murine models of Th1-driven chronic inflammatory and autoimmune disorders, including collagen-induced arthritis, inflammatory bowel disease, graft vs. host disease, and autoimmune uveitis (17–20). In these studies, the administration of Gal1 dramatically suppressed Th1-dependent responses and skewed toward Th2 cytokine profiles (17–20, 29). A careful examination of the mechanisms involved in Gal1-mediated Th2 skewing recently revealed that Th1 cells express the repertoire of cell surface glycans required for Gal1 binding and subsequent cell death, whereas Th2 cells are protected from Gal1 by differential sialylation of their cell surface glycoproteins (30).
In addition to the Th2 shift, we provide evidence showing that Gal1 treatment increases the relative abundance of CD4+CD25highFOXP3+ Treg cells that may blunt the host anti-cHL immune response. It is possible that the specific glycosylation pattern of Gal1 receptors on Treg cells renders them resistant to Gal1-induced apoptosis. In fact, Treg cells have been reported to exhibit increased α2,6 sialylation (compared with effector T cells) (31); this selective sialylation might interfere with Gal1 binding and cell death (32). This hypothesis is further supported by recent studies demonstrating that Treg cells overexpress Gal1 and remain resistant to Gal1-mediated apoptosis (33).
Taken together, our work provides insights into the biology of RS cells and identifies a key AP1-dependent mechanism regulating cHL-specific immune privilege. Because Gal1 blockade dramatically increased tumor rejection in recently described murine models (12), it is possible that Gal1 inhibition may augment host antitumor responses in primary cHL. Furthermore, this lectin is likely to have additional roles in the biology of cHL. Previous studies indicate that Gal1 also promotes tumor cell motility and enhances tumor angiogenesis (11, 34–36), processes critical for tumors such as cHL that spread by contiguous involvement of adjacent nodes and organs. For all of these reasons, Gal1 represents a promising rational therapeutic target in cHL.
Materials and Methods
Cell Lines.
Twenty-one DLBCL cell lines (Ly19, Ly18, Ly10, Ly8, Ly7, Ly4, Ly3, Ly1, Pleiffer, DHL10, DHL8, DHL7, DHL6, DHL5, DHL4, DB, HT, WSU, Karpas 422, Toledo, and Farage), one MLBCL cell line (Karpas 1106), and seven cHL cell lines (KMH2, HDLM2, SupHD1, L1236, L540, L428, and HD-MY-Z) were maintained as described previously (3, 37). Of note, the cHL cell lines were previously demonstrated to have constitutive AP1 activity and increased expression of c-Jun and JunB (Fig. 2 and SI Fig. 5).
Identification of Genes Overexpressed in cHL Cell Lines by Gene Expression Profiling.
Total RNAs from a panel of 21 DLBCL and 7 cHL cell lines were hybridized to U133A and B oligonucleotide microarrays (Affymetrix, Santa Clara, CA); the chips were scanned, and the data were analyzed as described previously (38). The top 9,586 genes that met threshold and variation index criteria were analyzed with the GenePattern program (www.broad.mit.edu/cancer/software/genepattern/CEF) to identify differentially expressed genes in cHL and DLBCL. Genes correlated with the class template (HL vs. DLBCL) were identified by ranking them according to their signal-to-noise ratio. For each gene, a specific P value based on permutation testing was calculated and corrected for false discovery rate (39, 40).
Analysis of Gal1 Expression in Cell Lines by Immunoblot.
DLBCL and cHL cell lines were maintained as described previously (3, 41). Cells were lysed, size-fractionated on NuPAGE Novex 4–12% Bistris gels (Invitrogen, Carlsbad, CA), and transferred to PVDF membranes (Millipore Corp., Bedford, MA). Membranes were immunostained with purified Gal1 rabbit IgG (12) and horseradish peroxidase-conjugated donkey anti-rabbit antibody (GE Healthcare, Piscataway, NJ) and developed by a chemiluminescent method (ECL; GE Healthcare).
Immunohistochemistry.
Immunohistochemistry was performed as described previously (42) with 5-μm-thick formalin-fixed, paraffin-embedded tissue sections of newly diagnosed primary cHL and DLBCL and purified Gal1 rabbit IgG.
Analysis of Regulatory Elements in the Gal1 Locus and Generation of Gal1 Enhancer Constructs.
Computational analysis of the Gal1 locus (chr22:36,400,510–36,406,802, alignment with Human NCBI Genome assembly v36, March 2006) was performed with the publicly available version of Genomatix suite (www.genomatix.de) (43) and rVISTA (http://rvista.dcode.org) (44), and a putative downstream regulatory element (enhancer) containing a conserved AP1-binding site was identified (1567–1675). To generate a series of Gal1 promoter–enhancer reporter constructs, we first PCR-amplified the Gal1 promoter region (−403 to +67) and ligated this sequence into the pGL3 promoterless reporter vector (Promega, Madison, WI), generating pGL3-Gal1−403+67-Luc. Thereafter, fragments spanning nucleotides 459-1746, 459–777, and 1346–1746 from the Gal1 transcription start site were PCR-amplified and cloned into pGL3-Gal1−403+67-Luc 3′ of the luciferase gene. Deletions in AP1 site (TGACTCA to TGxxxCA) were generated by using the pGL3-Gal1−403+67-Luc-e1346+1746 construct and the Gene Tailor site-directed mutagenesis system (Invitrogen) as recommended by the manufacturer. An additional set of constructs was generated with the candidate enhancer elements cloned upstream from the Gal1 promoter.
Generation of DN c-Jun Constructs.
DN c-Jun constructs were generated as described previously (45) with minor modifications. In brief, a c-Jun fragment that lacked the transactivation domain (amino acids 123–223) was PCR-amplified from intronless c-JUN genomic DNA and ligated in the pFLAG-CMV2 vector (pFLAG-CMV2-c-Jun-DN) (Sigma–Aldrich, St Louis, MO). All primer sequences are available upon request.
Analysis of Gal1 Promoter–Enhancer Constructs with Luciferase Assays.
The HD-MY-Z cHL and SU-DHL7 DLBCL cell lines were grown to 60–80% confluence on 24-well plates and cotransfected with 300 ng of the appropriate promoter–enhancer pGL3 construct (WT or mutant Gal1) per well and 100 ng of the control reporter plasmid, pRL-TK (Promega) per well by using FuGENE 6 transfection reagent (Roche Applied Science, Indianapolis, IN) according to the manufacturer's protocol. For cotransfection experiments with c-Jun-DN-FLAG, HD-MY-Z cells were transfected with 150 ng of pGL3-Gal1−403 +67-Luc-e1346 +1746, 250 ng of pFLAG-CMV2-c-Jun-DN, and 100 ng of pRL-TK. After 24 h of incubation, cells were lysed, and luciferase activities were determined by chemiluminescence assay with the dual luciferase assay kit (Promega) and Luminoskan Ascent luminometer (Thermo Lab Systems, Franklin, MA) as described (42).
Electrophoretic Mobility Shift Analyses of the AP1-Binding Site in the Gal1 Enhancer.
Nuclear extracts from three cHL cell lines (HD-MY-Z, L428, and SupHD1) and three DLBCL cell lines (SU-DHL7, SU-DHL4, and Toledo) were obtained as described previously (42). Double-stranded WT and mutant probes corresponding to the AP1-binding region in Gal1 enhancer (WT, 5′-TTTTCTGGGTGACTCACTTCCCCCG-3′; and mutant, 5′-TTTTCTGGGTtcagtACTTCCCCCG-3′) (mutant bases in are in lowercase letters) were end-labeled with [γ-32P]ATP, purified, and used in binding reactions as described (42). DNA binding was carried out by using 5 μg of nuclear extracts and ≈10,000 cpm of radiolabeled probe in 20 μl of binding buffer (42). After a 30-min incubation, reactions were loaded onto a 5% polyacrylamide gel and electrophoresed. Gels were vacuum dried and exposed to x-ray films overnight at −80°C. For competitor studies, a 100× molar excess of unlabeled WT or mutant probe was included in the binding reactions. For supershift studies, 1 μl of c-Jun antibody or β-actin [Santa Cruz Biotechnology (Santa Cruz, CA) and Sigma–Aldrich, respectively] was added to the reaction 15 min before the probe.
QPCR Analysis of Gal1 Transcript Abundance After AP1 Inhibition.
The HD-MY-Z cHL cell line was grown to 60–80% confluence on 100-mm plates and transiently transfected with 15 μg of pFLAG-CMV2 (empty vector) or pFLAG-CMV2-c-Jun-DN plasmids and FuGENE 6 transfection reagent (Roche Applied Science) according to the manufacturer's protocol. After 72 h in culture, RNA was extracted with TRIzol reagent (Invitrogen), and cDNA was synthesized from total RNA (3 μg) by using SuperScript II reverse transcriptase (Invitrogen) and random hexamer primers. Gal1 and GAPDH (housekeeping control) transcript abundance was evaluated by QPCR using Power SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA) and the following primers: GAPDH forward, GATTCCACCCATGGCAAATTC; GAPDH reverse, TGATTTTGGAGGGATCTCGCTC; Gal1 forward, TCGCCAGCAACCTGAATCTC; Gal1 reverse, GCACGAAGCTCTTAGCGTCA. PCR was performed using an ABI 7700 thermal cycler (Applied Biosystems), and threshold cycle (CT) values were generated with the Sequence Detection Software, version 1.2 (Applied Biosystems). Gal1 transcript abundance was calculated relative to the housekeeping control GAPDH by using the 2−(ΔCTGal1−ΔCTGAPDH) method according to the manufacturer's instructions. Standard deviations were calculated from triplicate ΔΔCT values.
RNAi-Mediated Gal1 Knockdown.
Gal1-specific siRNA was designed by the siRNA Selection Program (46) (http://jura.wi.mit.edu/bioc/siRNAext), synthesized as single-stranded DNA oligonucleotides by Integrated DNA Technologies (IDT, Inc., Coralville, IA) and annealed. Gal1-specific oligonucleotide (Gal1 RNAi, GATCCGCTGCCAGATGGATACGAATTCAAGAGATT C G T A T C CATCTGGCAGCTTTTTTG) or scrambled oligonucleotide (SCR, GATCCCCTCCATATCTCGCGCGTCTT C A A G A GAGACGCGCGAGATATGGAGGTTTTTTG) was ligated into the linearized pSIREN-RetroQ retroviral vector (BD Clontech, Mountain View, CA). Generation of recombinant retrovirus and infection of HD-MY-Z cells were performed as described previously (42). After infection, cells were subjected to puromycin selection (0.5 μg/ml) and subcloning by limiting dilution. Thereafter, whole-cell extracts of obtained subclones were prepared and screened for Gal1 expression by immunoblotting as described above. Of note, Gal1 knockdown did not alter the proliferation rate or viability of transduced HD-MY-Z cells.
Cocultures and Analyses of T Cell Responses in cHL Microenvironment.
Coculture.
Peripheral blood mononuclear cells were obtained from normal blood donors by Ficoll–Hypaque (GE Healthcare) gradient centrifugation. Thereafter, T cells were purified by using a Pan T cell isolation kit II (Miltenyi Biotec, Auburn, CA) and were activated with 1 μg/ml phytohemagglutinin (Sigma–Aldrich) for 64 h. Activated T cells (1 × 106) were then cocultured with monolayers (3 × 106 cells) of HD-MY-Z cells expressing either scrambled shRNA or Gal1-specific shRNA for 6 h at 37°C.
Analyses of viable T cells.
After coculture, all cells were harvested and sequentially stained with CD3-phycoerythrin (PE) and CD4-PE-Cy5 (Beckman Coulter, Fullerton, CA) followed by annexin V-FITC and analyzed with a Beckman Cytomics FC500 flow cytometer. The numbers of viable (annexin V and propidium iodide-negative) CD3+ and CD4+ T cells in Gal1 shRNA or scrambled shRNA HD-MY-Z/T cell cocultures were then compared.
Analyses of T-bet and GATA-3 expression in cocultured CD4+ T cells.
After phytohemagglutinin-activated T cells were cocultured with HD-MY-Z cells expressing either scrambled shRNA or Gal1-specific shRNA for 24–48 h, cells were collected, and nonviable cells were depleted by Ficoll–Hypaque gradient centrifugation. Remaining viable cells were washed, incubated with CD4 MACS microbeads, and purified by using a MACS LS column according to the manufacturer's protocol (Miltenyi Biotec). Thereafter, RNA was obtained from the isolated CD4+ cells, and cDNA was synthesized from total RNA (3 μg) as described above. GATA-3, T-bet, and GAPDH (housekeeping control) transcript abundance was evaluated by QPCR using Power SYBR Green PCR Master Mix (Applied Biosystems), the GAPDH primers listed above, and the following GATA-3 and T-bet primers: GATA-3 forward, TAACATCGACGGTCAAGGCA; GATA-3 reverse, ACACCTGGCTCCCGTGGT; T-bet forward, TGGACGTGGTCTTGGTGGACC; T-bet reverse, TGGACGTACAGGCGGTTTCC. PCR and transcript abundance analysis were performed as described above. GATA-3 and T-bet expression in purified CD4+ cells from SCR shRNA and Gal1 shRNA HD-MY-Z/T cell cocultures were then compared.
Cytokine production in T cell subpopulations treated with recombinant Gal1 (rGal1).
Recombinant Gal1 was obtained and purified essentially as described (18). T cells were purified as described above and simultaneously activated with anti-CD3- (0.1 μg/ml) and anti-CD28- (0.5 μg/ml) coated latex beads (Invitrogen) and treated with rGal1 (20 μM) in RPMI medium 1640 alone or rGal1 and the Gal1 inhibitor TDG (100 mM) (17) or left untreated for 16 h. Supernatants were then analyzed for IL-4, IL-5, IL-10, and IL-13 by using cytometric bead array Flex Set beads according to the manufacturer's protocol (BD Biosciences, San Jose, CA). In brief, multiplexed antibody-conjugated beads were incubated with culture supernatants or serial dilutions of cytokine standards for 1 h. Thereafter, the PE detection reagent was added, and samples were incubated for additional 2 h, washed, and analyzed by a FACS Aria flow cytometer (BD Biosciences). Results were captured with FACS Diva software and analyzed with the FCAP1.1 program (BD Biosciences).
Analyses of Treg cells.
T cells were purified, activated with anti-CD3- and anti-CD28- coated latex beads, and treated with rGal1 or rGal1 and TDG or left untreated for 24 h. Thereafter, cells were stained with CD4-FITC and CD25-allophycocyanin, washed, fixed, permeabilized, and stained with FOXP3-PE antibody or rat IgG2b isotype control as described previously (23) (human regulatory T cell staining kit; eBioscience, San Diego, CA). CD4+CD25highFOXP3+ Treg cells were identified and quantified by a Beckman Cytomics FC500 flow cytometer as described previously (23).
Statistical Analysis.
All statistical analyses were done by using Statistica 6.0 software (Statistica, Tulsa, OK). Student's t test was used for comparisons between two groups; ANOVA was used for multiple comparisons.
Supplementary Material
Abbreviations
- AP1
activator protein 1
- cHL
classical Hodgkin lymphoma
- DLBCL
diffuse large B cell lymphoma
- DN
dominant-negative
- Gal1
galectin-1
- MLBCL
mediastinal large B cell lymphoma
- PE
phycoerythrin
- QPCR
quantitative PCR
- rGal1
recombinant Gal1
- RS cell
Reed–Sternberg cell
- SCR
scrambled control shRNA
- shRNA
short hairpin RNA
- Treg
regulatory T cell
- TDG
thiodigalactoside
- Th cell
T helper cell.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0706017104/DC1.
References
- 1.Re D, Kuppers R, Diehl V. J Clin Oncol. 2005;23:6379–6386. doi: 10.1200/JCO.2005.55.013. [DOI] [PubMed] [Google Scholar]
- 2.Kanzler H, Kuppers R, Hansmann ML, Rajewsky K. J Exp Med. 1996;184:1495–1505. doi: 10.1084/jem.184.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, Bomment K, Mechta-Grigoriou F, Stein H, Dorken B, et al. EMBO J. 2002;21:4104–4113. doi: 10.1093/emboj/cdf389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuppers R, Schwering I, Brauinger A, Rajewsky K, Hansmann ML. Ann Oncol. 2002;13:11–18. doi: 10.1093/annonc/13.s1.11. [DOI] [PubMed] [Google Scholar]
- 5.Schwering I, Brauninger A, Klein U, Jungnickel B, Tinguely M, Diehl V, Hansmann ML, Dalla-Favera R, Rajewsky K, Kuppers R. Blood. 2003;101:1505–1512. doi: 10.1182/blood-2002-03-0839. [DOI] [PubMed] [Google Scholar]
- 6.Marshall NA, Christie LE, Munro LR, Culligan DJ, Johnston PW, Barker RN, Vickers MA. Blood. 2004;103:1755–1762. doi: 10.1182/blood-2003-07-2594. [DOI] [PubMed] [Google Scholar]
- 7.Gandhi MK, Lambley E, Duraiswamy J, Dua U, Smith C, Elliott S, Gill D, Marlton P, Seymour JF, Khanna R. Blood. 2006;108:2280–2289. doi: 10.1182/blood-2006-04-015164. [DOI] [PubMed] [Google Scholar]
- 8.Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, Inagaki H, Ueda R. Cancer Res. 2006;66:5716–5722. doi: 10.1158/0008-5472.CAN-06-0261. [DOI] [PubMed] [Google Scholar]
- 9.Skinnider BE, Kapp U, Mak TW. Leuk Lymphoma. 2002;43:1203–1210. doi: 10.1080/10428190290026259. [DOI] [PubMed] [Google Scholar]
- 10.Rabinovich GA, Baum LG, Tinari N, Paganelli R, Natoli C, Liu FT, Iacobelli S. Trends Immunol. 2002;23:313–320. doi: 10.1016/s1471-4906(02)02232-9. [DOI] [PubMed] [Google Scholar]
- 11.Liu F-T, Rabinovich GA. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 12.Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Llarregui JM, Bravo A, Mordoh J, Fainboim L, Poshajcer OL, Rabinovich GA. Cancer Cell. 2004;5:241–251. doi: 10.1016/s1535-6108(04)00024-8. [DOI] [PubMed] [Google Scholar]
- 13.Le Q-T, Shi G, Cao H, Nelson DW, Wang Y, Chen EY, Zhao S, Kong C, Richardson D, O'Byrne KJ, et al. J Clin Oncol. 2005;23:8932–8941. doi: 10.1200/JCO.2005.02.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vasta GR, Ahmed H, Odom EW. Curr Opin Struct Biol. 2004;14:617–630. doi: 10.1016/j.sbi.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson PJ, Liu F-T, Baum LG. J Immunol. 2006;176:778–789. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- 16.Perillo NL, Pace KE, Seihamer JJ, Baum LG. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 17.Rabinovich GA, Daly G, Dreja H, Tailor H, Riera CM, Hirabayashi J, Chernajovsky Y. J Exp Med. 1999;190:385–397. doi: 10.1084/jem.190.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toscano MA, Commodaro AG, Ilarregui JM, Bianco GA, Liberman A, Serra HM, Hirabayashi J, Rizzo LV, Rabinovich GA. J Immunol. 2006;176:6323–6332. doi: 10.4049/jimmunol.176.10.6323. [DOI] [PubMed] [Google Scholar]
- 19.Santucci L, Fiorucci S, Rubinstein N, Mencarelli A, Palazzetti B, Federici B, Rabinovich GA, Morelli A. Gastroenterology. 2003;124:1381–1394. doi: 10.1016/s0016-5085(03)00267-1. [DOI] [PubMed] [Google Scholar]
- 20.Baum LG, Blackall DP, Arias-Magallano S, Nanigian D, Uh SY, Browne JM, Hoffmann D, Emmanouilides CE, Territo MC, Baldwin GC. Clin Immunol. 2003;109:295–307. doi: 10.1016/j.clim.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 21.Salvatore P, Benvenuto G, Caporaso M, Bruni CB, Chiariotti L. FEBS Lett. 1998;421:152–158. doi: 10.1016/s0014-5793(97)01553-6. [DOI] [PubMed] [Google Scholar]
- 22.Gabrilovich DI. Curr Cancer Drug Targets. 2007;7:1. [PubMed] [Google Scholar]
- 23.Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, et al. Blood. 2006;108:1571–1579. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abramson J, Shipp M. Blood. 2005;106:1164–1174. doi: 10.1182/blood-2005-02-0687. [DOI] [PubMed] [Google Scholar]
- 25.Savage K, Monti S, Kutok J, Cattoretti G, Neuberg D, de Leval L, Kurtin P, Dal Cin P, Ladd C, Feuerhake F, et al. Blood. 2003;102:3871–3879. doi: 10.1182/blood-2003-06-1841. [DOI] [PubMed] [Google Scholar]
- 26.Gandhi MK, Moll G, Smith C, Dua J, Lambley E, Ramuz O, Gill D, Marlton P, Seymour JF, Khanna R. Blood. 2007 Apr 16; doi: 10.1182/blood-2007-01-066100. 10.1182/blood-2007-01-066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He J, Baum LG. J Biol Chem. 2004;279:4705–4712. doi: 10.1074/jbc.M311183200. [DOI] [PubMed] [Google Scholar]
- 28.von Wasielewski R, Seth S, Franklin J, Fischer R, Hubner K, Hansmann ML, Diehl V, Georgii A. Blood. 2000;95:1207–1213. [PubMed] [Google Scholar]
- 29.van der Leij J, van den Berg A, Harms G, Eschbach H, Vos H, Zwiers P, van Weeghel R, Groen H, Poppema S, Visser L. Mol Immunol. 2006;10:1–8. doi: 10.1016/j.molimm.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 30.Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, Zwirner NW, Poirier F, Riley EM, Baum LG, et al. Nat Immunol. 2007 Jun 24; doi: 10.1038/ni1482. 10.1038/ni 1482. [DOI] [PubMed] [Google Scholar]
- 31.Jenner J, Kerst G, Handgretinger R, Muller I. Exp Hematol. 2006;34:1211–1217. doi: 10.1016/j.exphem.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 32.Amano M, Galvan M, He J, Baum LG. J Biol Chem. 2003;278:7469–7475. doi: 10.1074/jbc.M209595200. [DOI] [PubMed] [Google Scholar]
- 33.Garin MI, Chu C-C, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Blood. 2007;109:2058–2065. doi: 10.1182/blood-2006-04-016451. [DOI] [PubMed] [Google Scholar]
- 34.Rabinovich GA. Br J Cancer. 2005;92:1188–1192. doi: 10.1038/sj.bjc.6602493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thijssen VL, Postel R, Brandwijk RJ, Dings RP, Nesmelova I, Satijn S, Verhofstad N, Nakabeppu Y, Baum LG, Bakkers J, et al. Proc Natl Acad Sci USA. 2006;103:15975–15980. doi: 10.1073/pnas.0603883103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camby I, Belot N, Lefranc F, Sadeghi N, de Launoit Y, Kaltner H, Musette S, Darro F, Danguy A, Salmon I, et al. J Neuropathol Exp Neurol. 2002;61:585–596. doi: 10.1093/jnen/61.7.585. [DOI] [PubMed] [Google Scholar]
- 37.Smith PG, Wang F, Wilkinson KN, Savage KJ, Klein U, Neuberg DS, Bollag G, Shipp MA, Aguiar RCT. Blood. 2005;105:308–316. doi: 10.1182/blood-2004-01-0240. [DOI] [PubMed] [Google Scholar]
- 38.Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, Wu B, Pasqualucci L, Neuberg D, Aguiar RC, et al. Blood. 2005;105:1851–1861. doi: 10.1182/blood-2004-07-2947. [DOI] [PubMed] [Google Scholar]
- 39.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Behav Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 40.Reiner A, Yekutieli D, Benjamini Y. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 41.Polo JM, Juszczynski P, Monti S, Cerchietti L, Ye K, Greally JM, Shipp M, Melnick A. Proc Natl Acad Sci USA. 2007;104:3207–3212. doi: 10.1073/pnas.0611399104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juszczynski P, Kutok JL, Li C, Mitra J, Aguiar RCT, Shipp MA. Mol Cell Biol. 2006;26:5348–5359. doi: 10.1128/MCB.02351-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scherf M, Klingenhoff A, Werner T. J Mol Biol. 2000;297:599–606. doi: 10.1006/jmbi.2000.3589. [DOI] [PubMed] [Google Scholar]
- 44.Loots GG, Ovcharenko I. Nucleic Acids Res. 2004;32:W217–W221. doi: 10.1093/nar/gkh383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ludes-Meyers JH, Liu Y, Munoz-Medellin D, Hilsenbeck SG, Brown PH. Oncogene. 2001;20:2771–2780. doi: 10.1038/sj.onc.1204377. [DOI] [PubMed] [Google Scholar]
- 46.Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. Nucleic Acids Res. 2004;32:W130–W134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}