

Abstract
Recessive mutations in GJA12/Cx47, the gene encoding the gap junction protein connexin47 (Cx47), cause Pelizaeus-Merzbacher-like disease (PMLD), which is characterized by severe CNS dysmyelination. Three missense PMLD mutations, P87S, Y269D and M283T, were expressed in communication-incompetent HeLa cells, and in each case the mutant proteins appeared to at least partially accumulate in the ER. Cells expressing each mutant did not pass Lucifer Yellow or neurobiotin in scrape loading assays, in contrast to robust transfer in cells expressing wild type Cx47. Dual whole-cell patch clamping of transfected Neuro2A cells demonstrated that none of the mutants formed functional channels, in contrast to wild type Cx47. Immunostaining sections of primate brains demonstrated that oligodendrocytes express Cx47, which is primarily localized to their cell bodies. Thus, the Cx47 mutants associated with PMLD likely disrupt the gap junction coupling between astrocytes and oligodendrocytes.
Gap junction communication (GJC) facilitates the diffusion of ions and small molecules (less than 1000 Da) between two apposed cells, thereby enabling electrical coupling, metabolic cooperation, and spatial buffering of ions (Bruzzone et al. 1996). The molecular basis of GJC is the gap junction plaque, an aggregation of tens to thousands of individual channels. Two hemichannels (or connexons) on apposing cell membranes form the channel. Hemichannels are composed of six connexins - a family of highly conserved integral membrane proteins that are usually named according to their predicted molecular mass (Willecke et al. 2002). Individual hemichannels can be composed of one (homomeric) or more than one (heteromeric) type of connexin. Similarly, channels can be composed of hemichannels containing the same (homotypic) or different (heterotypic) connexins (Kumar and Gilula 1996).
Anatomical and functional studies of the rodent CNS have demonstrated that astrocytes and oligodendrocytes are coupled by gap junctions (Massa and Mugnaini 1982; Ransom and Kettenmann 1990; Robinson et al. 1993; Rash et al. 1997), and that each cell type expresses different connexins (Dermietzel et al. 1989; Yamamoto et al. 1990; Scherer et al. 1995; Dermietzel et al. 1997; Ochalski et al. 1997; Nagy et al. 1999; Altevogt et al. 2002; Menichella et al. 2003; Odermatt et al. 2003; Kleopa et al. 2004). Astrocyte/astrocyte (A/A) coupling is likely mediated by Cx43/Cx43 and Cx30/Cx30 homotypic channels, and astrocyte/oligodendrocyte (A/O) coupling is most likely mediated by Cx43/Cx47 and Cx30/Cx32 heterotypic channels, with an uncertain contribution of Cx26/Cx32 channels (Rash et al. 2001; Nagy et al. 2003; Altevogt and Paul 2004; Li et al. 2004). Cx32/Cx32 homotypic channels appear to form gap junctions between layers of CNS myelin sheaths (Kamasawa et al. 2005), as previously described in PNS myelin sheaths (Bergoffen et al. 1993; Scherer et al. 1995; Balice-Gordon et al. 1998; Meier et al. 2004). A/O heterotypic channels appear to be spatially restricted, with Cx43/Cx47 outnumbering Cx30/Cx32 channels at oligodendrocyte somata (Kleopa et al. 2004; Kamasawa et al. 2005). Oligodendrocytes also express Cx29, which does not appear to form gap junction plaques (Altevogt et al. 2002; Kleopa et al. 2004). Mutations in GJB1, the gene encoding Cx32, cause the X-linked form of Charcot-Marie-Tooth disease (CMT1X), an inherited demyelinating neuropathy (Bergoffen et al. 1993; Scherer and Kleopa 2005). Clinical manifestations of CNS dysfunction in CMT1X are rare, associated with a few of the more than 300 different GJB1 mutations (Taylor et al. 2003), indicating that Cx30/Cx32 A/O coupling is not critical in humans.
Although CNS myelination in Gja12/cx47-null mice is minimally affected (Menichella et al. 2003; Odermatt et al. 2003), recessive GJA12/Cx47 mutations cause a devastating dysmyelinating disease in humans, called Pelizaeus-Merzbacher-like disease (PMLD; (Uhlenberg et al. 2004; Bugiani et al. 2006). PMD itself is an X-linked dysmyelinating disorder caused by mutations in Proteolipid Protein 1 (PLP1), which encodes the main intrinsic membrane protein of CNS myelin (Garbern et al. 1999). PMLD patients are clinically similar to those with PMD, with nystagmus, spasticity, and ataxia, as well as widespread changes in CNS white matter by MRI imaging (Hudson et al. 2004), but do not have PLP1 mutations. The clinical phenotype and MRI findings in patients with recessive GJA12/Cx47 mutations (Uhlenberg et al. 2004; Bugiani et al. 2006) provide provocative evidence that Cx47-mediated GJC is essential for the proper functioning of oligodendrocytes. We show here that Cx47 is expressed by oligodendrocytes in the primate brain and that recessive missense mutations in GJA12/Cx47 cause loss-of-function. Thus, the Cx47 mutants associated with PMLD likely disrupt the A/O coupling that is mediated by Cx43/Cx47 heterotypic channels.
RESULTS
The sequence of human Cx47
In their report describing the GJA12/Cx47 mutations that cause PMLD, Uhlenberg et al. (2004) used the amino acid sequence derived from a human Cx47 cDNA clone (NM_020435) that included part of the 5′untranslated region (UTR). Because this 5′UTR contains a potential additional start codon (ATG) that is 9 nucleotides upstream of the ATG conventionally considered to be the connexin start codon, Uhlenberg et al. (2004) added 3 amino acids in their description of the mutations. This alternative ATG is conserved in other mammalian Cx47 DNA sequences (human: AF014643, BC089439; mouse: AJ276435, NM_080454, NM_175452, AY394498, AY394499, AY394500; rat: AY23321; cow: XM_582393) and the most closely related bird sequence (red jungle fowl: XM_418503) but not in the most similar sequences from frogs (Xenopus: NM_205831) or teleost fish (zebrafish Cx47.1: NM_001004574; puffer fish: CAAB01003296).
Neither putative start codon resides in an optimal translation initiation context, or Kozak consensus sequence (GCC RCC ATG G; R is a purine), but the downstream ATG resides in a relatively more favorable site, with a purine (A) in the R location (Kozak 1989; Kozak 1996). Whether one or both start codons are translated when they are very close and the downstream codon is more favorable is unknown (Kozak 1996). The upstream ATG of Cx47, however, would encode an N-terminus with 24 amino acids, which is inconsistent with all other known connexins. Besides Cx47, the other 19 cloned human connexins are predicted to have a 21–23 amino acid N-terminus comparable to the one encoded by the downstream ATG of GJA12/Cx47 (Willecke et al. 2002). We also examined over 70 different human, dog, cow, mouse, rat, hamster, chicken, frog, and zebrafish connexin sequences, nearly all derived from mRNA, contained at least 9 bases of the 5′ UTR, and found only two other examples of one or more in frame potential upstream start codons. These would add 6 or 12 (hCx59, NM_030772) or 11 (mCx29, NM_080450) amino acids to the N-terminus. Given the highly conserved N-termini of connexins (Willecke et al. 2002), we conclude that all of these potential upstream alternative ATGs are unlikely to be translation initiation codons, including the one for human Cx47.
Cx47 mutant expression in communication-incompetent HeLa cells
To investigate the underlying molecular defects of the recessive Cx47 mutants we generated the three missense mutations described by Uhlenberg et al. (2004) – P87S, Y269D and M283T - by site-directed mutagenesis (Fig. 1A). Each mutation, as well as WT Cx47, was expressed in communication–incompetent HeLa cells. For this analysis, we used an affinity-purified rabbit antiserum raised against the C-terminus of mouse Cx47 (gift of Dr. David Paul) and two rabbit antisera against the C-terminus of human Cx47. These antisera recognized a ~47 kDa band in immunoblots of lysates collected from either bulk-selected (Fig. 1B) or transiently transfected (Suppl. Fig. 1) cells expressing WT or mutant Cx47, but not in parental cells or cells transfected with an “empty” vector.
Figure 1. Expression of GJA12/Cx47 missense mutations associated with PMLD.
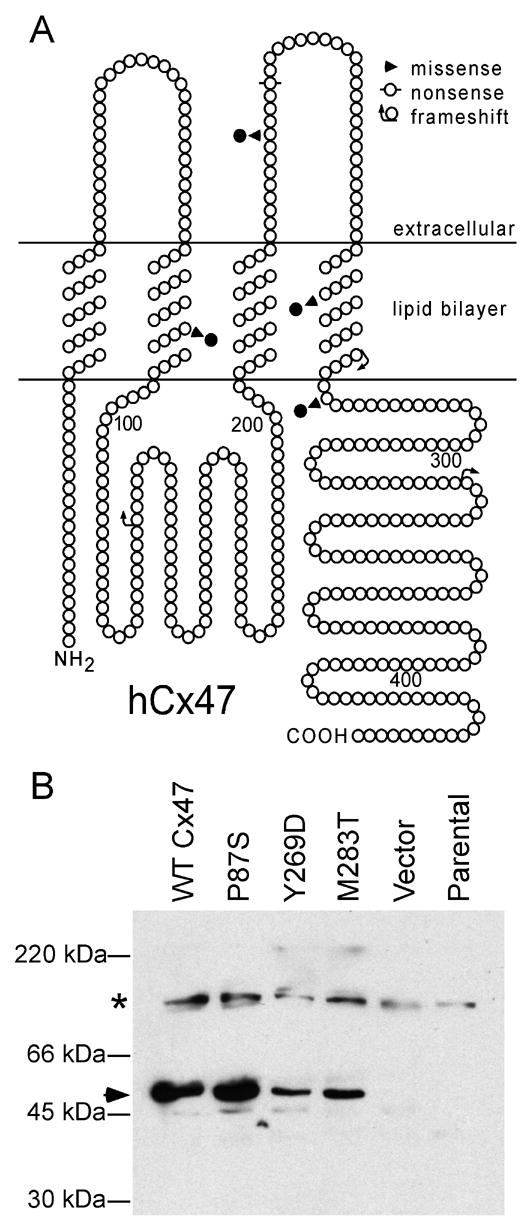
(A) This is a schematic drawing of human Cx47 that illustrates the position and nature of mutations associated with PMLD; note the four missense mutations P87S, G233S, Y269D, and M283T, as well as P128frameshift, R237stop, and L278frameshift, P327frameshift (Uhlenberg et al. 2004; Bugiani et al. 2006). The positions of the transmembrane domains are based on the work of Yeager and Nicholson (Yeager and Nicholson 1996). (B) Immunoblot analysis of Cx47. The immunoblot was hybridized with an affinity-purified rabbit antiserum against mouse Cx47. Untransfected HeLa parental cells and cells transfected with vector alone do not express Cx47; the specific band corresponding to 47 kDa was present in lysates collected from bulk-selected cells expressing WT Cx47 as well as the P87S, Y269D, and M283T mutants (arrowhead). The asterisk denotes a background band.
To examine the trafficking of the Cx47 mutants, we immunolabeled cells expressing WT Cx47 or one of the mutants with the three antisera against Cx47. Bulk-selected cells that expressed P87S or Y269D had dispersed cytoplasmic Cx47-immunoreactivity (Fig. 2B&C). In contrast, cells expressing WT Cx47 had Cx47-positive puncta at their cell borders, which we considered to be gap junction plaques (Fig. 2A). Bulk-selected cells expressing M283T (Fig. 2D) had both dispersed cytoplasmic staining (like cells expressing P87S or Y269D) and puncta (like cells expressing WT Cx47); these puncta were not seen in transiently transfected cells (data not shown). We confirmed the cell surface localization of WT Cx47 and M283T by double labeling with a monoclonal antibody that recognizes cadherins (Suppl. Fig. 2). The results with the antiserum against mouse Cx47 (Fig. 2) and the two antisera against human Cx47 (data not shown) were similar. Neither parental HeLa nor bulk-selected HeLa cells that had been transfected with an “empty” vector showed staining (data not shown).
Figure 2. PMLD-associated Cx47 mutants are retained intracellularly and colocalized with an ER marker.
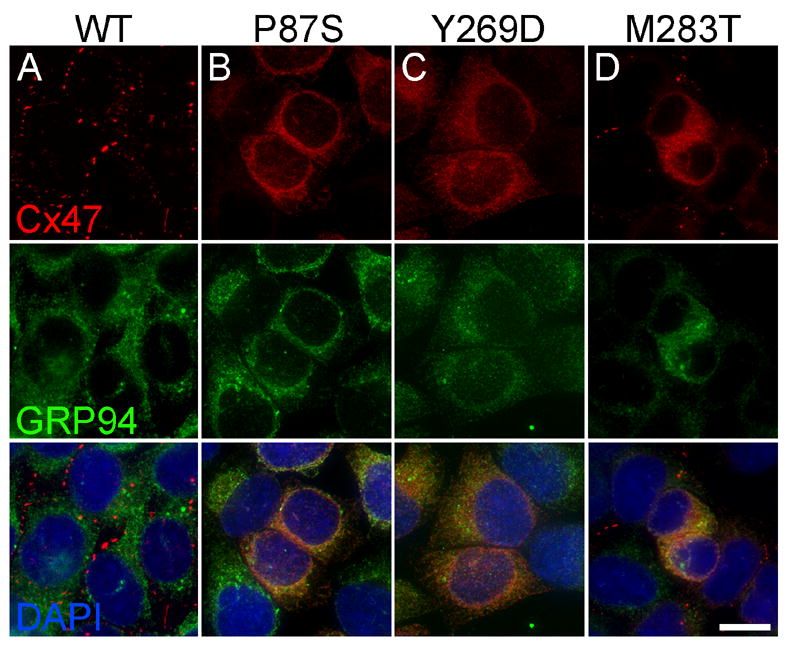
These are deconvolved images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against mouse Cx47 (red) and a rat monoclonal against the ER chaperone GRP94 (green), and counterstained with DAPI (blue). Note that WT Cx47 forms gap junction plaques at cell borders, whereas Cx47 mutants are mainly intracellularly localized, except for M283T, which also forms puncta, mainly at cell borders (D). The intracellular Cx47-immunoreactivity of the mutants (B–D) colocalizes with GRP94. Scale bar: 10μm.
Cx47 mutants localize to the endoplasmic reticulum
The dispersed intracellular pattern of Cx47-immunoreactivity looked like that of Cx32 mutants that were previously shown to be localized in the endoplasmic reticulum (ER) (Deschênes et al. 1997; VanSlyke et al. 2000; Kleopa et al. 2002; Yum et al. 2002). To determine whether Cx47 mutants were localized to the ER, we immunostained both transiently transfected and bulk-selected cells for Cx47 and GRP94, an ER chaperone. As shown in Figure 2, GRP94 staining overlaps with that of Cx47. Furthermore, the intracellular Cx47 staining of the mutants does not correspond to that of 58K, a component of the Golgi apparatus (Suppl. Fig. 3). As a further test of whether Cx47 mutants were localized in the ER, we treated bulk-selected cells for six hours with brefeldin A (BFA), a compound that disrupts the Golgi apparatus (Klausner et al. 1992). As expected, BFA treatment dispersed the 58K staining to an ER-like pattern (compare untreated and treated 58K panels in Fig. 3). BFA also caused the redistribution of WT Cx47 into an ER-like pattern, with fewer gap junction plaques (Fig. 3), but did not alter the appearance of P87S, Y269D, and M283T.
Figure 3. Disrupting the Golgi apparatus does not alter the localization of Cx47 mutants.
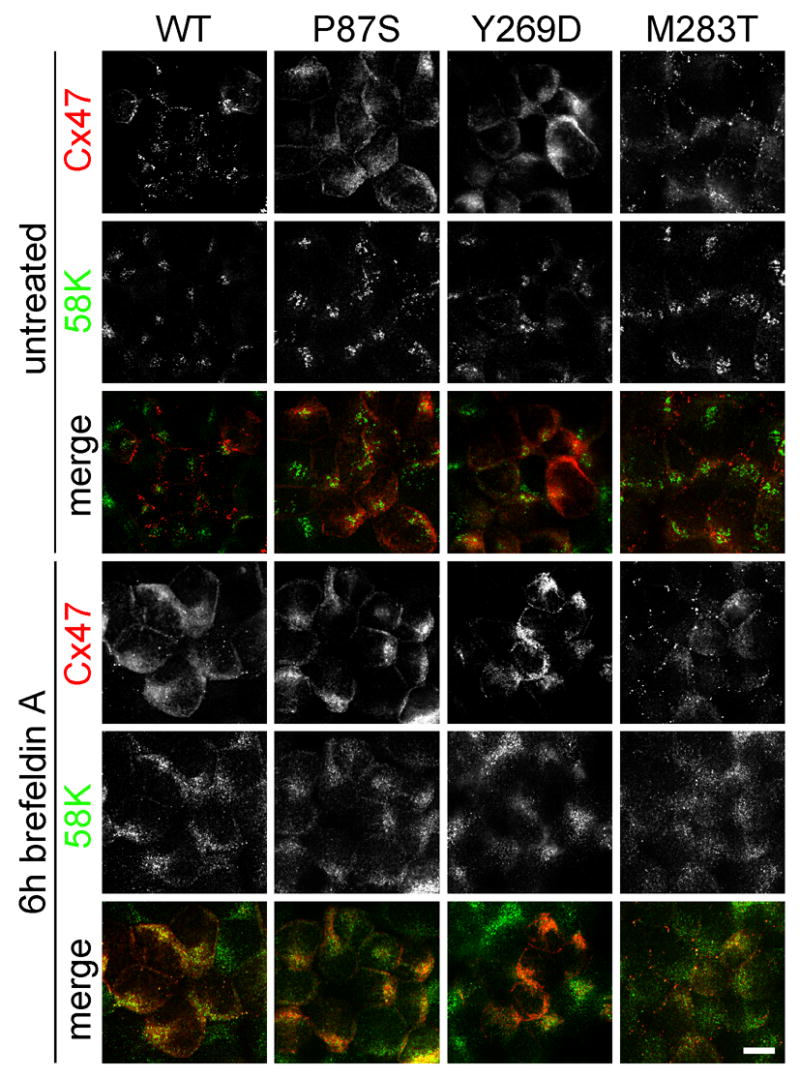
These are confocal (untreated WT example only) or deconvolved images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against mouse Cx47 (red) and a mouse monoclonal antibody against a 58K Golgi protein (green). Incubation in brefeldin A (BFA) for 6 hours causes the redistribution of 58K from a polarized, perinuclear aggregation to a dispersed ER-like pattern of staining. BFA treatment does not affect the localization of P87S or Y269D, both of which are localized in the ER. BFA appears to reduce the number of gap junction plaques and increase the amount of intracellular Cx47-immunoreactivity in cells expressing WT Cx47, but has no apparent effect on cells expressing M283T. All images were acquired from the same experiment with the same exposure time for each channel (except for the untreated WT example). Scale bar: 10μm.
Proteasomes and lysosomes degrade WT and mutant connexins (Laing et al. 1997; VanSlyke et al. 2000). To examine this issue in Cx47 mutants, we treated bulk-selected cells expressing WT or mutant Cx47 for six hours with lactacystin or chloroquine, which inhibit proteasomes or lysosomes, respectively. Lactacystin did not appear to alter the distribution or the intensity of GRP94 staining or the Cx47 staining of cells stably expressing WT Cx47, P87S, or Y269D; however, it did appear to increase the intracellular signal of M283T expressing cells (Suppl. Fig. 4). To confirm that lactacystin inhibited proteasomal degradation, cells were treated with cycloheximide, a blocker of protein synthesis, either alone or with lactacystin for six hours. Cycloheximide treatment alone eliminated most of the intracellular signal in cells expressing each of the Cx47 mutants, but low levels of intracellular protein was detected in cells treated with both cycloheximide and lactacystin (Suppl. Fig. 4). Chloroquine caused more prominent staining of LAMP-2, a lysosomal protein, and the redistribution of WT Cx47 to the lysosomes (Suppl. Fig. 5), but did not alter the pattern of Cx47 mutants, including the puncta of M283T.
Connexins are soluble in Triton X-100 prior to incorporation into gap junction plaques (Musil and Goodenough 1991, 1993; Das Sarma et al. 2001). To determine whether Cx47 mutants are soluble in Triton X-100, we incubated bulk-selected cells for 30 min in 1% Triton X-100, then immunostained them for Cx47. As shown in Figure 4, the gap junction plaques in cells expressing WT Cx47 and puncta in cells expressing M283T appeared to be unaffected, whereas the intracellular Cx47-immunoreactivity disappeared. These data, taken together, show that these Cx47 mutànts are largely localized to the ER and, furthermore, behave like ER-retained mutants in response to disruption of the Golgi apparatus, inhibition of proteasomes or lysosomes, and extraction with Triton X-100.
Figure 4. Intracellular Cx47 mutant protein is soluble in 1% Triton X-100.
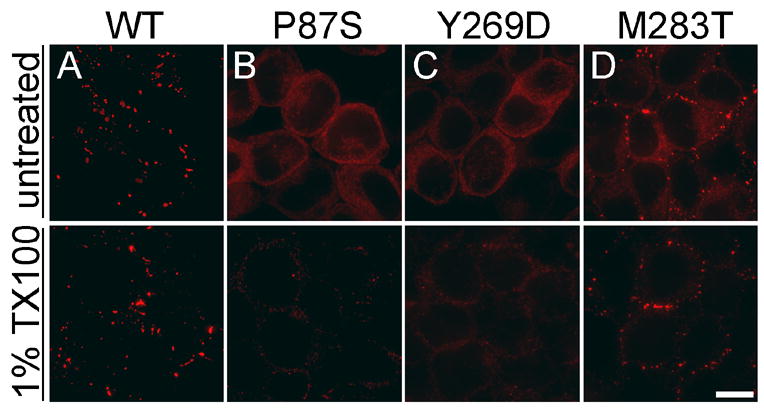
These are deconvolved images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants. The cells were incubated for 30 min in PBS containing 1% Triton X-100 (TX100), then immunostained with a rabbit antiserum against mouse Cx47 (red). The gap junction plaques formed by WT Cx47 and puncta of M283T are TX100-insoluble (A&D), whereas intracellular Cx47 of P87S (B), Y269D (C), and M283T (D) is TX100-soluble. All images were acquired from the same experiment with the same exposure time. Scale bar: 10μm.
We considered the possibility that ER retention of Cx47 mutants could be toxic to oligodendrocytes by inducing the unfolded protein response (UPR). The UPR consists of multiple signaling pathways that appear to protect the cell from ER-related stress, such as accumulation of misfolded protein, but may ultimately activate cell death programs (Gow 2004; Zhang and Kaufman 2006). CHOP is a downstream transcription factor of the PERK UPR pathway implicated in programmed cell death (Zinszner et al. 1998; Ma et al. 2002). Further, Southwood and colleagues (Southwood et al. 2002) have previously shown that ER-retained PLP mutants upregulate CHOP both in transfected cells and in oligodendrocytes of mice that express a dominant Plp mutation. To test whether Cx47 mutants activate the UPR, we electroporated L cells with either WT or mutant Cx47 using a bicistronic vector to express GFP as a surrogate marker of Cx47, and immunostained for CHOP. L cells treated for six hours with tunicamycin, an N-glycosylation inhibitor that potently stimulates the UPR, served as a positive control for CHOP expression (Wang et al. 1998). As summarized in Supplemental Figure 6, 92% (515/560) cells treated tunicamycin had CHOP+ nuclei as compared to 0.66% (4/602) of cells treated with DMSO alone. In contrast, none of the cells transfected with either WT Cx47, one of the mutations, or an “empty” vector, (GFP+ cells), had CHOP+ nuclei; only 0.14% (3/2092) of untransfected (GFP−) cells had a CHOP+ nucleus. We performed this experiment three times with similar results, and conclude that these Cx47 mutants do not induce the UPR, as measured by this assay.
Cx47 mutants do not form functional channels
To determine whether the Cx47 mutants can form functional gap junctions, we scrape loaded cells (el-Fouly et al. 1987; Trosko et al. 2000). In this assay, a confluent monolayer of cells is injured with a scalpel blade in media that contains 0.1% Lucifer Yellow (LY; MW 443) or 2% neurobiotin (NB; MW 287). No dye transfer was seen past the scrape line in cells expressing the Cx47 mutants (Fig. 5B–D & F–H), in parental cells, or in cells transfected with an “empty” vector (data not shown), whereas cells expressing WT Cx47 showed dye transfer to cells beyond the scrape line (Fig. 5A&E, Suppl. Fig. 7). This experiment was done four times with LY, and twice with NB, with similar results. We confirmed that cells that express WT Cx47 transfer NB to many cells layers by scrape loading cells with both NB and 10 kDa rhodamine-dextran, a molecule that is only taken up by injured cells but does not permeate gap junctions (Suppl. Fig. 7). Thus, Cx47 mutants, including M283T (which forms puncta that may represent gap junction plaques), do not appear to form functional gap junctions.
Figure 5. HeLa cells expressing Cx47 mutants are not dye-coupled.
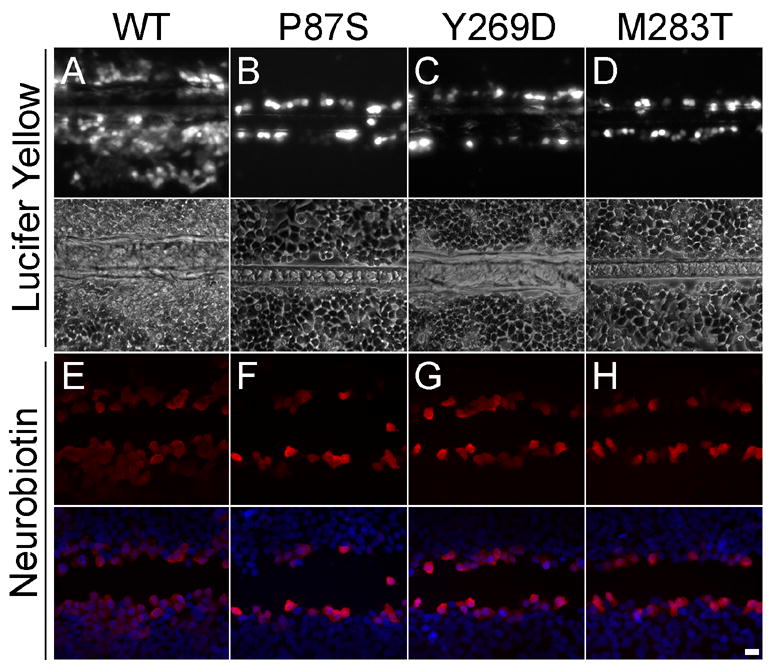
These are images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, scrape loaded with Lucifer Yellow (LY, A-D) or neurobiotin (NB, E–H). Panels A–D show both epifluorescence and phase contrast images from live cells 5 min after scrape loading with LY. Panels E–H show cells that were scrape loaded in NB, then visualized with rhodamine-conjugated streptavidin (red) and counterstained with DAPI (blue). Only HeLa cells expressing WT Cx47 (A&E) show transfer of LY or NB. Scale bar: 20μm.
To determine whether Cx47 mutants can form functional homotypic gap junctions using a more stringent assay, we performed dual whole-cell patch clamping on transiently transfected Neuro2a cells. As shown in Table 1, no cell pairs (except one, see below) expressing Cx47 mutants were electrically coupled (n=5 for each mutant), whereas all cell pairs expressing WT Cx47 were coupled, with a non-normalized average conductance (gj) of 10.29 ± 3.31 nS; the differences between WT and each mutant were statistically significant. We detected a single channel gj of 35 pS in one pair expressing P87S, which is consistent with the rarely observed endogenous channels in untransfected pairs of Neuro2A cells (Abrams, unpublished data).
Table 1.
Dual whole-cell patch clamp recordings.
| Homotypic Pair | Average Conductance (nS) |
|---|---|
| WT Cx47 (n=11) | 10.29 ± 3.31 |
| P87S (n=5) | 0.007 (p<0.001) |
| Y269D (n=5) | 0 (p<0.001) |
| M283T (n=5) | 0 (p<0.001) |
Neuro2a cells were transiently transfected to express WT or the indicated Cx47 mutant. All pairs of cells expressing WT Cx47 were coupled, whereas no pairs expressing any one of the mutants were coupled, except for one pair expressing P87S, that had a single channel conductance of ~35 pS, likely representing rare endogenous channels in Neuro2A cells. The data for each mutant were compared to WT Cx47 with a X2 test with Bonferroni correction for multiple comparisons.
Properties of the human Cx47 channel
We also used dual whole-cell patch clamping of Neuro2A pairs to determine the functional properties of homotypic WT Cx47 channels. Figure 6A shows representative current traces that demonstrate that junctional currents decayed more rapidly as the absolute value of transjunctional voltage (Vj) increased. The relation between the macroscopic normalized steady state junctional conductance (Gj) and Vj (the Gj-Vj relation) was calculated from macroscopic current traces and fit to a Boltzmann distribution (Fig. 6B). As the absolute value of Vj increased, the steady state Gj declined symmetrically about Vj=0 and came to a plateau at a Gmin of 0.13. These macroscopic characteristics are nearly identical to those described for mouse Cx47 homotypic channels expressed in HeLa cells (Teubner et al. 2001). Octanol (Fig. 6D) and CO2 (data not shown) reversibly decreased gj of homotypic WT Cx47 channels. Single channel gj was determined during recovery from octanol treatment by repeatedly applying voltage ramps to one cell of a pair, and measuring the transjunctional current in the second cell (Fig. 6C). The gj of the fully open human Cx47 channel was ~53 pS, with a residual conductance of ~8 pS, consistent with the gj of ~55 pS and ~8 pS residual conductance of mouse Cx47 (Teubner et al. 2001).
Figure 6. Functional properties of Cx47 in transiently transfected Neuro2A cells.
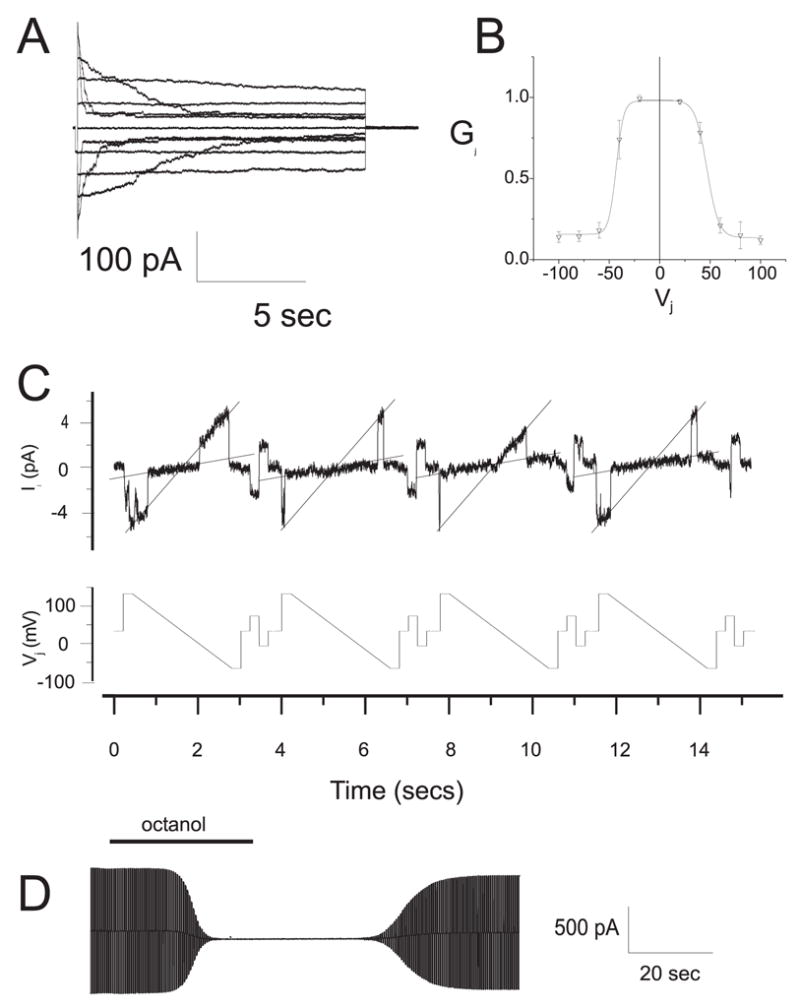
(A) Representative current traces for homotypic human Cx47 channels. At rest, both cells were voltage clamped at 0 mV, then cell 2 was stepped in 20 mV increments from −100 to +100 mV, and junctional currents (Ij) were recorded from cell 1. As the absolute value of applied voltage increased, the rate of Ij decay increased, especially at 60, 80, and 100 mV. (B) The Gj-Vj relation for homotypic Cx47 channels. The average normalized steady state junctional conductance (Gj) in relation to the junctional voltage (Vj) was calculated from the current trace at each voltage. The solid line represents the fit of the data to Boltzmann distributions. Parameters (+Vj, −Vj); Gmin: 0.13, 0.14; Gmax: 0.98, 0.99; K: 5.8, 5.0; V0: 46.7, −44.5. Note that Gj decreases symmetrically as the absolute value of Vj increases. (C) The single channel conductance (non-normalized, gj) of Cx47. gj was determined during recovery from octanol treatment by repeatedly applying voltage ramps from +100 to −100 mV to one cell of a pair (bottom record), and measuring the transjunctional current of single channels in cell 2 (upper record). The gj of the fully open channel is ~53 pS, with a predominant residual conductance of ~8 pS. (D) Chemical gating of Cx47 channels. Ij was measured during application of repeated voltage ramps of ± 30 mV, before, during, and after bath application of octanol (2 mM). Note that the reduction of Ij by octanol was more rapid than the recovery.
Primate oligodendrocytes express Cx47
In rodents, Cx47 is expressed by rat oligodendrocytes, but not by astrocytes or neurons (Menichella et al. 2003; Altevogt and Paul 2004; Kleopa et al. 2004). To determine whether human oligodendrocytes express Cx47 in the primate brain in a similar pattern, we immunostained cryosections of autopsied human brains, but poor preservation precluded any definitive conclusions. As an alternative, we immunostained cryosections of brainstem and optic nerve from rhesus monkeys. A mouse monoclonal antibody against Cx47 labeled small cell bodies in the optic nerve (Fig. 7C) and medulla (data not shown); these often formed chains in longitudinal sections like intrafascicular oligodendrocytes (Fig. 7B). Most of the Cx47-immunoreactivity was diffusely localized in the cell bodies (Fig. 7A, B, D), with few gap junction plaques (arrowhead, Fig. 7D). The rabbit antisera against Cx47 gave a similar pattern, but the background staining was higher (data not shown). Nearly all Cx47-positive cells both in the optic nerve (Fig. 7A) and in the medulla (data not shown) expressed aspartoacylase (ASPA), an enzyme that is localized to oligodendrocytes (Madhavarao et al. 2004), but not express GFAP, a marker of astrocytes (data not shown). A monoclonal antibody against Cx43, in contrast, labeled the cell bodies and processes of astrocytes (Fig. 7E), and Cx43-positive gap junction plaques often appear to surround small cell bodies (Fig. 7F), some of which were oligodendrocytes (Fig. 7B). These results indicate that Cx47 and Cx43 likely form A/O gap junctions on oligodendrocyte cell bodies in the primate brain.
Figure 7. Primate oligodendrocytes express Cx47.
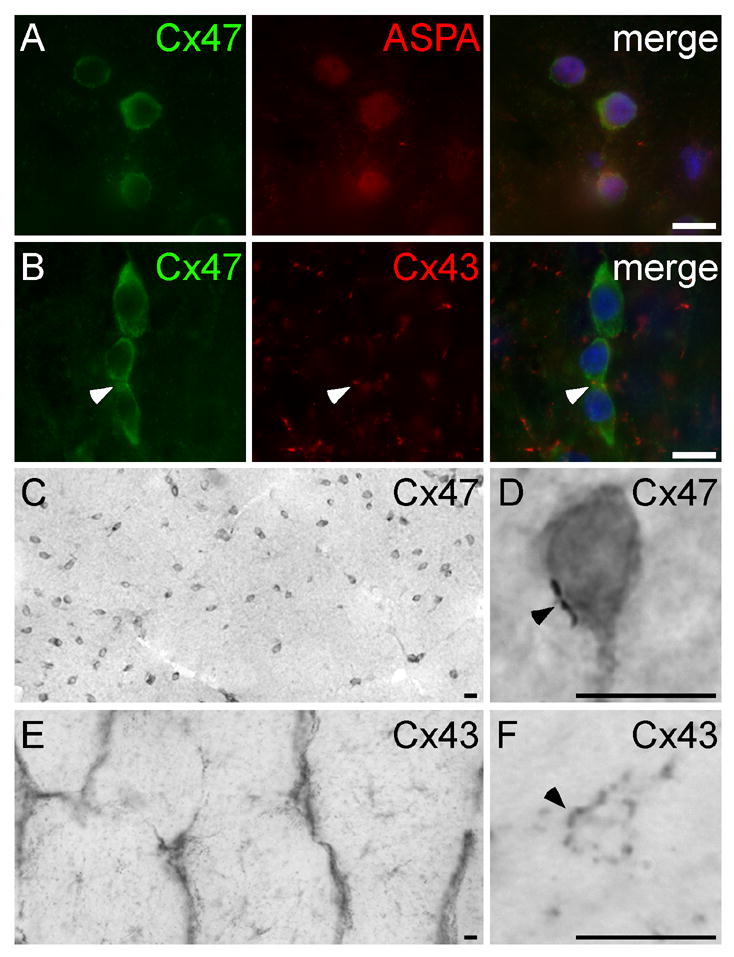
These are images of sections of rhesus monkey optic nerve, immunostained with mouse monoclonal antibodies against Cx47 (A–D) or Cx43 (E–F), a rabbit antisera against aspartoacylase (ASPA; A) or Cx43 (B), visualized with FITC- or TRITC- (A&B) or peroxidase-conjugated secondary antiserum (C–F), and counterstained with DAPI (A&B, merged panels). Cx47-immunoreactivity is diffusely dispersed within small cell bodies (A–B, green; C–D), with occasional gap junction plaques (arrowhead; D), and colocalizes with ASPA (A), a marker of oligodendrocytes. Cx43-immunoreactivity is found on astrocyte cell bodies and their proximal processes (E) and in gap junction plaques that are distributed throughout the optic nerve, including on small cell bodies that are likely to be oligodendrocytes (arrowheads, B&F). Scale bars: 10μm.
DISCUSSION
PMLD mutations appear to cause simple loss-of-function
The PMLD patients described to date (Uhlenberg et al. 2004; Bugiani et al. 2006) have similar phenotypes - nystagmus noted by 7 weeks, and impaired motor development noted by 15 months. Different genotypes - homozygous P128frameshift, M283T, G233S, or L278frameshift mutations, compound heterozygotes for P87S/P327frameshift or Y269D/R237stop mutations - cause the same phenotype, including alleles that would be predicted to disrupt the protein (P128frameshift, R237stop, and L278frameshift, P327frameshift), as well as ones that may not (P87S, G233S, Y269D, and M283T). Although the similar phenotypes caused by different mutations implies all these mutations cause loss-of-function, the rarity of PMLD, the finding that dominant mutations of other connexin genes cause disease in a cell autonomous manner (White and Paul 1999), and examples of monogenic recessive disorders whose phenotype is altered by other genes (Scriver and Waters 1999; Dipple and McCabe 2000), leave this issue unsettled.
In spite of these reservations, our results indicate that P87S, Y269D, and M283T mutants exhibit simple loss-of-function, and this appears to be sufficient to account for the recessive nature of PMLD. We show that these mutants fail to form functional gap junction channels when expressed in HeLa or Neuro2A cells, by both scrape loading and electrophysiology. All three mutants appear to at least partially accumulate in the ER, possibly contributing to their failure to form functional channels (although M283T may be an exception, as it also appears to form puncta that may be gap junction plaques). The failure of these mutants to upregulate CHOP, furthermore, indicates that they do not have the gain-of-function documented for some PLP mutants that cause the phenotypically similar disease, PMD (Southwood et al. 2002). However, we have not ruled out the possibility that the Cx47 mutants activate a different UPR-related pathway (Gass et al. 2002), that oligodendrocytes are more sensitive to ER accumulation of Cx47 mutants than the cells we used in our in vitro assay, or that Cx47 mutants have dominant effects on WT Cx47.
Cx47 mutants affect the panglial syncytium
Work that has been largely done in rodents indicates that there is a “panglial syncytium” between CNS glial cells. There are abundant A/A gap junctions, fewer A/O gap junctions, and few if any gap junctions between oligodendrocytes themselves or between neurons and glia (see Introduction). Cx43/Cx47 and Cx30/Cx32 likely form two parallel sets of heterotypic channels that constitute A/O coupling. These two pairs of connexins colocalize at A/O gap junction plaques in WT brains, but Cx30 (and not Cx43) is mislocalized in Gjb1/cx32-null brains (Nagy et al. 2003; Altevogt and Paul 2004). Cx30 and Cx32 can form functional heterotypic channels in oocytes (Dahl et al. 1996), but it remains to be shown whether Cx43 and Cx47 can also form functional heterotypic channels. Our finding that primate oligodendrocytes express Cx47, and are surrounded by Cx43-positive gap junctions, adds evidence to the argument that the recessive GJA12/Cx47 mutations that cause PMLD likely disrupt A/O gap junction coupling, probably by disrupting Cx43/Cx47 (but not Cx30/Cx32) channels.
Genotype-phenotype correlations
The marked discrepancy between the devastating CNS phenotype in people with PMLD (Uhlenberg et al. 2004) and the mild CNS phenotype of mice that are homozygous for a null Gja12/cx47 allele (Menichella et al. 2003; Odermatt et al. 2003) remains to be explained. The differences in the primary amino acid sequence of mouse and human Cx47 (Suppl. Fig. 8) may be relevant, but the most divergent regions (the intracellular loop and the C-terminus) also differ the most between all connexins (Willecke et al. 2002), and the only known functional motif, a C-terminal PDZ-binding domain (Li et al. 2004), is not affected. We have shown here that the electrophysiological properties of homotypic human Cx47 channels are nearly identical to mouse Cx47 (Teubner et al. 2001), but it is possible that the human and mouse Cx43/Cx47 heterotypic channels have different properties. Although we showed that Cx47 is localized on oligodendrocyte somata in the white matter of a primate, as previously described in rodents (see Introduction), the contribution of Cx43/Cx47 channels to A/O coupling in these different species remains to be determined. Perhaps Cx30/Cx32 coupling can compensate for the lack of Cx43/Cx47 coupling in mice to a greater degree than in humans, although the absence of Cx32 alone causes insignificant CNS abnormalities in Gjb1/cx32-null mice (Scherer et al. 1998; Sutor et al. 2000).
Our results for Cx47 mutants raise the possibility that ER retention of mutant connexins predict recessively inherited phenotypes. Many Cx32 mutants appear to be ER-retained (VanSlyke et al. 2000; Kleopa et al. 2002; Yum et al. 2002); however, they are not informative in this regard because CMT1X is an X-linked and not an autosomally inherited disease. The Cx26 (G12V, W77R, 235delC) and Cx31 (L34P, 141delI) mutants that appear to be retained intracellularly, possibly in the ER, are all associated with recessively inherited hearing loss (Martin et al. 1999; Choung et al. 2002; D’Andrea et al. 2002; Bruzzone et al. 2003; He et al. 2005). Conversely, most connexin mutants that cause dominantly inherited diseases are not localized to the ER, including the Cx43 mutants that cause oculodentodigital dysplasia (ODDD, Scherer, unpublished; (Seki et al. 2004; Roscoe et al. 2005; Shibayama et al. 2005; Lai et al. 2006), a Cx46 and a Cx50 mutant that each cause cataracts (Berthoud et al. 2003; Minogue et al. 2005), and most of the Cx26, Cx30, and Cx31 mutants that cause hearing loss and/or skin diseases (Martin et al. 1999; Rouan et al. 2001; Common et al. 2002; Di et al. 2002; Common et al. 2003; Marziano et al. 2003; Oshima et al. 2003; Thomas et al. 2003; Essenfelder et al. 2004; Thomas et al. 2004; Di et al. 2005; He et al. 2005; Oguchi et al. 2005; Piazza et al. 2005). On the other hand, two of the Cx30 mutants (G11R andA88V) that cause skin diseases (Common et al. 2002; Essenfelder et al. 2004), at least two of the Cx26 mutants (D50N, T55M), three of the Cx31 mutants (66delD, R180X, E183K) that cause dominantly inherited hearing loss (Di et al. 2002; Common et al. 2003; Di et al. 2005; He et al. 2005; Melchionda et al. 2005), and one Cx43 mutant that causes ODDD and skin disease (fs260, (Gong et al. 2006)), appear to be localized to the ER. Because the analysis of the mutants that were putatively localized to the ER was not done in detail (with the exception of the ODDD mutant fs260), we can only tentatively conclude that ER retention is not strictly associated with recessive inheritance.
EXPERIMENTAL METHODS
Mutant human Cx47 expression constructs
Using primers developed from a human Cx47 DNA sequence (GenBank accession number AF014643), we amplified the putative open reading frame of Cx47 by RT-PCR (SuperScript II, Invitrogen, Carlsbad, CA) from polyA RNA isolated from human brain, corpus callosum, or spinal cord (Clontech, Mountain View, CA), adding EcoRV and BamHI restriction sites at the 5′ and 3′ ends, respectively. The 5′ primer eliminated an unlikely alternative AUG that has been electronically translated in some sequences as the initiation codon (GenBank accession numbers NM_020435 and BC089439). We cloned the PCR sequence into pIRESpuro3 (Clontech, Mountain View, CA), and the resulting plasmid was used to transform DH5-α bacteria. For each PCR reaction, a large-scale plasmid preparation was made from a single colony (Sigma–Aldrich, St. Louis, MO), and analyzed at the Sequencing Core of the University of Pennsylvania. Because our sequence differed from AF014643 (the only one in the database at that time), we sequenced multiple PCR reactions from brain, corpus callosum, and spinal cord RNA. All of the sequences were identical; this sequence was deposited in GenBank (accession number AY285161), and has subsequently been found by others in human cDNA (NM_020435 and BC089439) and matches the genomic sequence (AL359510) as well.
We made the three missense GJA12/Cx47 mutations described by Uhlenberg et al. (Uhlenberg et al. 2004) by PCR site-directed mutagenesis using the QuikChange kit (Stratagene, La Jolla, CA). To generate the mutants, the following oligonucleotide primers were used (the underlined codon encodes the altered amino acid): P87S: 5′-ggtcatctccacgtcctcggtcatgtacc-3′; 5′-ggtacatgaccgaggacgtggagatgacc-3′; Y269D: 5′-cctgctggttatggacgtggtcagctgcc-3′; 5′-ggcagctgaccacgtccataaccagcagg-3′; M283T: 5′-caacctctgtgagacggcccacctgggc-3′; 5′-gcccaggtgggccgtctcacagaggttg-3′.
The mutagenic primers were incorporated using PfuTurbo or PfuUltra HF DNA polymerase, and the PCR products were digested by DpnI endonuclease to eliminate the DNA template. The resulting DNA was used to transform DH5-α bacteria, a large-scale plasmid preparation was made from a single colony (Sigma–Aldrich, St. Louis, MO) and analyzed at the Sequencing Core of the University of Pennsylvania.
Generation of rabbit antisera against human Cx47
A portion of the Cx47 intracellular cytoplasmic tail (amino acids 344–399, encoded by nucleotides 1030–1197) was amplified by PCR using wild type (WT) human Cx47 plasmid as the template, primers (5′-cgcggatccgaccagaacctggcaaacct–3′ and 5′–ccggaattcagtgcccgcagaggtagc–3′), and the Pfu Turbo Polymerase Kit (Stratagene, La Jolla, CA). The PCR product was ligated with the T4 Rapid Ligation Kit (Roche, Indianapolis, IN) into the pGEX-2TK vector (Amersham Biosciences, Uppsala, Sweden), which encodes a glutathione S-transferase moiety. The resulting construct was used to transform DH5-α bacteria, a large-scale plasmid preparation was made from a single colony (Sigma–Aldrich, St. Louis, MO) and analyzed at the Sequencing Core of the University of Pennsylvania. The construct was transformed into BL21 codon + RP Competent Cells (Stratagene, La Jolla, CA) for protein expression. The GST-fusion protein was purified using Glutathione-Sepharose beads (Amersham Biosciences, Uppsala, Sweden), then sent to Covance Research Products (Denver, PA) to develop polyclonal antisera from two rabbits using incomplete Freund’s Adjuvant.
Transfections
Communication-incompetent HeLa cells (Elfgang et al. 1995) were obtained from Dr. Klaus Willecke (University of Bonn, Bonn, Germany), and grown in low-glucose Dulbecco’s modified Eagle’s Medium (DMEM) supplemented by 10% fetal bovine serum (FBS) and antibiotics (100 units/ml penicillin & 100 μg/ml streptomycin; GIBCO Invitrogen, Carlsbad, CA) in a humidified atmosphere containing 5% CO2 at 37°C. For transfection, 10 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and 4 μg of plasmid DNA were incubated separately in Optimem (GIBCO Invitrogen, Carlsbad, CA) for 15 min at RT, then combined for another 20 min. HeLa cells (approximately 80–90% confluent) were washed with Hank’s Buffered Saline Solution (HBSS), incubated with the combined Lipofectamine 2000/DNA solution in Optimem for 5 h at 37°C, then fed with DMEM supplemented with 20% FBS. After 2 days, transfected cells were either processed for immunostaining or immunoblotting (for transient transfection) or fed with 1 μg/ml puromycin-supplemented media (Sigma-Aldrich, St. Louis, MO). Selection was continued for 2 weeks, with medium changes every 3–4 days, until colonies with stable growth were obtained. Untransfected HeLa cells were also treated with puromycin and did not survive after two weeks. Bulk selection was performed after trypsinization of all colonies. Transfected cells were expanded for immunocytochemistry and immunoblotting. Transient transfections were repeated at least three times.
L cells were purchased from ATCC (CRL-2648; Manassas, VA) and grown in DMEM (ATCC, Manassas, VA) supplemented by 10% fetal bovine serum (FBS) and antibiotics (100 units/ml penicillin & 100 μg/ml streptomycin; GIBCO Invitrogen, Carlsbad, CA) in a humidified atmosphere containing 5% CO2 at 37°C. For electroporation, 5 × 106 cells/condition were pelleted and resuspended in 5 μg DNA (WT or mutant Cx47 subcloned into the bicistronic vector pIRES2-egfp (Clontech, Mountain View, CA)) and 100 μl Nucleofector solution (Amaxa, Gaithersburg, MD), then electroporated using a Nucleofector device (Amaxa, Gaithersburg, MD) set to program U-30. Cells were then resuspended in media and plated on sterile coverslips for 48 hours before processing for immunofluorescence.
Immunocytochemistry and immunohistochemistry
Cells were trypsinized, plated onto 4-chamber glass slides (Nalge Nunc International, Rochester, NY), and incubated for 1–3 days to approximately 90% confluency. The cells were washed in PBS, fixed in acetone at −20°C for 10 min, and blocked for 1 h at RT with 5% fish skin gelatin in PBS containing 0.1% Triton X-100 (TX100). Primary antibodies - a rabbit antiserum (gift of Dr. David Paul, Harvard University, Boston, MA; diluted 1:1500) raised against the C-terminus of mouse Cx47 (Menichella et al. 2003), and the two rabbit antisera raised against the C-terminus of human Cx47 described above (diluted 1:2000) - were added to this blocking solution and the samples were incubated overnight at 4°C. Some slides were double-labeled with monoclonal antibodies GRP94 (Abcam Inc., Cambridge, MA, diluted 1:250), 58K protein (Sigma-Aldrich, St. Louis, MO, diluted 1:100), LAMP-2 (RDI division of Fitzgerald Industries Intl., Concord, MA, diluted 1:100) or pancadherin (Abcam Inc., Cambridge, MA, diluted 1:200), which are ER, Golgi, lysosomal and cell surface markers, respectively. After washing in PBS, the TRITC-, FITC- and Cy5-conjugated donkey anti-rabbit, anti-mouse and anti-rat antisera (Jackson ImmunoResearch, West Grove, PA; diluted 1:200) were added to the same blocking solution and incubated at RT for 1 h. The slides were washed, counterstained with DAPI, coverslips were mounted with Vectashield (Vector Laboratories Inc., Burlingame, CA), and imaged with a Leica fluorescence microscope with a Hamamatsu digital camera C4742-95 connected to a G5 Mac computer, using Openlab 3.1.7 and deconvolution software, or confocal microscopy using a Leica laser scanning microscope.
For inhibitor experiments, confluent bulk-selected HeLa cells were fed with medium containing one of the following inhibitors for 6 h, then processed for immunofluorescence: 6 μg/ml brefeldin A (Sigma-Aldrich, St. Louis, MO), 200 μM chloroquine (Sigma-Aldrich, St. Louis, MO), 10 μM lactacystin (EMD Biosciences, San Diego, CA) or 20 μg/ml cycloheximide (Sigma-Aldrich, St. Louis, MO). Stock aliquots of 5 mg/ml brefeldin A (in ethanol), 1 mM lactacystin (in DMSO) and 2 mg/ml cycloheximide (in ddH2O) were stored at −20°C. 10 mM chloroquine (in ddH2O) was made before each experiment (VanSlyke et al. 2000). For in situ Triton X-100 extraction, confluent bulk-selected HeLa cells were plated on 12 mm glass coverslips (Fisher Scientific, Hampton, NH), then washed and incubated in 1% Triton X-100 solution in 1xPBS containing 0.675 mM CaCl2 and 0.2 mM MgCl2 for 30 min at 15°C (Musil and Goodenough 1991; Das Sarma et al. 2001). Cells were then processed for immunofluorescence. The inhibitor and Triton X-100 experiments were repeated three and two times, respectively.
Subconfluent L cells on glass coverslips were incubated in 20 μg/ml of tunicamycin (Sigma-Aldrich, St. Louis, MO) diluted in DMSO, or an equivalent volume of DMSO only, for four to six hours. Tunicamycin-treated cells, as well as cells that had been electroporated 48 hours earlier, were immunostained for CHOP as previously described (Gow 2003). Briefly, cells were fixed in 2% paraformaldehyde diluted in DMEM, washed and permeabilized in 0.1% saponin in 1xPBS for 30 min at RT, then immunostained with a rabbit polyclonal raised against mouse CHOP (diluted 1:2000; gift of Dr. Alexander Gow; (Sharma and Gow 2006), and counterstained with DAPI. About 20 epifluorescent images were acquired by an observer blinded to each of 5 conditions (WT Cx47, P87S, Y269D, M283T, and pIRES2-egfp); then these images were randomized. For a positive control, we similarly prepared 48 images from untransfected cells that were treated with tunicamycin or DMSO. For cells treated with tunicamycin or DMSO, an observer blinded to the treatment type determined whether CHOP immunostaining was primarily cytoplasmic, nuclear, or indeterminate, by comparing to the nuclear DAPI counterstain. Cells scored as indeterminate were not included in the analysis. For the transfected cells, a blinded observer determined whether the CHOP− immunoreactivity was nuclear or not in cells that were clearly GFP+ or GFP−. This experiment was performed 3 times, with similar results, but counts were performed on only one such experiment.
Frozen paraformaldehyde-fixed rhesus monkey optic nerve and medulla (gift of Dr. Douglas Rosene, (Hinman et al. 2006) was embedded in OCT and processed for immunofluorescence and immunoperoxidase staining as previously described (Kleopa et al. 2004). Sections immunostained with the above Cx47 antibodies were double-labeled with rabbit antisera raised against aspartoacylase (ASPA; gift of Dr. James Garbern, (Madhavarao et al. 2005); diluted 1:1000), Cx43 (Chemicon, Intl., Temecula, CA; diluted 1:2000), or monoclonal antibodies against GFAP (gift of Dr. Virginia Lee, University of Pennsylvania, Philadelphia, PA; diluted 1:10) or Cx43 (Chemicon, Intl., Temecula, CA; diluted 1:250).
Immunoblots
Transfected HeLa cells were grown to confluence in 6-well dishes and lysed directly in ice-cold RIPA buffer (10 mM sodium phosphate pH 7.0, 150 mM sodium chloride, 2 mM EDTA, 50 mM sodium fluoride, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) for 15 min at 4°C. Samples were collected with a cell scraper and centrifuged for 30 min at 4°C at 13,000 rpm. The supernatant was then either stored at −80°C or mixed directly with loading buffer and loaded onto a 12.3% SDS-polyacrylamide/0.1% SDS gel, electrophoresed, and transferred to an Immobilon-PVDF membrane (Millipore, Billerica, MA) over 1.5 h using a semidry transfer unit (Fisher Scientific, Hampton, NH). The blots were blocked (5% powdered skim milk and 0.5% Tween-20 in Tris-buffered saline) for 1 h at RT and incubated overnight at 4°C in a rabbit antiserum against mouse or human Cx47 (diluted 1:10,000 or 1:5000, respectively). After washing in blocking solution, the blots were incubated in peroxidase-coupled secondary antibodies against rabbit IgG (Jackson ImmunoResearch, West Grove, PA, diluted 1:10,000) for 1 h at RT. After washing in blocking solution and Tris-buffered saline, blots were visualized by detection of enhanced chemiluminescence (ECL kit, Amersham Biosciences, Uppsala, Sweden) with X-ray film, according to the manufacturer’s protocols.
Scrape loading
Confluent 60 mm plates of bulk-selected HeLa cells were washed with HBSS without Ca2+ or Mg2+, then incubated with 0.1% Lucifer Yellow (Sigma-Aldrich, St. Louis, MO) in PBS or HBSS without Ca2+ or Mg2+, and multiple scrape lines were made with a scalpel blade. The cells were incubated for 5 min, washed with HBSS without Ca2+ or Mg2+, and maintained in HBSS containing Ca2+ and Mg2+. Cells were viewed immediately with a Nikon Eclipse TE 2000 inverted epifluorescence phase microscope with a Blue long pass filter (500 nm) using a 20x objective and images were acquired with Spot Advance RT Camera/Software. For scrape loading with neurobiotin, confluent bulk-selected HeLa cells were plated on 4-chamber Permanox slides (Nalge Nunc International, Rochester, NY), washed with PBS, incubated in 2% Neurobiotin (Vector Laboratories Inc., Burlingame, CA) with or without 0.2% 10 kDa tetramethyl-rhodamine dextran (Molecular Probes, Invitrogen Corp., Carlsbad, CA) in PBS or HBSS without Ca2+ or Mg2+, and injured with a scalpel blade as above. After 5 min, the cells were washed in PBS with or without Ca2+ or Mg2+, fixed with ice cold 4% paraformaldehyde for 10 min, and incubated in blocking solution for 1 h at RT. Cells were then immunostained as above for Cx47 at RT for 1 hour, then incubated in TRITC-conjugated streptavidin or FITC-conjugated extravidin (Jackson ImmunoResearch, West Grove, PA, diluted 1:300 in blocking solution) for 1 h at RT, washed, counterstained with DAPI, mounted with Vectashield, and imaged.
Recording from transfected mammalian cell lines
WT and mutant Cx47 were subcloned into pIRES-EGFP2 (Clontech, Mountain View, CA) to identify transfected cells with fluorescence microscopy. Neuro2a cells were transiently transfected using Lipofectamine 2000 (GIBCO Invitrogen, Carlsbad, CA) as described above, except that Lipofectamine 2000/DNA complexes were added to antibiotic-free media rather than Optimem. Dual whole-cell voltage clamping and analysis was performed as previously described (Abrams et al. 2003). Recording solutions: pipette solution, 145 mM CsCl, 5 mM EGTA, 0.5 mM CaCl2, 10 mM HEPES pH 7.2; bath solution: 150 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM Dextrose, 2 mM Pyruvate, 10 mM HEPES; pH 7.4. Junctional conductances were determined from isolated pairs by measuring instantaneous junctional current responses to junctional voltage pulses from 0 to ± 40 or ± 100 mV and applying Ohm’s law. Cytoplasmic bridges were excluded by demonstrating the sensitivity of the junctional conductances to application of bath solution containing 2 mM octanol. Values are presented as mean ± SEM. Frequencies of coupling were compared using a chi square test with Bonferroni correction.
Supplementary Material
Supplemental Figure 1. Immunoblot analysis with three different rabbit antisera against Cx47. Triplicate immunoblots were prepared from transiently transfected HeLa cells, and individually hybridized with an affinity-purified rabbit antiserum (from Dr. David Paul) against mouse Cx47, or a non-affinity purified rabbit antiserum against human Cx47 (935 and 936). Untransfected parental cells and cells transfected with vector alone do not express Cx47; the specific band corresponding to 47 kDa was present in lysates collected from transiently transfected cells expressing WT Cx47 as well as the P87S, Y269D, and M283T mutants (arrowhead). Asterisks denote background bands, which differ between the different antisera.
Supplemental Figure 2. Gap junction plaques localize to the cell surface of HeLa cells. These are confocal images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against mouse Cx47 (red) and a mouse monoclonal antibody against pan-cadherin (green). The pan-cadherin staining at cell borders surrounds the intracellular Cx47 staining of the P87S (B) Y269D (D), and M283T (C) mutants, and interdigitates with the cell surface staining of Cx47 in cells that express WT Cx47 (A) or M283T (C). Scale bar: 10μm.
Supplemental Figure 3. Cx47 mutants do not accumulate in the Golgi.. These are deconvolved images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with rabbit antiserum against mouse Cx47 (red) and a mouse monoclonal antibody against a 58K Golgi protein (green), and counterstained with DAPI (blue). Neither WT Cx47 (A) nor the Cx47 mutants (B–D) are colocalized with 58K. Scale bar: 10μm.
Supplemental Figure 4. Proteasomal inhibition does not alter the localization of Cx47 mutants. These are images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against mouse Cx47. Incubation in lactacystin for 6 hours appeared to increase the intracellular Cx47 staining in cells expressing M283T, but did not alter the localization or the level of intracellular Cx47 staining in cells expressing WT Cx47, P87S or Y269D. Incubation with cycloheximide (CHX) for 6 hours reduced the level of intracellular Cx47 staining for cells expressing Cx47 mutants and the size of GJ plaques for cells expressing WT Cx47. Incubation in both lactacystin and cycloheximide (lacta + CHX) results in levels of Cx47 signal intermediate between untreated and cycloheximide treatment, demonstrating that lactacystin inhibited proteasomal degradation. All images were acquired from the same experiment, the exposure time was the same for all images of cells expressing the Cx47 mutants; a shorter exposure time was used for all of WT Cx47 images. This experiment was repeated three times with similar results. Scale bar: 10μm.
Supplemental Figure 5. Lysosomal inhibition does not alter the localization of Cx47 mutants. These are deconvolved images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against mouse Cx47 (red) and a mouse monoclonal antibody against the lysosomal protein LAMP-2 (green). Incubation in chloroquine for 6 hours results in more prominent lysosomal staining (compare LAMP-2 staining in untreated and treated cells), and WT Cx47 redistributes in a punctate pattern that partially overlaps with that of LAMP-2 (compare merged panels for WT Cx47). In contrast, the localization of the Cx47 mutants does not appear to be altered by chloroquine. All images were acquired from the same experiment with the same exposure time for each channel. Scale bar: 10μm.
Supplemental Figure 6. Expression of Cx47 mutants does not relocalize CHOP, a marker of the UPR. These are representative images from one experiment of L cells that were either incubated in tunicamycin for 6 hours (A), or transiently transfected to express WT Cx47 (B) or one the indicated mutants (C–E) with a bicistronic vector that also contains GFP. The cells were fixed with paraformaldehyde, immunostained with a rabbit antiserum against CHOP (red), and counterstained with DAPI (blue). The right column shows the proportion of CHOP+ nuclei for each condition; the raw numbers of CHOP+/DAPI+ nuclei is given in parentheses. In panels B–E, note that nuclei are rarely CHOP+, regardless of whether the cell also expresses GFP, a surrogate marker of Cx47 expression. In contrast, there were many CHOP+ nuclei in cells were incubated in tunicamycin. Scale bar: 10μm.
Supplemental Figure 7. HeLa cells expressing Cx47 mutants are not dye-coupled. These are representative images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants. The cells were scrape-loaded with neurobiotin (NB) and 10 kDa rhodamine-dextran (dextran, red), then labeled with a rabbit antiserum against mouse Cx47, followed by FITC-conjugated extravidin (NB, green) and Cy5-conjugated donkey anti-rabbit antiserum and DAPI counterstain (blue, merge row). Only HeLa cells expressing WT Cx47 showed transfer of NB to cells that were not also loaded with rhodamine-dextran. Note that there are abundant GJ plaques in the cells that transferred NB (A). This experiment was repeated twice with similar results; panel D is from a different experiment than panels A–C. Scale bar: 20μm.
Supplemental Figure 8. Comparison of human and mouse Cx47. This is a schematic drawing of human Cx47; the amino acid substitutions, deletions, and insertions that are found in mouse Cx47 are indicated.
Acknowledgments
We thank Jonathan Lee, Julia Beamesderfer, and Kate Hawk for technical assistance, Dr. Mike Koval for advice, Drs. Klaus Willecke and Bruce Nicholson for the HeLa cells, Dr. David Paul for the Cx47 antibody, Dr. Doug Rosene for the rhesus monkey tissue, and Dr. Jim Garbern for the ASPA antibody, Dr. Virginia Lee for the GFAP antibody, and Dr. Alex Gow for the CHOP antibody. This work was supported by the NIH/NIA AG 00255 “Training in Age Related Neurodegenerative Disease” and NS054363 (to J.L.O-M.), NS050345 (to C.K.A.), and NS42878 and NS043560 (to S.S.S.), and by a grant from the National Multiple Sclerosis Society (to S.S.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams CK, Freidin M, Bukauskas F, Dobrenis K, Bargiello TA, Verselis VK, Bennett MVL, Chen L, Sahenk Z. Pathogenesis of X-linked Charcot-Marie-Tooth disease: differential effects of two mutations in connexin 32. J Neurosci. 2003;23:10548–10558. doi: 10.1523/JNEUROSCI.23-33-10548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J Neurosci. 2004;24:4313–4323. doi: 10.1523/JNEUROSCI.3303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL. Cx29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci. 2002;22:6458–6470. doi: 10.1523/JNEUROSCI.22-15-06458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Bone LJ, Scherer SS. Functional gap junctions in the Schwann cell myelin sheath. J Cell Biol. 1998;142:1095–1104. doi: 10.1083/jcb.142.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergoffen J, Scherer SS, Wang S, Oronzi-Scott M, Bone L, Paul DL, Chen K, Lensch MW, Chance P, Fischbeck K. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- Berthoud VM, Minogue PJ, Guo J, Williamson EK, Xu X, Ebihara L, Beyer EC. Loss of function and impaired degradation of a cataract-associated mutant connexin50. Eur J Cell Biol. 2003;82:209–221. doi: 10.1078/0171-9335-00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D’Andrea P, White TW. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003;533:79–88. doi: 10.1016/s0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- Bugiani M, Al Shahwan S, Lamantea E, Bizzi A, Bakhsh E, Moroni I, Balestrini MR, Uziel G, Zebiani M. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology. 2006;67:273–279. doi: 10.1212/01.wnl.0000223832.66286.e4. [DOI] [PubMed] [Google Scholar]
- Choung YH, Moon SK, Park HJ. Functional study of GJB2 in hereditary hearing loss. Laryngoscope. 2002;112:1667–1671. doi: 10.1097/00005537-200209000-00026. [DOI] [PubMed] [Google Scholar]
- Common JEA, Becker D, Di WL, Leight IM, O’Toole EA, Kelsell DP. Functional studies of human skin disease- and deafness-associated connexin 30 mutations. Biochem Biophys Res Commun. 2002;298:651–656. doi: 10.1016/s0006-291x(02)02517-2. [DOI] [PubMed] [Google Scholar]
- Common JEA, Di WL, Davies D, Galvin H, Leigh IM, O’Toole EA, Kelsell DP. Cellular mechanisms of mutant connexins in skin disease and hearing loss. Cell Commun Adhes. 2003;10:347–351. doi: 10.1080/cac.10.4-6.347.351. [DOI] [PubMed] [Google Scholar]
- D’Andrea P, Veronesi V, Bicego M, Melchionda S, Zelante L, Di Iorio E, Bruzzone R, Gasparini P. Hearing loss: frequency and functional studies of the most common connexin26 alleles. Biochem Biophys Res Commun. 2002;296:685–691. doi: 10.1016/s0006-291x(02)00891-4. [DOI] [PubMed] [Google Scholar]
- Dahl E, Manthey D, Chen Y, Schwarz HJ, Chang YS, Lalley PA, Nicholson BJ, Willecke K. Molecular cloning and functional expression of mouse connexin-30, a gap junction gene highly expressed in adult brain and skin. J Biol Chem. 1996;271:17903–17910. doi: 10.1074/jbc.271.30.17903. [DOI] [PubMed] [Google Scholar]
- Das Sarma J, Meyer RA, Wang FS, Abraham V, Lo CW, Koval M. Multimeric connexin interactions prior to the trans-Golgi network. J Cell Sci. 2001;114:4013–4024. doi: 10.1242/jcs.114.22.4013. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Farooq M, Kessler JA, Althaus H, Hertzberg EL, Spray DC. Oligodendrocytes express gap junction proteins connexin32 and connexin45. Glia. 1997;20:101–114. [PubMed] [Google Scholar]
- Dermietzel R, Traub O, Hwang TK, Beyer E, Bennett MVL, Spray DC, Willecke K. Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci USA. 1989;86:10148–10152. doi: 10.1073/pnas.86.24.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes SM, Walcott JL, Wexler TL, Scherer SS, Fischbeck KH. Altered trafficking of mutant connexin32. J Neurosci. 1997;17:9077–9084. doi: 10.1523/JNEUROSCI.17-23-09077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di WL, Gu Y, Common JEA, Aasen T, O’Toole EA, Kelsell DP, Zicha D. Connexin interaction patterns in keratinocytes revealed morphologically and by FRET analysis. J Cell Sci. 2005;118:1505–1514. doi: 10.1242/jcs.01733. [DOI] [PubMed] [Google Scholar]
- Di WL, Monypenny J, Common JEA, Kennedy CTC, Holland KA, Leigh IM, Rugg EL, Zicha D, Kelsell DP. Defective trafficking and cell death is characteristic of skin disease-associated connexin 31 mutations. Hum Mol Genet. 2002;11:2005–2014. doi: 10.1093/hmg/11.17.2005. [DOI] [PubMed] [Google Scholar]
- Dipple KM, McCabe ERB. Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000;66:1729–1735. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Fouly MH, Trosko JE, Chang C. Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp Cell Res. 1987;168:422–430. doi: 10.1016/0014-4827(87)90014-0. [DOI] [PubMed] [Google Scholar]
- Elfgang C, Eckert R, Lichternberg-Frate H, Butterweck A, Traub O, Klein RA, Hulser DF, Willecke K. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J Cell Biol. 1995;129:805–817. doi: 10.1083/jcb.129.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essenfelder GM, Bruzzone R, Lamartine J, Charollais A, Blanchet-Bardon C, Barbe MT, Meda P, Waksman G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum Mol Genet. 2004;13:1703–1714. doi: 10.1093/hmg/ddh191. [DOI] [PubMed] [Google Scholar]
- Garbern J, Cambi F, Shy M, Kamholz J. The molecular pathogenesis of Pelizaeus-Merzbacher disease. Arch Neurol. 1999;56:1210–1214. doi: 10.1001/archneur.56.10.1210. [DOI] [PubMed] [Google Scholar]
- Gass JN, Gifford NM, Brewer JW. Activation of an Unfolded Protein Response during Differentiation of Antibody-secreting B Cells. J Biol Chem. 2002;277:49047–49054. doi: 10.1074/jbc.M205011200. [DOI] [PubMed] [Google Scholar]
- Gong X-Q, Shao Q, Lounsbury CS, Bai D, Laird DW. Functional characterization of a GJA1 frame-shift mutation causing oculodentodigital dysplasia and palmoplantar keratoderma. J Biol Chem. 2006:M605961200. doi: 10.1074/jbc.M605961200. [DOI] [PubMed] [Google Scholar]
- Gow A. The COS-7 cell in vitro paradigm to study myelin proteolipid protein 1 gene mutations. In: Potter NT, editor. Neurogenetics: Methods and Protocols. Vol. 217. Humana Press: Totowa, NJ; 2003. pp. 263–275. [DOI] [PubMed] [Google Scholar]
- Gow A. Protein misfolding as a disease determinant. In: Lazzarini RA, editor. Myelin Biology and Disorders. Vol. 2. Elsevier; San Diego: 2004. pp. 1009–1036. [Google Scholar]
- He LQ, Liu Y, Cai F, Tan ZP, Pan Q, Liang DS, Long ZG, Wu LQ, Huang LQ, Dai HP, Xia K, Xia JH, Zhang ZH. Intracellular distribution, assembly and effect of disease-associated connexin 31 mutants in HeLa cells. Acta Biochim et Biophys Sin. 2005;37:547–554. doi: 10.1111/j.1745-7270.2005.00080.x. [DOI] [PubMed] [Google Scholar]
- Hinman JD, Peters A, Cabral H, Rosene DL, Hollander W, Rasband MN, Abraham CR. Age-related molecular reorganization at the node of Ranvier. J Comp Neurol. 2006;495:351–362. doi: 10.1002/cne.20886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson LD, Garbern JY, Kamholz JA. Pelizaeus-Merzbacher Disease. In: Lazzarini RA, editor. Myelin Biology and Disorders. Vol. 2. Elsevier; San Diego: 2004. pp. 867–885. [Google Scholar]
- Kamasawa N, Sik A, Morita M, Yasumura T, Davidson KGV, Nagy JI, Rash JE. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: Implications for ionic homeostasis and potassium siphoning. Neuroscience. 2005;136:65–86. doi: 10.1016/j.neuroscience.2005.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleopa KA, Yum SW, Scherer SS. Cellular mechanisms of connexin32 mutations associated with CNS manifestations. J Neurosci Res. 2002;68:522–534. doi: 10.1002/jnr.10255. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Orthmann JL, Enriquez A, Paul DL, Scherer SS. Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia. 2004;47:346–357. doi: 10.1002/glia.20043. [DOI] [PubMed] [Google Scholar]
- Kozak M. The scanning model for translation: an update. J Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Interpreting cDNA sequences: some insights from studies. Mamm Gen. 1996;7:563–574. doi: 10.1007/s003359900171. [DOI] [PubMed] [Google Scholar]
- Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–389. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- Lai A, Le DN, Paznekas WA, Gifford WD, Jabs EW, Charles AC. Oculodentodigital dysplasia connexin43 mutations result in non-functional connexin hemichannels and gap junctions in C6 glioma cells. J Cell Sci. 2006;119:532–541. doi: 10.1242/jcs.02770. [DOI] [PubMed] [Google Scholar]
- Laing JG, Tadros PN, Westphale EM, Beyer EC. Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp Cell Res. 1997;236:482–492. doi: 10.1006/excr.1997.3747. [DOI] [PubMed] [Google Scholar]
- Li X, Ionescu AV, Lynn BD, Lu S, Kamasawa N, Morita M, Davidson KGV, Yasumura T, Rash JE, Nagy JI. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience. 2004;126:611–630. doi: 10.1016/j.neuroscience.2004.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two Distinct Stress Signaling Pathways Converge Upon the CHOP Promoter During the Mammalian Unfolded Protein Response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MAA, Jacobowitz DM. Immunohistochemical localization of aspartoacylase in the rat central nervous system. J Comp Neurol. 2004;472:318–329. doi: 10.1002/cne.20080. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Arun P, Moffett JR, Szucs S, Surendran S, Matalon R, Garbern J, Hristova D, Johnson A, Jiang W, Namboodiri MAA. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc Nat Acad Sci USA. 2005;102:5221–5226. doi: 10.1073/pnas.0409184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PEM, Coleman SL, Casalotti SO, Forge A, Evans WH. Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal hereditary deafness. Hum Mol Genet. 1999;8:2369–2376. doi: 10.1093/hmg/8.13.2369. [DOI] [PubMed] [Google Scholar]
- Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet. 2003;12:805–812. doi: 10.1093/hmg/ddg076. [DOI] [PubMed] [Google Scholar]
- Massa PT, Mugnaini E. Cell junctions and intramembrane particles of astrocytes and oligodendrocytes: a freeze-fracture study. Neuroscience. 1982;7:523–538. doi: 10.1016/0306-4522(82)90285-8. [DOI] [PubMed] [Google Scholar]
- Meier C, Dermietzel R, Davidson KGV, Yasumura T, Rash JE. Connexin32-containing gap junctions in Schwann cells at the internodal zone of partial myelin compaction and in Schmidt-Lanterman incisures. J Neurosci. 2004;24:3186–3198. doi: 10.1523/JNEUROSCI.5146-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionda S, Bicego M, Marciano E, Franze A, Morgutti M, Bortone G, Zelante L, Carella M, D’Andrea P. Functional characterization of a novel Cx26 (T55N) mutation associated to non-syndromic hearing loss. Biochem Biophys Res Commun. 2005;337:799–805. doi: 10.1016/j.bbrc.2005.09.116. [DOI] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the central nervous system. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogue PJ, Liu X, Ebihara L, Beyer EC, Berthoud VA. An aberrant sequence in a connexin46 mutant underlies congenital cataracts. J Biol Chem. 2005;280:40788–40795. doi: 10.1074/jbc.M504765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1075–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Patel D, Ochalski PAY, Stelmack GL. Connexin30 in rodent, cat and human brain: selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience. 1999;88:447–468. doi: 10.1016/s0306-4522(98)00191-2. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Ionescu AV, Lynn BD, Rash JE. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: implications from normal and connexin32 knockout mice. Glia. 2003;44:205–218. doi: 10.1002/glia.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochalski PAY, Frankenstein UN, Hertzberg EL, Nagy JI. Connexin-43 in rat spinal cord: localization in astrocytes and identification of heterotypic astro-oligodendrocytic gap junctions. Neuroscience. 1997;76:931–945. doi: 10.1016/s0306-4522(96)00394-6. [DOI] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen G, Euwens C, Fuss B, Bussow H, Schilling K, Stenhauser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguchi T, Ohtsuka A, Hashimoto S, Oshima A, Abe S, Kobayashi Y, Nagai K, Matsunaga T, Iwasaki S, Nakagawa T, Usami S. Clinical features of patients with GJB2 (connexin 26) mutations: severity of hearing loss is correlated with genotypes and protein expression patterns. J Hum Genet. 2005;50:76–83. doi: 10.1007/s10038-004-0223-7. [DOI] [PubMed] [Google Scholar]
- Oshima A, Doi T, Mitsuoka K, Maeda S, Fujiyoshi Y. Roles of Met-34, Cys-64, and Arg-75 in the assembly of human connexin 26. Implication for key amino acid residues for channel formation and function. J Biol Chem. 2003;278:1807–1816. doi: 10.1074/jbc.M207713200. [DOI] [PubMed] [Google Scholar]
- Piazza V, Beltramello M, Menniti M, Colao E, Malatesta P, Argento R, Chiarella G, Gallo LV, Catalano M, Perottii N, Mammano F, Cassandro E. Functional analysis of R75Q mutation in the gene coding for connexin 26 identified in a family with nonsyndromic hearing loss. Clin Genet. 2005;68:161–166. doi: 10.1111/j.1399-0004.2005.00468.x. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Kettenmann H. Electrical coupling, without dye coupling, between mammalian astrocytes and oligodendrocytes in cell culture. Glia. 1990;3:258–266. doi: 10.1002/glia.440030405. [DOI] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Dudek FE, Nagy JI. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J Neurosci. 2001;21:1983–2000. doi: 10.1523/JNEUROSCI.21-06-01983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Duffy HS, Dudek FE, Bilhartz BL, Whalen LR, Yasumura T. Grid-mapped freeze-fracture analysis of gap junctions in gray and white matter of adult rat central nervous system, with evidence for a “panglial syncytium” that is not coupled to neurons. J Comp Neurol. 1997;388:265–292. doi: 10.1002/(sici)1096-9861(19971117)388:2<265::aid-cne6>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Robinson SR, Hampson ECGM, Munro MN, Vaney DI. Unidirectional coupling of gap junctions between neuroglia. Science. 1993;262:1072–1074. doi: 10.1126/science.8093125. [DOI] [PubMed] [Google Scholar]
- Roscoe W, Veitch GIL, Gong XQ, Pellegrino E, Bai D, McLachlan E, Shao Q, Kidder GM, Laird DW. Oculodentodigital dysplasia-causing connexin43 mutants are non-functional and exhibit dominant effects on wild-type connexin43. J Biol Chem. 2005;280:11458–11466. doi: 10.1074/jbc.M409564200. [DOI] [PubMed] [Google Scholar]
- Rouan F, White TW, Brown N, Taylor AV, Lucke TW, Paul DL, Munro CS, Uitto J, Hodgins MB, Richard G. Trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci. 2001;114:2105–2113. doi: 10.1242/jcs.114.11.2105. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Kleopa KA. X-linked Charcot-Marie-Tooth disease. In: Dyck PJ, Thomas PK, editors. Peripheral Neuropathy. 2. Vol. 4. Saunders; Philadelphia: 2005. pp. 1791–1804. [Google Scholar]
- Scherer SS, Deschênes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci. 1995;15:8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ. Connexin32-null mice develop a demyelinating peripheral neuropathy. Glia. 1998;24:8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Scriver CR, Waters PJ. Monogenic traits are not simple: lessons from phenylketonuria. Trends Genet. 1999;15:267–272. doi: 10.1016/s0168-9525(99)01761-8. [DOI] [PubMed] [Google Scholar]
- Seki A, Coombs W, Taffet S, Delmar M. Loss of electrical communication, but not plaque formation, after mutations in the cytoplasmic loop of connexin43. Heart Rhy. 2004;1:227–233. doi: 10.1016/j.hrthm.2004.03.066. [DOI] [PubMed] [Google Scholar]
- Sharma RN, Gow A. Minimal role for caspase-12 in the Unfolded Protein Response in oligodendrocytes in vivo. J Neurochem. 2006 doi: 10.1111/j.1471-4159.2007.04541.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibayama J, Paznekas W, Seki A, Taffet S, Wang Jabs E, Delmar M, Musa H. Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ Res. 2005;96:e83–e91. doi: 10.1161/01.RES.0000168369.79972.d2. [DOI] [PubMed] [Google Scholar]
- Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/s0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutor B, Schmolke C, Teubner B, Schirmer C, Willecke K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb Cortex. 2000;10:684–697. doi: 10.1093/cercor/10.7.684. [DOI] [PubMed] [Google Scholar]
- Taylor RA, Simon EM, Marks HG, Scherer SS. The CNS phenotype of X-linked Charcot-Marie-Tooth disease: more than a peripheral problem. Neurology. 2003;61:1475–1478. doi: 10.1212/01.wnl.0000095960.48964.25. [DOI] [PubMed] [Google Scholar]
- Teubner B, Odermatt B, Guldenagel M, Söhl G, Degen J, Bukauskas FF, Kronengold J, Verselis VK, Jung YT, Kozak CA, Schilling K, Willecke K. Functional expression of the new gap junction gene connexin47 transcribed in mouse brain and spinal cord neurons. J Neurosci. 2001;21:1117–1126. doi: 10.1523/JNEUROSCI.21-04-01117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Telford D, Laird DW. Functional domain mapping and selective trans-dominant effects exhibited by Cx26 disease-causing mutations. J Biol Chem. 2004;279:19157–19168. doi: 10.1074/jbc.M314117200. [DOI] [PubMed] [Google Scholar]
- Thomas T, Aasen T, Hodgins M, Laird DW. Transport and function of Cx26 mutants involved in skin and deafness disorders. Cell Commun Adhes. 2003;10:353–358. doi: 10.1080/cac.10.4-6.353.358. [DOI] [PubMed] [Google Scholar]
- Trosko JE, Change CC, Wilson MR, Upham B, Hayashi T, Wade M. Gap junctions and the regulation of cellular functions of stem cells during development and differentiation. Methods. 2000;20:245–264. doi: 10.1006/meth.1999.0941. [DOI] [PubMed] [Google Scholar]
- Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, StoltenburgDidinger G, Aksu F, Topaloglu H, Nurnberg P, Hubner C, Weschke B, Gartner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Amer J Hum Genet. 2004;75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSlyke JK, Deschênes SM, Musil LS. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol Biol Cell. 2000;11:1933–1946. doi: 10.1091/mbc.11.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, Zinszner H, Ron D. Identification of novel stress-induced genes downstream of chop. EMBO J. 1998;17:3619–3630. doi: 10.1093/emboj/17.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TW, Paul DL. Genetic diseases and gene knockouts reveal diverse connexin functions. Annu Rev Physiol. 1999;61:283–310. doi: 10.1146/annurev.physiol.61.1.283. [DOI] [PubMed] [Google Scholar]
- Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Söhl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383:725–737. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Ochalski A, Hertzberg EL, Nagy JL. On the organization of astrocytic gap junctions in rat brain as suggested by LM and EM immunohistochemistry of connexin43 expression. J Comp Neurol. 1990;302:853–883. doi: 10.1002/cne.903020414. [DOI] [PubMed] [Google Scholar]
- Yeager M, Nicholson BJ. Structure of gap junction intercellular channels. Curr Opin Struct Biol. 1996;6:183–192. doi: 10.1016/s0959-440x(96)80073-x. [DOI] [PubMed] [Google Scholar]
- Yum SW, Kleopa KA, Shumas S, Scherer SS. Diverse trafficking abnormalities for connexin32 mutants causing CMTX. Neurobiol Dis. 2002;11:43–52. doi: 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. The unfolded protein response: A stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti HJLS, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Immunoblot analysis with three different rabbit antisera against Cx47. Triplicate immunoblots were prepared from transiently transfected HeLa cells, and individually hybridized with an affinity-purified rabbit antiserum (from Dr. David Paul) against mouse Cx47, or a non-affinity purified rabbit antiserum against human Cx47 (935 and 936). Untransfected parental cells and cells transfected with vector alone do not express Cx47; the specific band corresponding to 47 kDa was present in lysates collected from transiently transfected cells expressing WT Cx47 as well as the P87S, Y269D, and M283T mutants (arrowhead). Asterisks denote background bands, which differ between the different antisera.
Supplemental Figure 2. Gap junction plaques localize to the cell surface of HeLa cells. These are confocal images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against mouse Cx47 (red) and a mouse monoclonal antibody against pan-cadherin (green). The pan-cadherin staining at cell borders surrounds the intracellular Cx47 staining of the P87S (B) Y269D (D), and M283T (C) mutants, and interdigitates with the cell surface staining of Cx47 in cells that express WT Cx47 (A) or M283T (C). Scale bar: 10μm.
Supplemental Figure 3. Cx47 mutants do not accumulate in the Golgi.. These are deconvolved images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with rabbit antiserum against mouse Cx47 (red) and a mouse monoclonal antibody against a 58K Golgi protein (green), and counterstained with DAPI (blue). Neither WT Cx47 (A) nor the Cx47 mutants (B–D) are colocalized with 58K. Scale bar: 10μm.
Supplemental Figure 4. Proteasomal inhibition does not alter the localization of Cx47 mutants. These are images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against mouse Cx47. Incubation in lactacystin for 6 hours appeared to increase the intracellular Cx47 staining in cells expressing M283T, but did not alter the localization or the level of intracellular Cx47 staining in cells expressing WT Cx47, P87S or Y269D. Incubation with cycloheximide (CHX) for 6 hours reduced the level of intracellular Cx47 staining for cells expressing Cx47 mutants and the size of GJ plaques for cells expressing WT Cx47. Incubation in both lactacystin and cycloheximide (lacta + CHX) results in levels of Cx47 signal intermediate between untreated and cycloheximide treatment, demonstrating that lactacystin inhibited proteasomal degradation. All images were acquired from the same experiment, the exposure time was the same for all images of cells expressing the Cx47 mutants; a shorter exposure time was used for all of WT Cx47 images. This experiment was repeated three times with similar results. Scale bar: 10μm.
Supplemental Figure 5. Lysosomal inhibition does not alter the localization of Cx47 mutants. These are deconvolved images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against mouse Cx47 (red) and a mouse monoclonal antibody against the lysosomal protein LAMP-2 (green). Incubation in chloroquine for 6 hours results in more prominent lysosomal staining (compare LAMP-2 staining in untreated and treated cells), and WT Cx47 redistributes in a punctate pattern that partially overlaps with that of LAMP-2 (compare merged panels for WT Cx47). In contrast, the localization of the Cx47 mutants does not appear to be altered by chloroquine. All images were acquired from the same experiment with the same exposure time for each channel. Scale bar: 10μm.
Supplemental Figure 6. Expression of Cx47 mutants does not relocalize CHOP, a marker of the UPR. These are representative images from one experiment of L cells that were either incubated in tunicamycin for 6 hours (A), or transiently transfected to express WT Cx47 (B) or one the indicated mutants (C–E) with a bicistronic vector that also contains GFP. The cells were fixed with paraformaldehyde, immunostained with a rabbit antiserum against CHOP (red), and counterstained with DAPI (blue). The right column shows the proportion of CHOP+ nuclei for each condition; the raw numbers of CHOP+/DAPI+ nuclei is given in parentheses. In panels B–E, note that nuclei are rarely CHOP+, regardless of whether the cell also expresses GFP, a surrogate marker of Cx47 expression. In contrast, there were many CHOP+ nuclei in cells were incubated in tunicamycin. Scale bar: 10μm.
Supplemental Figure 7. HeLa cells expressing Cx47 mutants are not dye-coupled. These are representative images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants. The cells were scrape-loaded with neurobiotin (NB) and 10 kDa rhodamine-dextran (dextran, red), then labeled with a rabbit antiserum against mouse Cx47, followed by FITC-conjugated extravidin (NB, green) and Cy5-conjugated donkey anti-rabbit antiserum and DAPI counterstain (blue, merge row). Only HeLa cells expressing WT Cx47 showed transfer of NB to cells that were not also loaded with rhodamine-dextran. Note that there are abundant GJ plaques in the cells that transferred NB (A). This experiment was repeated twice with similar results; panel D is from a different experiment than panels A–C. Scale bar: 20μm.
Supplemental Figure 8. Comparison of human and mouse Cx47. This is a schematic drawing of human Cx47; the amino acid substitutions, deletions, and insertions that are found in mouse Cx47 are indicated.