

Abstract
To improve our knowledge on the pathophysiology of rheumatoid arthritis (RA), we investigated gene expression patterns in synovial tissue from RA and osteoarthritis (OA) patients. DNA oligonucleotide microarray analysis was employed to identify differentially expressed genes in synovial tissue from pathologically classified tissue samples from RA (n = 20) and OA patients (n = 10). From 7131 gene sets displayed on the microarray chip, 101 genes were found to be upregulated and 300 genes to be downregulated in RA as compared with OA. Semiquantitative reverse-transcription polymerase chain reaction, Western blotting and immunohistochemistry were used to validate microarray expression levels. These experiments revealed that Cys–X–Cys receptor (CXCR)1, CXCR2 and CXCR3 mRNAs, as well as Cys–X–Cys ligand (CXCL)9 (monokine induced by IFN-γ) and CXCL10 (IFN-γ inducible protein 10) mRNAs, were significantly upregulated in RA as compared with OA disease. Elevated protein levels in RA synovial tissue were detected for CXCR1 and CXCR3 by Western blotting. Using immunohistochemistry, CXCR3 protein was found to be preferentially expressed on mast cells within synovial tissue from RA patients. These findings suggest that substantial expression of CXCR3 protein on mast cells within synovial tissue from RA patients plays a significant role in the pathophysiology of RA, accompanied by elevated levels of the chemokines CXCL9 and CXCL10. Mature mast cells are likely to contribute to and sustain the inflamed state in arthritic lesions (e.g. by production of inflammatory mediators such as histamine, proteinases, arachidonic acid metabolites and cytokines). Thus, the mast cell could become a potential target in therapeutic intervention.
Keywords: chemokines, CXCR3, inflammation, mast cells, rheumatoid arthritis, synovial tissue
Introduction
Rheumatoid arthritis (RA) is a chronic disease of joints that is characterized by three main manifestations, namely inflammation, abnormal cellular and humoral immunoresponse, and synovial hyperplasia. Eventually the interplay between these pathologic processes leads to complete joint destruction [1].
A hallmark of RA is infiltration of leukocytes into synovial tissue, mediated by a complex network of cytokines, adhesion molecules and chemoattractants [2-6]. The presence of activated leukocytes contributes to persistence of destructive synovitis [6,7]. Nevertheless, leukocyte recruitment to the joint is not yet fully understood. The presence of specific functional and inflammatory T-cell subsets that express a characteristic pattern of cell surface markers, such as T-cell receptor (TCR), T-cell associated proteins as well as adhesion molecules [8], are of particular significance. Other cell types that are involved in disease manifestation in the synovial tissue include macrophages and neutrophilic granulocytes, as well as tissue mast cells (MCs) [9,10].
Migration of T cells to sites of inflammation is mediated by selectins and their ligands [11,12]. Regulation of leukocyte migration is orchestrated by activating cytokines and adhesion molecules. Furthermore, recruitment of leukocytes to sites of inflammation is driven and mediated by the effects of chemoattractants [13,14]. These molecules, termed inducible chemokines, are members of the large superfamily of IFN-γ inducible small cytokines (8–10 kDa), which can be divided into four groups (CXC, CX3C, CC, and C), according to a conserved structural motif of the first two closely paired cysteines within their amino acid sequence [4,6]. Two major families of chemokines have been reported: CC chemokines, which contain the first two of four conserved cysteines in adjacent positions; and CXC chemokines, with a single amino acid separating the first two cysteines. Cys–X–Cys ligand (CXCL)9 and CXCL10 are members of the small cytokine (intercrine/chemokine) CXC subfamily and represent the specific ligands of the Cys–X–Cys receptor (CXCR)3 [6,15,16]. It has been shown that Th1 and Th2 cells respond differently to several chemokines and express different chemokine receptors [17]. Production of chemokines such as CXCL9 (monokine induced by IFN-γ) and CXCL10 (IFN-γ inducible protein 10) is dependent on release of IFN-γ, corresponding to a Th1 shifted ST compartment in RA disease [18,19].
Receptors of IFN-γ inducible chemokines are members of the seven-transmembrane-spanning, G-protein-coupled receptor family, and are thought to mediate inflammatory effects of chemoattractants within RA synovial tissue [6,20]. Chemokines and their receptors are molecules that may manage selective migration of particular T-cell subsets. Lymphocytes that shift to IFN-γ producing Th1 effector cells express chemokine receptors such as CCR5 and CXCR3 [12,18,21]. High CXCR3 expression was originally shown to be restricted to activated T lymphocytes [5,22,23] and could be observed in resting T lymphocytes, B lymphocytes, monocytes or granulocytes [20,24]. In contrast, Th2 lymphocytes were reported to produce CCR3, CCR4, and CCR8 [5,12,13,18,25]. However, in other investigations additional expression of CXCR3 was detected in endothelial cells and dendritic cells, as well as in eosinophils within Th1 dominated tissues, including RA synovial tissue [19,26,27]. Thus, CXCR3 expression does not appear to be restricted to activated T lymphocytes, and chemokines may attract more than just T lymphocytes.
Differential expression of CXC chemokines and their receptors has been associated with numerous disease stages [28,29]. In a recent study it was demonstrated that increasing levels of CXCL8 (IL-8) are responsible for activation of neutrophils and T lymphocytes that migrate into the epidermis of arthritis patients. CXCL8 was shown to induce the expression of HLA-DR and to be chemotactic and mitogenic for keratinocytes [30,31]. Another group demonstrated that mRNA levels of the CXCL8 receptors CXCR1 and CXCR2 were 10-fold elevated in injured psoriatic epidermis as compared with normal skin, suggesting a role for high expression of CXCL8 receptors in epidermal hyperplasia, leukocyte infiltration, and increased HLA-DR expression in psoriasis [7,32]. Moreover, it has been shown that increased synthesis of CXCL8 is linked to particular signs and symptoms of RA [33,34].
Chemokines and their receptors probably play important roles in directing the migration of immunocompetent cells to sites of inflammation and in determining the pathohistologic outcome of chronic inflammation and synovial hyperplasia [4,6]. Th1 cytokines such as IFN-γ induced chemokines (e.g. CXCL9 and CXCL10, as well as their receptor CXCR3) are thought to contribute to the documented morphologic and clinical features of RA [35,36].
In the present study, DNA oligonucleotide microarray analysis was performed to search for differentially expressed genes that might represent diagnostic as well as therapeutic markers for pathogenesis and treatment of RA. Transcriptome data, together with our recent observations, that indicated a shift in the Th1/Th2 balance within synovial tissue of RA patients [37] prompted us to validate expression and distribution of selected chemokine receptors, mainly CXCR3, in RA versus osteoarthritis (OA) synovial tissue. Significantly increased levels of CXCR1, CXCR2, and CXCR3 mRNA, as well as highly abundant CXCR1 and CXCR3 protein levels, were found in synovial tissue from RA as compared with that from OA patients. Concomitantly, significantly increased mRNA levels of CXCL9 and CXCL10 were also detected in RA synovial tissue. Our immunohistochemical analysis demonstrated high expression of CXCR3 protein on tissue MCs within rheumatoid synovial tissue samples.
Materials and methods
Patients
Synovial membranes from patients with RA (n = 20) and OA (n = 10) were obtained by synovectomy at the Department of Orthopaedic Surgery, University of Leipzig, Germany. All samples were collected with the approval of the Ethics Board of the University of Leipzig. Clinical, biologic and demographic characteristics of the patients are summarized in Table 1.
Table 1.
Demographic and clinical data for the 20 representative patients included in the study
| Patient number | Age (years) | F/M | Duration of disease (years) | Source of synovial tissue | CRP (mg/l) | RF | DMARDs | NSAIDs | Corticosteroid |
| Rheumatoid arthritis patients | |||||||||
| 1 | 32 | M | 5 | TJR, knee joint left | 82.7 | + | + | - | + |
| 2 | 49 | F | 6 | Expiration baker cystis, knee joint right | 32.5 | + | + | - | + |
| 3 | 73 | M | 10 | TJR, knee joint right | 74.8 | + | - | + | - |
| 4 | 65 | F | 16 | TJR, thigh joint right | 29.5 | ? | + | + | + |
| 5 | 60 | M | 9 | SE, knee joint right | 84.6 | + | - | + | - |
| 6 | 55 | F | 10 | TJR, knee joint right | 62.2 | - | - | + | - |
| 7 | 57 | F | 10 | TJR, knee joint left | 17.4 | + | - | + | + |
| 8 | 55 | M | 10 | TJR, knee joint left | 49.5 | - | - | + | - |
| 9 | 46 | M | 8 | SE, wrist joint left | 15.9 | + | + | - | - |
| 10 | 49 | F | 12 | SE, wrist joint right | 40.7 | + | + | + | - |
| Osteoarthritis patients | |||||||||
| 1 | 52 | M | 2 | SE, knee joint left | 96.0 | - | - | - | - |
| 2 | 31 | F | 5 | SE, knee joint left | 36.0 | + | - | - | - |
| 3 | 37 | M | 1 | SE, knee joint right | <5.0 | - | - | - | - |
| 4 | 70 | M | 5 | TJR, knee joint right | <5.0 | - | - | + | - |
| 5 | 77 | M | 8 | TJR, knee joint right | <5.0 | - | - | + | - |
| 6 | 62 | F | 16 | TJR, knee joint right | <5.0 | - | - | + | - |
| 7 | 74 | F | 20 | TJR, knee joint left | <5.0 | - | - | + | - |
| 8 | 69 | F | 10 | TJR, knee joint right | 12.2 | ? | - | + | - |
| 9 | 71 | F | 10 | TJR, knee joint left | 15.9 | + | - | + | - |
| 10 | 67 | F | 1.5 | TJR, knee joint left | <5.0 | - | - | + | - |
CRP, C-reactive protein; DMARD, disease-modifying antirheumatic drug; F/M, female/male; NSAID, nonsteroidal anti-inflammatory drug; RF, rheumatoid factor; SE, synovectomy; TJR, total joint replacement.
All RA patients had chronic disease of at least 5 years' duration and met the American College of Rheumatology 1987 classification criteria [38]. All had active disease with typical properties (i.e. increased number of infiltrating immunocompetent cells, characteristic number and size of lymphatic follicles, proliferating fibroblasts, and extension of fibrin exudation) [39]. All patients were receiving treatment that included disease-modifying antirheumatic and/or nonsteroidal anti-inflammatory drugs, as well as steroids (Table 1). Diagnosis of OA was based on clinical and radiologic examination, typical symptoms and serologic differences from RA.
All biopsies from RA and OA patients were histopathologically assessed to confirm the clinical diagnosis and to ensure typical pathologic characteristics of RA and OA. Infiltration of T as well as B cells and their organization into lymphatic aggregates and follicular structures were the commonest histopathologic characteristics of synovial tissue from RA patients. In contrast, only a small number of lymphocytes, sometimes with single plasma cells and very small lymphocytic aggregates, lack of fibrin exudation and indications for detritus synovialitis, as well as a mild or higher degree of fibrosis, were the histopathologic hallmarks of synovial tissue from patients with OA. Histologic assessment of RA and OA synovial membranes was conducted by one of the investigators (PS), who has diagnosed more than 2500 synovial tissue samples of RA.
DNA microarray analysis
A global expression analysis of synovial tissue from patients suffering from RA and OA was performed using Affymetrix GeneChip technology (Affymetrix Inc., Santa Clara, CA, USA). Patient material was chosen on the basis of similar patient and disease characteristics. Standardized amounts of total RNA from cryoconserved synovial tissue from either the 10 RA or the 10 OA patients were pooled. The RNA pools were treated, labelled, and hybridized to Affymetrix 5600 HuGeneFL Arrays (Affymetrix Inc.), according to the manufacturer's instructions. Scans of the arrays were evaluated using Affymetrix Microarray Suite 5.0 (Affymetrix Inc.).
RNA isolation and semiquantitative reverse transcription polymerase chain reaction
All synovial tissue samples were obtained directly during the surgical procedure. The tissue material was transferred into liquid nitrogen immediately and stored [40,41]. Total RNA was prepared from 30 mg cryoconserved synovial tissue from each patient using the RNeasy-Mini kit (Qiagen, Hilden, Germany). All RNA samples were subjected to digestion with 1 U DNase I (Life Technologies, Eggenstein, Germany). Quality of all total RNA samples was controlled by a 2100 bioanalyzer according to a RNA 6000 Nano-LabChip Kit procedure (Agilent Technologies, Palo Alto, CA, USA), using 0.3 μg of each total RNA. cDNA was synthesized from 1 μg total RNA in a 20 μl reaction using 200 U Superscript™ II reverse transcriptase (Life Technologies), 500 μmol/l of each deoxynucleotide, 5 mmol/l DTT and 0.5 μg of oligo(dT)15 (Invitek, Berlin, Germany).
Polymerase chain reaction (PCR) was performed using a 20 μl volume with 0.5 U InViTAQ™ DNA polymerase (Invitek), 1 μl single-stranded cDNA, 100 μmol/l dNTPs, 125 nmol/l of each primer (BioTez, Berlin, Germany) in 50 mmol/l Tris-HCl (pH 8.8), 16 mmol/l (NH4)2SO4, 2.5 mmol/l MgCl2, and 0.01% Triton X-100. All PCRs were performed using cDNA samples adjusted to equal glyceraldehyde-3-phosphate dehydrogenase (G3PDH) inputs under conditions that permit exponential accumulation of PCR products. PCR cycle number was chosen after amplification of cDNA derived from samples with the highest concentrations of the gene under study. One cycle consisted of a 30 s denaturation at 94°C, annealing for 30 s at a gene specific temperature (see below), and extension at 72°C for 1 min. Control samples without reverse transcription (RT) input RNA were included in all experiments.
The primer sequence and PCR conditions for IL-6 were 5'-TAG CCG CCC CAC ACA GAC AG-3' and 5'-GGC TGG CAT TTG TGG TTG GG-3', used at 68°C annealing temperature over 36 cycles. CXCR1-specific PCR was done using 38 cycles with the primers 5'-ACA CAG CAA AAT GGC GGA TGG-3' and 5'-CGA TGA AGG CGT AGA TGA TGG-3', at 60°C annealing temperature. The primer pairs 5'-TGG GCA ACA ATA CAG CAA ACT-3' and 5'-GAG CAG GAA GAT GAG GAC GAC-3', at 58°C annealing temperature and for 33 cycles, were used for CXCR2-specific amplification; and 5'-GCT TTG ACC GCT ACC TGA ACA-3' and 5'-GGC CAC CAC GAC CAC CAC CAC-3', at 62°C and for 32 cycles, were used for CXCR3-specific amplification. CXCL9 mRNA was detected after 29 cycles with the primers 5'-GGA GTG CAA GGA ACC CCA GTA-3' and 5'-CTT TTG GCT GAC CTG TTT CTC-3', and CXCL10 mRNA was amplified using 26 cycles with the primers 5'-ATT TGC TGC CTT ATC TTT CTG-3' and 5'-GAC ATC TCT TCT CAC CCT TCT-3', at annealing temperatures of 52°C and 55°C, respectively.
To determine G3PDH levels, G3PDH cDNA was amplified with 27 cycles in the presence of a competitor and the primer pair 5'-GCA GGG GGG AGC CAA AAG GG-3' and 5'-TGC CAG CCC CAG CGT CAA AG-3', at 59°C annealing temperature. The amplified region from the competitor (851 bp) was 285 bp longer than the amplicons derived from G3PDH cDNA samples.
PCR products were separated by electrophoresis on a 1.8% agarose gel. Ethidium bromide-stained agarose gels were subjected to densitometry using the documentation system 1000 (Biorad, Hercules, CA, USA). In order to facilitate comparison of the results obtained from different experiments, mRNA levels were expressed in relative units. Specific mRNA level from each patient is given in arbitrary units representing integrated peak areas (adjusted volumes [counts × mm2]) of amplified cDNA, analyzed by densitometric measurement.
Immunohistochemistry
For immunohistologic analysis of distribution of CXCR1, CXCR2, and CXCR3, synovial tissue from patients with RA and OA was fixed in 4% formaldehyde immediately after surgery and subsequently embedded in paraffin wax. Tissue from patients was cut in 2–5 μm thick sections. Sections were dewaxed with xylol three times for 5 min and hydrated with decreasing concentrations of ethanol (100% for 5 min, 75% for 5 min, and finally aqua destillata for 5 min). Afterward, the slides were treated with 3% H2O2 in phosphate buffered saline (PBS) to quench endogenous peroxidase. For demasking of CXCR1, CXCR2, CD3, and CD68, sections were subjected to three 5-min heating cycles in citrate buffer using a microwave oven at 560 W. Slides stained for prolyl-4-hydroxylase were covered with the same buffer and incubated for 30 min in the microwave oven. Pretreatment for MC tryptase staining involved 5 min incubation with 0.1% pronase (Sigma, St. Louis, MO, USA) in PBS.
All sections were blocked in PBS, 5% goat serum albumin (blocking buffer) for 20 min, and staining was performed with the following primary antibodies at the given dilution in blocking buffer (1 hour, room temperature): mouse monoclonal antibodies against CXCR1 (Clone 42705.111, 1:40; R&D Systems, Minneapolis, MN, USA), CXCR2 (Clone 48311.211, 1:10; R&D), CXCR3 (Clone 49801.111, 1:100; R&D), MC tryptase (Clone AA1, 1:50; Dako, Hamburg, Germany), CD68 (Clone KP1, 1:80; Dako), fibroblast prolyl-4-hydroxylase (Clone 5B5, 1:10; Dako), and CD3 (Clone F7.2.38, 1:50; Dako). After four washes of 10 min each with PBS, secondary reagents were applied for 30 min at room temperature. Primary antibodies were detected in general using a biotinylated goat antimouse IgG (Biogenex, San Ramon, CA, USA). After extensive washing in PBS as above, sections were incubated with peroxidase-conjugated streptavidin for 30 min at room temperature. Antigen-antibody complexes were visualized by incubation with substrate solution containing 0.5 mg/ml 3-amino-9-ethylcarbazole (Sigma) and 3% H2O2 in 0.1 mol/l sodium acetate buffer pH 5.2 for 5 min at room temperature. Subsequently, the slides were rinsed in distilled water, counterstained with Mayer's hematoxylin (Merck, Darmstadt, Germany), and mounted in Aquatex (Merck). In order to identify the cell type of CXCR-positive cells, serial sectioning was performed and subsequent sections were stained for the particular CXCR proteins and the cell type marker. Antibody staining specificity was verified using isotype controls. CXCR3 antibody was confirmed using IgG1 isotype matched control (Sigma). The slides were examined and scored independently by two of us (PR, PS) without knowledge of the clinical and pathologic data for the particular sample.
Western blotting
Protein levels of CXCR1, CXCR2, and CXCR3 in RA versus OA synovial tissue were examined by Western blotting of tissue extracts. Extracts were obtained using Mem-PER® mammalian membrane protein extraction kit (Pierce, Rockford, IL, USA), as detailed in the manufacturer's protocol. Protein concentrations were determined using the DC protein assay (Biorad). Each sample, equivalent to 10 μg total protein, was separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequently transferred to Hybond-N nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, USA) by standard procedures. The blotting membrane was blocked for 2 hours with PBS, 6% nonfat milk powder, 0.1% Tween (for CXCR1 staining) or TBS, 1% bovine serum albumin, and 0.05% Tween (for CXCR2 and CXCR3). The primary antibodies against CXCR1, CXCR2, and CXCR3 were the same as above and used at 1:100 (CXCR1 and CXCR2) and 1:80 (CXCR3) in the respective blocking buffer at 4°C overnight. To assess equal loading of protein lysate for each sample, a parallel blot was incubated with an anti-β-actin antibody (Clone AC-15, 1:50000; Sigma). Bound primary antibodies were detected using biotinylated goat antimouse IgG secondary antibody (Dako) and subsequently incubated with streptavidin-conjugated peroxidase (Dako), each for 1 hour at room temperature. After each incubation, blots were washed with PBS–Tween 0.05%. Signals were developed with ECL chemiluminescence reagent and recorded on Hyperfilm™-ECL™(Amersham Biosciences). The signals were subjected to densitometric measurements using the Chemi Doc system (Biorad).
Statistical analysis
Statistically significant differences were determined by the Student's t-test and Mann–Whitney rank sum test as indicated in the figure legends. P < 0.05 was considered statistically significant. The analysis was conducted using SigmaStat for Windows 2.0 (Jandel Cooperation Inc., San Rafael, CA, USA).
Results
CXCR mRNA expression
To unravel disease-specific differences that are characteristic for synovial tissue from patients with RA versus OA disease, total RNA from 30 mg synovial tissue was isolated. Quality of all samples was controlled in a 2100 bioanalyzer (Fig. 1). In the first pilot experiment we used Affymetrix HuGene FL DNA oligonucleotide microarrays (7131 gene sets) and two pools of RNAs from 10 patients, each with RA or OA disease. In total, 101 genes were found to be elevated whereas 300 genes were decreased in RA in comparison with OA (data not shown). This initial experiment showed that levels of the IFN-γ inducible chemokine receptor CXCR3 and of its ligands CXCL9 and CXCL10 are strongly upregulated in RA as compared with OA (Table 2). CXCR3 exhibited 2.3-fold, CXCL9 4.6-fold, and CXCL10 9.8-fold increased levels in RA samples. Signals on the chip for the related chemokine receptors CXCR1 (IL-8 receptor α) and CXCR2 (IL-8 receptor β) were either scored as absent in both situations or scored as not changed.
Figure 1.
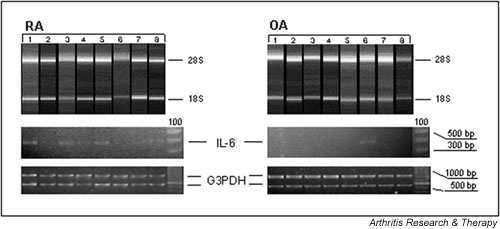
Analysis of IL-6 mRNA levels within synovial tissue from rheumatoid arthritis (RA) as compared with that from osteoarthritis (OA) patients. Upper panels: quality control of total RNA preparations. Aliquots (300 ng) of total RNA extracted from synovial tissue from RA and OA patients were plotted on a RNA 6000 Nano-LabChip. Quality of RNA was scanned using a 2100 bioanalyzer. RNA gel electropherograms show the presence of 28S and 18S ribosomal units, indicating intact RNA of the investigated samples. Lower panels: differential IL-6 mRNA levels were determined by semiquantitative reverse transcription polymerase chain reaction (PCR). The figure shows a representative analysis of eight cDNA samples derived from patients with RA and of eight cDNA samples from patients with OA. cDNA samples were adjusted to equal glyceraldehyde-3-phosphate dehydrogenase (G3PDH) levels, performed by competitive PCR using an internal standard (see Materials and methods). Numbered lanes correspond to individual patients within Table 1.
Table 2.
Selected RNA profiling data
| Accession number | Gene | Signal OA chip | Detection OA chip | Signal RA chip | Detection RA chip | Signal log ratio | Fold change | Change | P (for change) |
| U11870 | CXCR1 | 119.6 | A | 163.5 | A | 0.5 | NA | NC | 0.5 |
| U11872 | CXCR1splice variant | 180.7 | A | 232.5 | A | -0.0 | NA | NC | 0.5 |
| L19593 | CXCR2 | 34.9 | P | 41.3 | A | -0.2 | NA | NC | 0.5 |
| X95876 | CXCR3 | 478.6 | A | 1295.6 | P | 1.2 | 2.3 | I | 0.000051 |
| X72755 | CXCL9 (Mig) | 177.5 | P | 1988.1 | P | 3.3 | 9.8 | I | <0.000001 |
| X02530 | CXCL10 (IP-10) | 189.3 | P | 656.6 | P | 2.2 | 4.6 | I | <0.000001 |
| J04132 | TCR-ζ (CD247) | 146.3 | P | 345 | P | 1.5 | 2.8 | I | 0.000133 |
RNA pools from patients suffering from rheumatoid arthritis (RA) or osteoarthritis (OA) were analyzed using Affymetrix HuGeneFL microarrays. Data assessment was done using Affymetrix Microarray Suite 5.0. CXCL, Cys–X–Cys ligand; CXCR, Cys–X–Cys receptor; NA, not applicable; TCR, T-cell receptor.
Because pooled samples may sometimes produce obscure findings and PCR-based methods are known to be more sensitive than the Affymetrix gene chip technology, semiquantitative RT-PCR was introduced to validate Affymetrix-derived mRNA expression levels in individual patient samples (RA, n = 20; OA, n = 10). First, IL-6 mRNA levels were quantified to provide a positive control for unregulated gene expression in RA versus OA. As expected, levels of IL-6 transcript were significantly higher in RA samples than in those derived from OA synovial tissue, which apparently did not exhibit detectable IL-6 transcripts (Fig. 1). Then, mRNA levels of chemokine receptors were investigated. RT-PCR revealed increased CXCR3 mRNA levels (P < 0.001) in RA as compared with OA synovial tissue (Fig. 2a). This an increase of 3.6-fold in CXCR3 transcript levels was found in synovial tissue of RA patients (Fig. 2a,2b). Similarly, levels of CXCR1 and CXCR2 transcripts were increased by 10-fold (P < 0.05) and approximately sixfold (P < 0.05) in RA versus OA synovial samples (Fig. 2b), respectively. RT-PCR analyses for the CXCR3 ligands CXCL9 and CXCL10 revealed large increases (i.e. 135-fold [P < 0.001] and 340-fold [P < 0.05], respectively) in RA as compared with OA synovial tissue (Fig. 2b). Altogether, we confirmed that the chemokine receptors CXCR1, CXCR2 and CXCR3, as well as the CXCR3 ligands CXCL9 and CXCL10, are more abundantly expressed at the mRNA level in RA synovial tissue than in OA synovial tissue.
Figure 2.
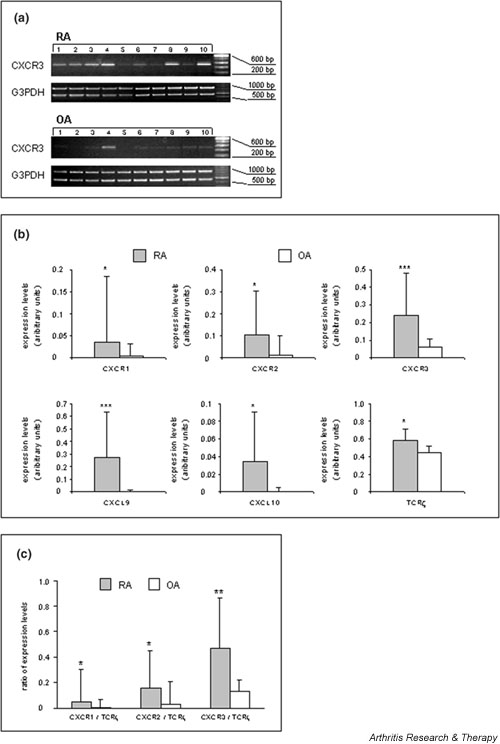
Analysis of mRNA levels of selected genes in synovial tissue from rheumatoid arthritis (RA) as compared to that from osteoarthritis (OA) patients by semiquantitative reverse transcription polymerase chain reaction (RT-PCR). Bars represent means ± SD of signal intensities after amplification of samples (see Materials and methods). The data from one representative experiment with one determination per patient sample are shown. Differences between RA and OA sample groups were statistically evaluated using the Student's t-test (*P < 0.05, **P < 0.01, ***P < 0.001). (a) RT-PCR analysis of 10 cDNA samples derived from patients with RA and of 10 cDNA samples from patients with OA. cDNA samples were adjusted to equal glyceraldehyde-3-phosphate dehydrogenase (G3PDH) levels, performed by competitive PCR using an internal standard (see Materials and methods). Numbered lanes correspond to individual patients within Table 1. (b) Quantitation of the expression of Cys–X–Cys receptor (CXCR)1, CXCR2, CXCR3, T-cell receptor (TCR)-ζ, Cys–X–Cys ligand (CXCL)9, and CXCL10 mRNAs in RA and OA synovial tissues. (c) CXCR/TCR-ζ mRNA ratios in RA versus OA synovial tissues.
It was previously found that T cells are present in approximately 50% of RA synovial tissue [42]. According to our own observations, nearly 20% T cells in the synovial tissue of RA patients can be readily demonstrated (data not shown). In order to consider the degree of differential infiltration of T lymphocytes as well as their influence on inflammation-induced CXCR3 expression between RA and OA, we analyzed the expression of TCR-ζ (CD247). DNA microarray data (Table 2) and RT-PCR experiments in individual patient samples (Fig. 2b) clearly corroborated higher levels of TCR-ζ transcripts within the RA than in the OA samples. However, calculation of ratios between the respective mean CXCR mRNA and the mean TCR-ζ mRNA levels of each disease group revealed higher values for the three analyzed CXCR transcripts in the RA synovial tissue (CXCR1, P < 0.05; CXCR2, P < 0.05; CXCR3, P < 0.01), suggesting higher CXCR expression levels in non-T cells in RA synovial tissue (Fig. 2c).
Evaluation of CXCR3 protein expression
To confirm the increase in CXCR3 expression at the protein level, Western blot experiments in selected extracts from synovial tissue of RA and OA patients were conducted (Fig. 3a). Staining for CXCR1 (P < 0.05) and CXCR3 (P < 0.01) revealed a higher level of expression for each protein in RA than in OA synovial tissue (Fig. 3b). CXCR2 protein levels were rather low, and signals were not significantly different between the two disease situations. Thus, in agreement with differential mRNA expression, CXCR1 and CXCR3 proteins were expressed in synovial tissue from patients with RA at higher levels than in tissues from patients with OA.
Figure 3.
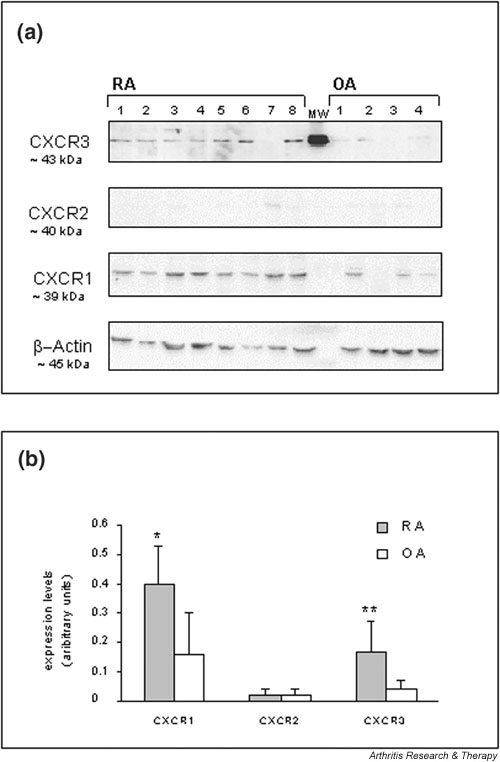
Western blot analysis of Cys–X–Cys receptor (CXCR)1, CXCR2, and CXCR3 protein expression in selected rheumatoid arthritis (RA) and osteoarthritis (OA) synovial tissues. (a) Tissue extracts from RA (n = 8) and from OA patients (n = 4) were analyzed. Numbered lanes correspond to individual patients within Table 1. Staining of the indicated proteins on parallel blots is shown. Equal loading of tissue extracts was controlled by β-actin protein staining. MW indicates a protein from ECL molecular weight markers. (b) Western blot signals on Hyperfilm™ ECL™ after the chemiluminescence reactions were analyzed semiquantitatively using densitometric scanning. Expression is given in arbitrary units and the means ± SD of the RA and OA groups are plotted. Differences between RA and OA groups were assessed statistically using the Student's t-test (*P < 0.05, **P < 0.01).
Distribution and cellular assignment of CXCR1, CXCR2, and CXCR3 to different cellular subsets in RA and OA tissues
Initial immunohistochemical analyses revealed over-expression of IL-6 protein within RA tissue sections (data not shown). Next, we investigated cellular distribution of the CXCR1, CXCR2, and CXCR3 proteins. Among the RA synovial tissue samples examined for CXCR1, CXCR2, and CXCR3 immunoreactivity, 8 out of 20 specimens exhibited heterogeneous histologic changes in terms of inflammatory infiltration in sublining regions. Twelve samples showed a high number of infiltrating lymphocytes as well as macrophages, and exhibited a destroyed synovial intima, including fibrin exudation. All RA synovial tissue samples exhibited medium to strong CXCR1 as well as CXCR3 immunoreactivity. In contrast, signals for CXCR2 were undetectable in all RA synovial tissue samples.
CXCR1+ and CXCR3+ cells varied from region to region and from patient to patient (ranging from 20% to 60%) and were assigned to specific cellular subsets by differential antibody staining of sequential sections. The CXCR1 protein was weakly expressed on CD68+ macrophages in a diffuse manner and showed a consistent distribution pattern within all sections of RA patients (data not shown). Unexpectedly, in all samples inspected prominent staining for CXCR3 was found on scattered MCs within sublining layers and interstitial areas, as well as in perivascular compartments of the rheumatoid synovial tissue (Fig. 4). In agreement with earlier reports, CXCR3 protein was also observed on CD3+ T lymphocytes (data not shown). Strong staining of MCs suggested a high density of CXCR3 antigen expression. Longer color development during immunohistochemical staining revealed weak and more diffuse signals for CXCR3 protein, appearing in all areas of the rheumatoid tissue. By sequential sectioning, these signals could be attributed to synovial fibroblasts, identified by an antibody against prolyl-4-hydroxylase (data not shown). In 10 OA samples examined, there was staining for CXCR1 protein on a few macrophages within subintimal regions of OA synovial tissue and a subset of resident mononuclear phagocytes (synovial macrophages or histocytes) in all areas of synovial tissue. Signals for CXCR3 protein were low and diffuse and could be assigned to synovial fibroblasts – but not to tissue MCs – in a wide range of sublining compartments (data not shown).
Figure 4.
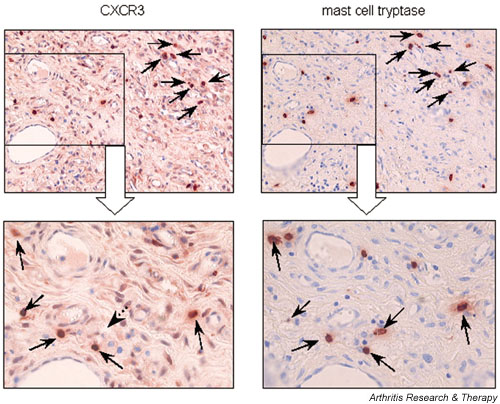
Cellular distribution of Cys–X–Cys receptor (CXCR)3 protein in synovial tissue from rheumatoid arthritis (RA) patients. Localization of strong CXCR3 protein signals in mast cells within the sublining areas of rheumatoid synovial tissues was found. Sequential sections of paraffin-embedded tissue were stained for CXCR3 and mast cell tryptase proteins or using an IgG1 isotype-matched control. Each arrow refers the same cell that was positively stained for CXCR3 and mast cell tryptase (original magnification: upper panel × 200; lower panel × 400).
Discussion
Using differential display of gene expression by microarray analysis, one set of 101 upregulated RA-related genes and one set of 300 gene transcripts considered to be downregulated in RA were detected and are now available for further research.
A comparative analysis of synovial tissue pools from RA versus OA patients and our earlier studies on Th1/Th2 balance in RA [37] prompted us to validate and to confirm the expression of chemokines and their receptors in RA versus OA synovial tissue.
Our initial experiments revealed higher levels of chemokine ligand (CXCL9, CXCL10) and receptor (CXCR1, CXCR2, CXCR3) mRNAs in RA than in OA synovial tissue. Similar to other diseases [12,18], high expression of CXCR3 suggests the presence of an inflammatory trigger and of chemotactic recruitment of T-cell subsets to the sites of inflammation in RA. Because activated CD3+ T cells have been found to be the major cell type expressing chemokine receptors, the increase in CXCR3 expression could be due, at least in part, to higher levels of T cells in RA than in OA synovial tissue samples [4,22]. There is an established relationship between joint-specific manifestations of RA and recruitment of leukocytes derived from the blood in response to chemokines [5,6,20]. In comparison with OA, more pronounced T cell infiltration can be observed in RA synovial tissue [43]. Therefore, the present study showed significantly increased expression of TCR-ζ mRNA in RA as compared with OA tissues. However, CXCR3/TCR-ζ mRNA ratio was higher in RA than in OA. Although CXCR3 expression was previously demonstrated in synovial tissue of RA patients, high CXCR3 mRNA levels in synovial MCs has not yet been described [5,17]. Increased CXCR3 mRNA expression within synovial tissue from RA versus OA patients is reflected by higher CXCR3/TCR-ζ mRNA ratios and is apparently associated with high CXCR3 mRNA levels on MCs within RA synovial tissue.
At the protein level, we observed abundant expression of CXCR1 and CXCR3 in RA synovial tissue. Thus, we identified CXCR1 protein expression on synovial macrophages in RA as well as in OA patients. In this respect, our report confirms increased CXCR1 protein expression on synovial macrophages, which has been considered to cause a chemotactic influx of mononuclear cells into RA synovial tissue in response to CXCL8 (IL-8) [33,34].
The most exciting observation was the strong CXCR3 protein expression on tissue MCs in RA synovial tissue. These data indicate that increasing CXCR3 protein levels are most likely due to enhanced recruitment of MCs that express CXCR3 in RA synovial tissue. To our knowledge, this is the first report to demonstrate expression of CXCR3 in MCs within synovial tissue of RA patients. Additional expression of CXCR3 protein on synovial fibroblasts in both RA and OA points possibly to an increased level of activation among these cells. The chemokine receptor CXCR3 was previously found to be strongly expressed on activated T lymphocytes, exhibiting lower or no detectable expression in resting T cells, B cells, monocytes, or granulocytes [6]. Other authors assigned CXCR3 and CCR5 proteins predominantly to Th1 lymphocytes, whereas Th2 lymphocytes produced CCR3 and CCR4 [12,13,18,26]. In RA, CXCR3 expression was also found to be restricted to lymphocytic cells in perivascular inflammatory infiltrates within active lesions of synovial tissue [5,20,25]. The ligands of CXCR3 (CXCL9 and CXCL10) do not chemotactically attract granulocytes, but appear to promote T-cell adhesion to endothelial cells [44]. A recent report by Qin and coworkers [5] showed that more than 80% of perivascular T lymphocytes within rheumatoid synovial tissue were immunoreactive for CXCR3. Disparity in findings may arise from study of various stages and different histopathologic subtypes of RA [1,2,36,38].
Similar to another report that implicated recruitment of eosinophils via CXCR3 [28], we suggest that MC precursors are recruited to sites of inflammation through CXCR3 by chemoattractants. Indeed, apart from macrophages, lymphocytes, fibroblasts and neutrophils, which are considered to be important contributors to the pathogenesis of RA, increased numbers of MCs are found in the synovial tissue and synovial fluid of RA patients [44,45]. MC-associated CXCR3 expression may indicate that additional mechanism exist that result in an amplified proinflammatory stimulus, by secretion of proteinases, chemotactic factors, and vasoactive material [46]. The contributions made by MCs to the events of inflammation and degradation of extracellular matrix were recently pointed out [47]. Interestingly, the zymogen forms of the matrix metalloproteinases prostromelysin and procollagenase are activated by specific MC subsets that either express tryptase (MCT) or tryptase and chymase (MCTC) [48,49]. Distinct functional differences between these MC subsets are reflected by differential expression of IL-4, IL-5 and IL-6 in MCT, and IL-4 in MCTC, which can also be observed in rheumatic tissue [50]. The cytokine profile expressed by different MC subsets, including the proinflammatory mediators tumor necrosis factor-α and IL-1β [46,51], fits well into our model of active recruitment of MC precursors into rheumatoid lesions via CXCR3 [52].
MCs mature from circulating CD34+, c-kit+, and CD13+progenitors after moving into peripheral tissues [35,53,54]. It is likely that MC precursors can also be recruited to sites of inflammation through their additional CXCR3 surface expression and support the characteristic features of RA. The impact on inflammatory and erosive arthritis by MCs was recently demonstrated in an animal model [55]. There was no evidence for arthritis in one MC-deficient mice strain (W/WV) after arthritogenic serum was transferred from K/B×N mice, although control mice exhibited all of the clinical and histological features of inflammatory and erosive arthritis. A hallmark of MC activation in the effector phases of inflammatory arthritis included degranulation (release of histamine, proteases, tumor necrosis factor-α and IL-1) in synovial tissue but not in other tissues. The authors concluded further that tissue MCs exhibit a synovial tissue-specific role, and that they represent a cellular link between soluble mediators and both erosive and degenerative events in inflammatory arthritis. In this context, the functionality of chemokine receptors was shown by the decreased recruitment/migration of CXCR3-expressing mononuclear cells, including MCs, after treatment with self-specific anti-CXCL10 and antimurine CXCR3 in animal models [56,57]. Antimurine CXCR3 treatment within a collagen-induced arthritis mouse model should be a valid model with which to analyze the recruitment/migration of inflammatory MCs in RA [58].
Our observations suggest that the proinflammatory character of RA is mediated through continuous recruitment and activation and/or presence of various immunocompetent cells, including tissue MCs.
The present study suggests that Th1-associated CXCR3 expression in synovial tissue is associated with distinct biologic functions of MCs in RA. It appears that the actions of CXCL9 and CXCL10 are not restricted to promoting recruitment of activated T lymphocytes and their migration to sites of inflammation, but that they may also serve to recruit MC precursors into rheumatoid synovial tissue. Finally, we suggest that either vessel-derived MC precursors express CXCR3 a priori and become recruited to sites of inflammation, or that mature tissue MCs become activated within RA synovial tissue and upregulate CXCR3 secondarily in response to signals from the proinflammatory trigger. Activated MCs are characterized by degranulation of inflammatory and proteolytic molecules (histamine, proteases, tumor necrosis factor-α) and thus might represent an effector cell subset for degradation and destruction in RA synovial tissue.
Conclusion
Microarray analysis is a valuable tool with which to detect differential expression of genes in RA and OA. One gene whose expression is increased in RA synovial tissue encodes the chemokine receptor CXCR3. Importantly, the CXCR3 ligands CXCL9 and CXCL10 are also upregulated in RA. Tissue MCs are largely responsible for CXCR3 expression. We propose a novel regulatory aspect of joint destruction comprising MCs that transmit the effects of soluble cytokines, including chemokines. Thus, MCs may represent a new target for therapeutic intervention in RA.
Competing interests
None declared.
Abbreviations
CXCL = Cys–X–Cys ligand; CXCR = Cys–X–Cys receptor; G3PDH = glyceraldehyde-3-phosphate dehydrogenase; IFN = interferon; IL = interleukin; MC = mast cell; OA = osteoarthritis; PBS = phosphate buffered saline; PCR = polymerase chain reaction; RA = rheumatoid arthritis; RT = reverse transcription; TCR = T-cell receptor; Th = T-helper (cell).
Acknowledgments
Acknowledgements
The present study was performed as part of the 'BMBF-Leitprojekt Molekulare Medizin: Proteomanalyse des Menschen' initiative supported by the German government (Bundesministerium für Forschung und Technologie, 'FKZ: 01GG9835/4'). We thank Dr G Aust for the IL-6 primers. We thank Mrs A Gronemann for skilled technical assistance.
References
- Gay S, Gay RE, Koopman WJ. Molecular and cellular mechanisms of joint destruction in rheumatoid arthritis: two cellular mechanisms explain joint destruction? Ann Rheum Dis. 1993;52:39–47. doi: 10.1136/ard.52.suppl_1.s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Ladner U. Molecular and cellular interactions in rheumatoid synovium. Curr Opin Rheumatol. 1996;8:210–220. doi: 10.1097/00002281-199605000-00008. [DOI] [PubMed] [Google Scholar]
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Kunkel SL. Through the looking glass: the diverse in vivo activities of chemokines. J Clin Invest. 1999;104:1333–1334. doi: 10.1172/JCI8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;95:3032–3043. [PubMed] [Google Scholar]
- Murray K, Thompson SD, Glass DN. Pathogenesis of juvenile chronic arthritis: genetic and environmental factors. Arch Dis Child. 1997;77:530–534. doi: 10.1136/adc.77.6.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzalis C, Kingsley G, Haskard D, Panayi G. The preferential accumulation of helper-inducer T lymphocytes in inflammatory lesions: evidence for regulation by selective endothelial and homotypic adhesion. Eur J Immunol. 1988;18:1397–1404. doi: 10.1002/eji.1830180915. [DOI] [PubMed] [Google Scholar]
- Godfrey HP, Ilardi C, Engber W, Graziano FM. Quantitation of human synovial mast cells in rheumatoid arthritis and other rheumatic diseases. Arthritis Rheum. 1984;27:852–856. doi: 10.1002/art.1780270803. [DOI] [PubMed] [Google Scholar]
- Okada Y. The mast cells in synovial membrane of patients with joint disease. Japanese J Ortho Surg. 1973;47:657–674. [Google Scholar]
- Austrup F, Vestweber D, Borges E, Lohning M, Brauer R, Herz U, Renz H, Hallmann R, Scheffold A, Radbruch A, Hamann A. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature. 1997;385:81–83. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- Siveke JT, Hamann A. T helper 1 and T helper 2 cells respond differentially to chemokines. J Immunol. 1998;160:550–554. [PubMed] [Google Scholar]
- Kelner GS, Kennedy J, Bacon KB, Kleyensteuber S, Largaespada DA, Jenkins NA, Copeland NG, Bazan JF, Moore KW, Schall TJ, et al. Lymphotactin: a cytokine that represents a new class of chemokine. Science. 1994;266:1395–1399. doi: 10.1126/science.7973732. [DOI] [PubMed] [Google Scholar]
- Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10) J Exp Med. 1987;166:1084–1097. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Nakajima A, Yoshino S, Matsushima K, Yagita H, Okumura K. Selective accumulation of CCR5+ T lymphocytes into inflamed joints of rheumatoid arthritis. Int Immunol. 1999;11:553–559. doi: 10.1093/intimm/11.4.553. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187:875–883. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedderburn LR, Robinson N, Patel A, Varsani H, Woo P. Selective recruitment of polarized T cells expressing CCR5 and CXCR3 to the inflamed joints of children with juvenile idiopathic arthritis. Arthritis Rheum. 2000;43:765–774. doi: 10.1002/1529-0131(200004)43:4<765::AID-ANR7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28:3696–3705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Farber JM. HuMig: a new human member of the chemokine family of cytokines. Biochem Biophys Res Commun. 1993;192:223–230. doi: 10.1006/bbrc.1993.1403. [DOI] [PubMed] [Google Scholar]
- Piali L, Weber C, LaRosa G, Mackay CR, Springer TA, Clark-Lewis I, Moser B. The chemokine receptor CXCR3 mediates rapid and shear-resistant adhesion-induction of effector T lymphocytes by the chemokines IP10 and Mig. Eur J Immunol. 1998;28:961–972. doi: 10.1002/(SICI)1521-4141(199803)28:03<961::AID-IMMU961>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Ruth JH, Rottman JB, Katschke KJ, Jr, Qin S, Wu L, LaRosa G, Ponath P, Pope RM, Koch AE. Selective lymphocyte chemokine receptor expression in the rheumatoid joint. Arthritis Rheum. 2001;44:2750–2760. doi: 10.1002/1529-0131(200112)44:12<2750::aid-art462>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Imai T, Nagira M, Takagi S, Kakizaki M, Nishimura M, Wang J, Gray PW, Matsushima K, Yoshie O. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int Immunol. 1999;11:81–88. doi: 10.1093/intimm/11.1.81. [DOI] [PubMed] [Google Scholar]
- Garcia-Lopez MA, Sanchez-Madrid F, Rodriguez-Frade JM, Mellado M, Acevedo A, Garcia MI, Albar JP, Martinez C, Marazuela M. CXCR3 chemokine receptor distribution in normal and inflamed tissues: expression on activated lymphocytes, endothelial cells, and dendritic cells. Lab Invest. 2001;81:409–418. doi: 10.1038/labinvest.3780248. [DOI] [PubMed] [Google Scholar]
- Jinquan T, Jing C, Jacobi HH, Reimert CM, Millner A, Quan S, Hansen JB, Dissing S, Malling HJ, Skov PS, Poulsen LK. CXCR3 expression and activation of eosinophils: role of IFN-gamma-inducible protein-10 and monokine induced by IFN-gamma. J Immunol. 2000;165:1548–1556. doi: 10.4049/jimmunol.165.3.1548. [DOI] [PubMed] [Google Scholar]
- Kunkel SL, Lukacs N, Kasama T, Strieter RM. The role of chemokines in inflammatory joint disease. J Leukoc Biol. 1996;59:6–12. doi: 10.1002/jlb.59.1.6. [DOI] [PubMed] [Google Scholar]
- Lukacs NW, Kunkel SL. Chemokines and their role in disease. Int J Clin Lab Res. 1998;28:91–95. doi: 10.1007/s005990050025. [DOI] [PubMed] [Google Scholar]
- Tuschil A, Lam C, Haslberger A, Lindley I. Interleukin-8 stimulates calcium transients and promotes epidermal cell proliferation. J Invest Dermatol. 1992;99:294–298. doi: 10.1111/1523-1747.ep12616634. [DOI] [PubMed] [Google Scholar]
- Schonbeck U, Brandt E, Petersen F, Flad HD, Loppnow H. IL-8 specifically binds to endothelial but not to smooth muscle cells. J Immunol. 1995;154:2375–2383. [PubMed] [Google Scholar]
- Schulz BS, Michel G, Wagner S, Suss R, Beetz A, Peter RU, Kemeny L, Ruzicka T. Increased expression of epidermal IL-8 receptor in psoriasis. Down-regulation by FK-506 in vitro. J Immunol. 1993;151:4399–4406. [PubMed] [Google Scholar]
- Kraan MC, Patel DD, Haringman JJ, Smith MD, Weedon H, Ahern MJ, Breedveld FC, Tak PP. The development of clinical signs of rheumatoid synovial inflammation is associated with increased synthesis of the chemokine CXCL8 (interleukin-8) Arthritis Res. 2001;3:65–71. doi: 10.1186/ar141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- Dolhain RJ, ter Haar NT, Hoefakker S, Tak PP, de Ley M, Claassen E, Breedveld FC, Miltenburg AM. Increased expression of interferon (IFN)-gamma together with IFN-gamma receptor in the rheumatoid synovial membrane compared with synovium of patients with osteoarthritis. Br J Rheumatol. 1996;35:24–32. doi: 10.1093/rheumatology/35.1.24. [DOI] [PubMed] [Google Scholar]
- Stiehl P. Histologie der Rheumatoid-Arthritis. Beitrag zur diagnostischen und pathogenetischen Heterogenität, zur Aktivitätsdiagnose und Prognose. In: Sack U, editor. Arthritiden. Vol. 5. Lengerich: Pabst Science Publishers; 1997. pp. 188–200. [Google Scholar]
- Ruschpler P, Stiehl P. Shift in Th1 (IL-2 and IFN-gamma) and Th2 (IL-10 and IL-4) cytokine mRNA balance within two new histological main-types of rheumatoid-arthritis (RA) Cell Mol Biol. 2002;48:285–293. [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Geiler G, Stiehl P. Significance of the concepts basic activity and actual activity in the morphological evaluation of synovial membranes in rheumatoid arthritis. Z Rheumatol. 1974;33:73–86. [PubMed] [Google Scholar]
- Heller RA, Schena M, Chai A, Shalon D, Bedilion T, Gilmore J, Woolley DE, Davis RW. Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci USA. 1997;94:2150–2155. doi: 10.1073/pnas.94.6.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotzer MA, Patti R, Geoerger B, Eggert A, Chou TT, Phillips PC. Biological stability of RNA isolated from RNAlater-treated brain tumor and neuroblastoma xenografts. Med Pediatr Oncol. 2000;34:438–442. doi: 10.1002/(sici)1096-911x(200006)34:6<438::aid-mpo12>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Konttinen YT, Reitamo S, Ranki A, Hayry P, Kankaanapaa U, Wegelius O. Characterization of the immunocompetent cells of rheumatoid synovium from tissue sections and eluates. Arthritis Rheum. 1981;24:71–79. doi: 10.1002/art.1780240112. [DOI] [PubMed] [Google Scholar]
- Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetlow LC, Woolley DE. Distribution, activation and tryptase/ chymase phenotype of mast cells in the rheumatoid lesion. Ann Rheum Dis. 1995;54:549–555. doi: 10.1136/ard.54.7.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber BL, Kaplan AP. Mast cells and rheumatic diseases. In: McCarty DJ, Koopmann WJ, editor. Arthritis and Allied Conditions. Philadelphia: Lea & Febiger; 1993. pp. 417–429. [Google Scholar]
- Galli SJ. New concepts about the mast cell. N Engl J Med. 1993;328:257–265. doi: 10.1056/NEJM199301283280408. [DOI] [PubMed] [Google Scholar]
- Tetlow LC, Woolley DE. Mast cells, cytokines, and metalloproteinases at the rheumatoid lesion: dual immunolocalisation studies. Ann Rheum Dis. 1995;54:896–903. doi: 10.1136/ard.54.11.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees M, Taylor DJ, Woolley DE. Mast cell proteinases activate precursor forms of collagenase and stromelysin, but not of gelatinases A and B. Eur J Biochem. 1994;223:171–177. doi: 10.1111/j.1432-1033.1994.tb18980.x. [DOI] [PubMed] [Google Scholar]
- Saarinen J, Kalkkinen N, Welgus HG, Kovanen PT. Activation of human interstitial procollagenase through direct cleavage of the Leu83-Thr84 bond by mast cell chymase. J Biol Chem. 1994;269:18134–18140. [PubMed] [Google Scholar]
- Gotis-Graham I, McNeil HP. Mast cell responses in rheumatoid synovium. Association of the MCTC subset with matrix turnover and clinical progression. Arthritis Rheum. 1997;40:479–489. doi: 10.1002/art.1780400314. [DOI] [PubMed] [Google Scholar]
- Woolley DE. Mast cells in rheumatoid lesion: ringleaders or innocent bystanders? Ann Rheum Dis. 1995;54:533–534. doi: 10.1136/ard.54.7.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Tedla N, Lloyd AR, Wakefield D, McNeil PH. Mast cell activation and migration to lymph nodes during induction of an immune response in mice. J Clin Invest. 1998;102:1617–1626. doi: 10.1172/JCI3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. 1978;52:447–452. [PubMed] [Google Scholar]
- Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13) Blood. 1999;94:2333–2342. [PubMed] [Google Scholar]
- Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- Belperio JA, Keane MP, Burdick MD, Lynch JP, 3rd, Xue YY, Li K, Ross DJ, Strieter RM. Critical role for CXCR3 chemokine biology in the pathogenesis of bronchiolitis obliterans syndrome. J Immunol. 2002;169:1037–1049. doi: 10.4049/jimmunol.169.2.1037. [DOI] [PubMed] [Google Scholar]
- Salomon I, Netzer N, Wildbaum G, Schif-Zuck S, Maor G, Karin N. Targeting the function of IFN-gamma-inducible protein 10 suppresses ongoing adjuvant arthritis. J Immunol. 2002;169:2685–2693. doi: 10.4049/jimmunol.169.5.2685. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Okunishi H. Mast cells as a target of rheumatoid arthritis treatment. Jpn J Pharmacol. 2002;90:7–11. doi: 10.1254/jjp.90.7. [DOI] [PubMed] [Google Scholar]