

Abstract
Rheumatoid arthritis (RA), like many other autoimmune syndromes, is a disease of adults, with the highest incidence rates reported in the elderly. The immune system undergoes profound changes with advancing age that are beginning to be understood and that need to be incorporated into the pathogenetic models of RA. The age-related decline in thymic function causes extensive remodeling of the T-cell system. Age-dependent changes in T-cell homeostasis are accelerated in patients with RA. The repertoire of naive and memory T cells is less diverse, possibly as a result of thymic insufficiency, and it is biased towards autoreactive cells. Presenescent T cells emerge that are resistant to apoptosis and that often expand to large clonal populations. These cells are under the regulatory control of nonconventional costimulatory molecules, display potent effector functions, and appear to be critical in the synovial and extra-articular manifestations of RA.
Keywords: costimulation, immunosenescence, pathogenesis, rheumatoid arthritis, T-cell homeostasis
Introduction
During thymic development, large arrays of clonally distributed α–β TCRs are generated that mediate the recognition of foreign peptides in the context of the appropriate MHC molecule. The theoretical diversity of the TCR repertoire is between 1015 and 1018 TCRs [1]. Thymic selection mechanisms impose significant restrictions on this diversity [2]; however, the resulting functional TCR repertoire is still extensive. Arstila and colleagues [3] have estimated that the functional T-cell repertoire in the human adult is composed of > 2 × 106 different TCR β-chains, each of which may combine with > 100 TCR α-chains. Wagner and colleagues [4] established even higher estimates of 2 × 107 different TCR β-chains in the naive T-cell compartment of young human adults. Given that the human body harbors ~ 1011 T cells, these estimates imply that each naive T cell has a clonal size of 100–1000 cells (Table 1).
Table 1.
Parameters of T-cell homeostasis in humans
| T-cell population | ||||
| CD4+ naive | CD4+ memory | CD8+ naive* | CD8+ memory* | |
| Pool size (n) | ~ 1 × 1011 | ~ 1 × 1011 | ~ 5 × 1010 | ~ 5 × 1010 |
| Diversity | (2–20) × 106 TCR β-chains, each paired with > 25 α-chains [3,4]† | (2–20) × 105 TCR β-chains, each paired with 1–2 α-chains [3,4]† | Not determined | Not determined |
| Oligoclonality | None | Infrequent | None | Frequent in the elderly |
| Frequency of Ki67+ T cells (%) | ~ 0.2 | 1–2 | ~ 0.3 | ~ 3 |
| Daily replacement rate by stable isotope labeling of DNA (%) | ~ 0.1 [25] | ~ 0.8 [87] | ~ 0.1 [25] | ~ 0.9 [87] |
| Half-life (days) | ~ 700 | ~ 80 | ~ 700 | ~ 70 |
| Daily production rates (n) | 1.5 × 108 | 1.5 × 109 | 0.8 × 108 | 0.8 × 109 |
* Phenotypic distinction imperfect. †Data from [3] for total T cells.
Studies using the frequency of TCR excision circle (TREC)-positive T cells as an indirect measure of diversity are consistent with the higher estimates of diversity [5-7]. TRECs are generated during TCR rearrangement, are not replicated, and are diluted during subsequent cell divisions [8,9]. The frequency of TREC+ cells within the naive T-cell compartment can, therefore, be taken as an indirect measure of clonal size. Studies have suggested that this clonal size is strictly regulated at 10–20 cells per clonotype in the newborn and that it then slowly but steadily increases with age [7]. Compared with the naive population of T cells, the memory compartment is clearly contracted in diversity. However, even memory T cells are very diverse. Estimates of diversity within the memory compartment range from 1 × 105 to 1 × 106 different TCR β-chains, each combined with one or very few different TCR α-chains [3,4].
It is generally assumed that this high degree of TCR diversity is necessary to guarantee recognition of the universe of antigenic peptides. In fact, the T-cell repertoire is capable of responding to virtually any foreign organism. In spite of its structural diversity, however, the repertoire of functional TCR is still greatly outnumbered by potential antigenic peptides, particularly in small mammals such as the mouse. Plasticity in the TCR–peptide–MHC complex may account for the recognition of multiple antigenic peptides by the same TCR [10,11].
T-cell diversity, tolerance, and autoimmunity
Recent studies have interpreted the need for repertoire diversity within the T-cell compartment from a totally different perspective, namely, one of regulation of immune responsiveness [12,13]. The immune system is under strict homeostatic control [14,15]. T-cell responses to self-antigens are prevented in the majority of individuals. Also, the magnitude of T-cell responses to foreign antigens is regulated. Generally accepted control mechanisms include the induction of apoptosis in the responding T-cell population, and feedback control by inhibitory receptors and regulatory T cells. Remarkably, diversity of the repertoire of naive and memory T cells has now been established as a major additional way to control unwanted clonal expansions, presumably functioning by means of clonal competition for space and resources.
A characteristic example of a breakdown in this control mechanism is the lymphopenic mouse [16-20]. Thymectomy shortly after birth is generally sufficient to induce an autoimmune syndrome. Similarly, adoptive transfer of small numbers of naive T cells into a T-cell-deficient host induces a wasting disease that has many features of the autoimmune disease, inflammatory bowel disorder. These autoimmune phenomena have been initially attributed to the absence of regulatory cells in the thymectomized mouse or in the lymphopenic host [16]. Experiments by Barthlott and colleagues [12], however, have shown that these autoimmune manifestations can be prevented by naive T cells that lack any features of regulatory cells but that have the potential of homeostatic expansion. Clonal competition is in part antigen specific, and clonal T-cell populations can selectively inhibit the division of T cells of their own specificity [21]. Equally important, regulatory control can also be exerted by T-cell populations of completely unrelated specificities, so long as these populations have the propensity for homeostatic proliferation [12]. These studies emphasize the intrinsic regulatory mechanism that is inherent in a diverse population of T cells and that keeps autoreactive T-cell responses in check while not curtailing immune responses to exogenous antigens.
Threats to T-cell diversity
T-cell diversity is continuously challenged [2]. Antigenic stimulation induces rapid expansion of antigen-specific T cells that expand to large clonal sizes. This expansion is counterbalanced by subsequent clonal contraction, which appears to be preprogrammed. Clonal contraction is robust and is usually sufficient to maintain a diverse memory T-cell compartment. However, clonal T-cell populations can emerge, and they have been associated with chronic infection such as cytomegalovirus or HIV [22]. These clonal expansions are usually limited to the memory T-cell compartment and do not affect the diversity of naive T cells because naive and memory T cells underlie different homeostatic control mechanisms and compete for different resources [14].
One additional biological variable that has a profound impact on T-cell homeostasis is age. The generation of new T cells in the thymus is highest in the newborn and then progressively declines [23]. Thymic involution progresses at the rate of ~ 3% per year, and individuals older than 50 years have < 15% of their thymic tissue remaining [24]. However, the demand for production of new T cells remains high in the adult.
In studies using endogenous labeling of DNA, the daily fractional replacement rate is 0.1–0.6% for naive T cells, and memory T cells turn over at a daily rate of 0.9–3.1% [25]. In essence, adults need to produce 1.5 × 108 naive T cells and 1.5 × 109 memory T cells every day (Table 1). New naive T cells are only produced in the thymus. Therefore, the formation of new T cells declines sharply with age. The frequency of TREC+ cells, which gives an upper estimate of all (intrathymic and extrathymic) newly generated T cells, declines by > 95% between the ages of 20 and 60 years. This decline demonstrates that thymic production in a 60-year old is, at most, 5% of the capacity that existed at the age of 20 years [5,26]. Consequently, the need for the replenishment of naive T cells must come from the autoproliferation of existing T cells [27]. Homeostatic proliferation of naive T cells is dependent on the recognition of self-antigen [28-30]. As a result, the generation of 'new' naive T cells by autoproliferation is under selective pressure and ultimately leads to TCR diversity contraction.
Studies on the impact of age on the repertoire diversity of naive T cells are not available; however, the continuous decline in the frequency of TREC+ cells indicates a steady increase in the average clonal size. Preliminary evidence suggests that the contraction accelerates markedly at approximately age 65 years, after which 95% of the CD4+ T-cell diversity is lost (unpublished observations). Data for CD8+ naive and memory T cells are not available because of the lack of a reliable phenotypic marker to distinguish these subsets.
The mechanisms underlying this accelerated contraction are unknown. Uneven homeostatic proliferation, which favors CD4+ T cells with higher avidity for self-antigens, may be one factor. An additional factor may be increasing competitive pressure from memory cells and a breakdown of distinct naive and memory cell compartments. Also, the phenotypic distinction of naive and memory cells based on CD45 isoforms, which is relatively reliable for CD4+ T cells, may be less distinct with age. The observed repertoire contraction may, in part, represent a shrinkage in size of the naive compartment.
Contraction in diversity and dominance of clonal T-cell populations is a relatively common finding in the memory compartment of elderly healthy individuals [31-33]. These clonal expansions predominantly involve CD8+ T cells, but they can also be found in CD4+ T cells [33,34]. These clonal expansions appear to resemble T-cell oligoclonality that is associated with chronic infections. Indeed, clonally expanded CD8+ T cells in otherwise healthy individuals may be specific for cytomegalovirus [22].
T-cell diversity in rheumatoid arthritis
Early evidence that T-cell homeostasis is not intact in patients with rheumatoid arthritis (RA) came from the observation that these patients carried large clonally expanded populations of CD4+ and CD8+ T cells [35-37]. TCR studies demonstrated some degree of preference for certain TCR variable region β-chains [38,39]. However, sharing of the third complementary determining region of the TCRs among different patients was not found, suggesting that these T cells were not specific for a common antigen. Also, the expanded T-cell clones were present in the circulation as well as in inflamed tissues. Frequencies of expanded clonotypes were independent of disease activity and were stable over time, again suggesting that these clonal expansions were not simply a consequence of an antigen-driven activation event in the synovial tissue [40].
Studies by Wagner and colleagues [4] and by Koetz and colleagues [26] examined whether the clonal expansions were indicators of a more profound defect in T-cell homeostasis (Fig. 1). Specifically, these authors examined whether repertoire contraction also involved the naive T-cell compartment. Koetz and colleagues [26] stated that the frequency of TREC+ T cells was significantly lower in patients with RA compared with age-matched controls. One possible interpretation of these data is that patients with RA have a premature diminution of thymic production. In this model, the immune system in patients with RA would be prematurely aged by 20–30 years and would increasingly rely on autoproliferation to fill the void.
Figure 1.
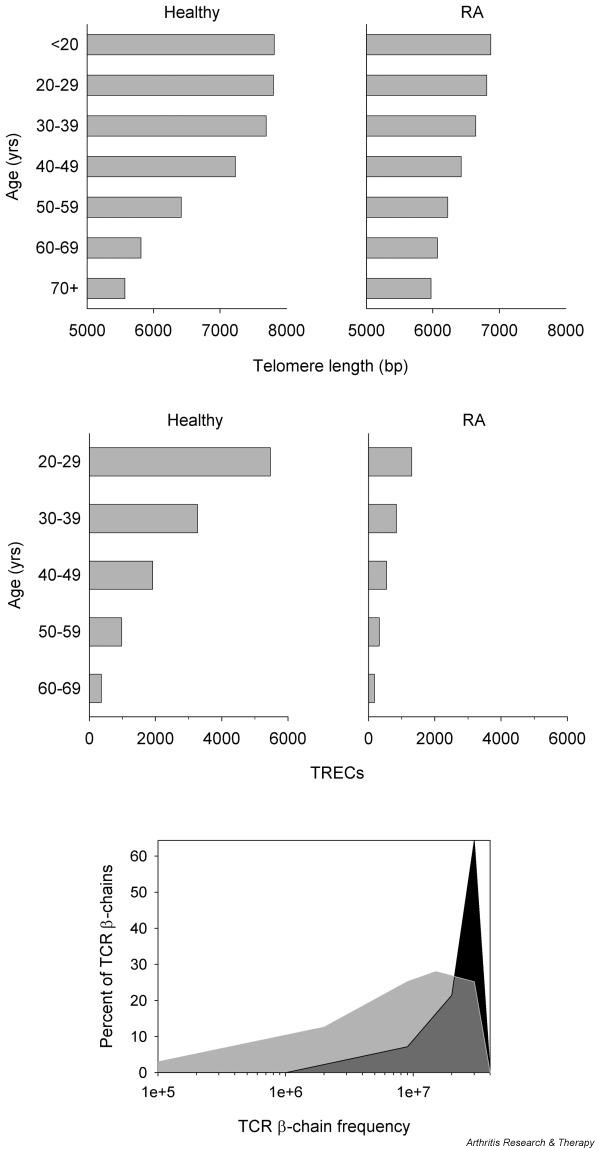
Replicative stress and contraction of TCR diversity. (a) With normal aging, peripheral T cells develop progressive telomeric erosion as evidence of replicative stress. (b) Frequencies of TCR excision circle (TREC)-positive T cells decline as a consequence of thymic dysfunction and cumulative peripheral turnover. Both processes are accelerated in patients with rheumatoid arthritis (RA). (c) The TCR repertoire of naive T cells in RA (light-shaded area) is markedly contracted compared with age-matched controls (dark-shaded area). Individual naive T cells in RA are present at higher frequencies and are of larger clonal sizes, resulting in a lower number of different TCRs. bp, base pairs.
de Boer and colleagues [9] proposed an alternative model; namely, these findings may be the consequence of a primary increase in the turnover of naive T cells that would result in dilution of TREC+ T cells. The time of increased turnover must have preceded the onset of RA. By the time the patients have developed RA, they have reached a steady state as indicated by two observations. First, the frequency of cycling Ki-67+ T cells in the peripheral blood of patients with RA is not increased, but is even slightly decreased, indicating a reduced peripheral turnover. The second observation is that the concentrations of TREC+ cells are already reduced in 20-year old patients with RA, and the subsequent age-dependent annual loss is not different from age-matched healthy controls. This again suggests that the turnover at the time of disease is not increased [26]. Ponchel and colleagues [41] have confirmed the reduction in TREC+ T cells in patients with RA, and have correlated this with phenotypic changes of naive T cells that may be the consequences of increased homeostatic proliferation.
Irrespective of the primary defect, these data suggest that patients with RA have a history of increased homeostatic proliferation of naive T cells that predated their disease, that may have occurred to compensate for a lymphopenic state, and that has imposed major phenotypic changes. Increased homeostatic proliferation should lead to repertoire contraction and to signs of replicative stress; indeed, this is the case.
The history of replicative stress can be assessed by measuring the telomere length. Telomeres in CD4+ T cells in healthy individuals are relatively intact until the age of 40 years, when they begin to progressively erode until they plateau at a rather short length at the age of 65 years [26,42]. In contrast, patients with RA have nearly complete erosion of their telomeric ends in their early twenties. Most notably, the telomeric erosion in patients with RA affects naive T cells as well as memory T cells. Memory T cells in healthy individuals have lost ~ 1000 base pairs in telomeric length compared with naive T cells, which is consistent with an increased replicative history of more than 20 generations. In contrast, the telomeric lengths of naive T cells from patients with RA are only slightly longer than those of their own memory cells, and these telomeres are as short as those in memory cells of healthy age-matched individuals.
This increased replicative history is associated with a significant contraction in TCR diversity [4]. A contraction in diversity is to be expected if T-cell loss from the naive compartment is compensated by homeostatic proliferation, and this is further accelerated if homeostatic proliferation is not random. Diversity of the TCR was estimated by determining the frequency of arbitrarily selected TCR β-chain sequences derived from either CD45RO- (naive) or CD45RO+ (memory) CD4+ T cells. Compared with age-matched controls, the diversity of TCR β-chains was contracted approximately 10-fold (median frequency of a TCR β-chain of 2 × 10-6 compared with 2 × 10-7 in controls). The naive T-cell compartment, which is the primary contributor to TCR diversity, was affected in addition to the memory T cells. Contraction of diversity in the naive T-cell compartment could not be attributed to contamination of memory cells that reverted to the CD45RA phenotype. Based on sequence analysis, the distinction between naive CD4+ T cells and memory CD4+ T cells was maintained. The impact of a relative lymphopenia with subsequent increased homeostatic proliferation and repertoire contraction in RA is unclear but, in light of the experiments in the lymphopenic mouse, it is tempting to speculate that this scenario represents a major risk factor for breaking tolerance and developing autoimmune diseases such as RA.
Cellular T-cell senescence: a gain and loss in function
The immune system is a highly proliferative system because of homeostatic proliferation as well as antigen-specific responses. It is not surprising that, with advancing age, the immune system has evidence of high replicative stress. Multicellular organisms have evolved a mechanism to prevent the dysregulated growth and transformation of proliferating cells. One such mechanism, cellular senescence, was first described as a process that limits the proliferation of senescent fibroblasts.
Based on these studies, three cardinal features of cellular senescence have been defined [43]. The first is that, after repeated divisions, the proliferative capacity of a cell starts to dwindle and eventually ceases. One reason for this proliferative arrest is the shortening of telomeres. T cells have the ability to upregulate telomerase and they are able to prolong their lifespan; however, they are not resistant to telomere erosion. The second cardinal feature is that senescent cells develop resistance to apoptotic cell death. Finally, senescent cells undergo multiple phenotypic and functional changes. Notably, these changes are not necessarily a consequence of loss of gene expression, but they are frequently associated with a gain in function, such as the production of inflammatory cytokines in senescent fibroblasts. This latter finding has led to a model of senescence, the evolutionary theory of antagonistic pleiotropy [44]. This model implies that genes selected to enhance the fitness of young organisms have unselected deleterious effects in the aged organism if aberrantly expressed.
Consistent with this model, replicatively stressed CD4+ and CD8+ T cells undergo multiple phenotypic and functional changes (Fig. 2) [45]. The most widely acknowledged phenotypic change is the loss of CD28, which increases in frequency in the CD8+ T-cell population with age but which also occurs in CD4+ T cells to a lesser degree [46-48]. CD28 expression is regulated at the level of a CD28-specific initiator complex that includes the nuclear proteins nucleolin and hnRPD [49,50]. Replicative senescence and chronic exposure to tumor necrosis factor alpha induce a loss of this initiator complex, particularly in CD8+ T cells [51]. This loss is partially reversible by IL-12 [52]. However, CD28 loss is not the only, and possibly not the most prominent, change in gene expression in senescent T cells. Senescent CD4+ and CD8+ T cells acquire the expression of many genes that are generally expressed on natural killer (NK) cells and that are associated with effector functions [53]. Even CD4+ T cells can acquire cytotoxic activity through the expression of perforin and granzymes [54,55]. Also, senescent CD4+ T cells express a number of new regulatory molecules instead of the traditional ones, such as CD28 and CTLA-4, that control their activation or inhibition.
Figure 2.
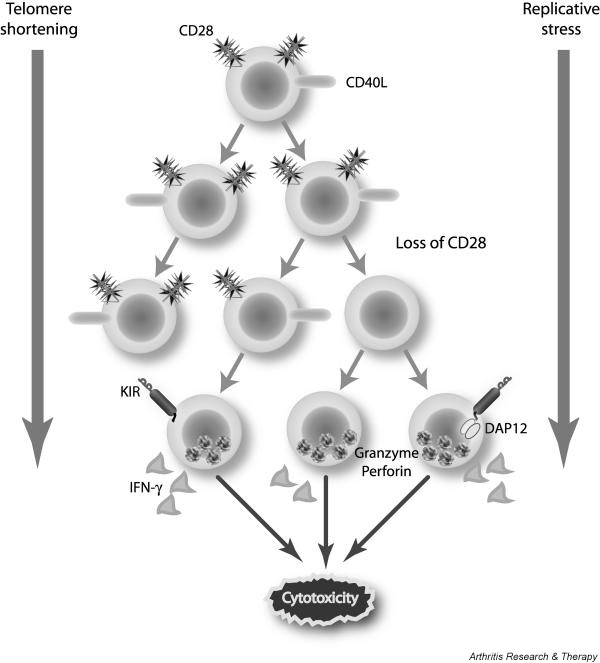
Replicative senescence and shifts in gene expression. Cumulative replication of T cells is associated with telomeric erosion and loss of CD28 and CD40L expression, consistent with cellular senescence. Presenescent CD4+ T cells gain effector functions such as high production of cytokines and cytotoxic ability through a perforin/granzyme mechanism. These cells are under the regulatory control of MHC class I-recognizing receptors, such as killer immunoglobulin-like receptors (KIRs), that can provide costimulatory signals or, if coexpressed with the appropriate adapter molecule DAP12, form an independent, fully competent recognition unit.
In particular, CD4+CD28null T cells express immunoreceptors of the killer immunoglobulin-like receptor (KIR) family [53,56-58]. This receptor family is usually expressed on NK cells and often displays specificity for MHC class I molecules. The family is highly polymorphic, and individuals differ in the number of genes as well as allelic polymorphisms. The KIR family includes stimulatory and inhibitory members. The stimulatory receptors require an adapter molecule (DAP12) to be functional, but they then constitute an independent recognition unit. T cells lack this adapter molecule, and KIRs expressed on T cells are not stimulatory on their own. However, the KIRs are able to provide a costimulatory signal for T-cell effector functions in the absence of DAP12 [59]. This costimulatory signal functions through the activation of the c-Jun N-terminal kinase pathway, and it is important in lowering the threshold in response to TCR stimulation.
In essence, the aging T-cell compartment is characterized by the increased frequency of highly competent effector T cells that are under the control of regulatory molecules found on NK cells. It can be envisioned, based on their unique properties, that these T-cell populations play an important role in tissue injury and in loss of self-tolerance as the biological system ages.
Senescent T cells: facilitators of inflammation
Expansion of CD4+ and CD8+ T cells that have lost the expression of CD28, and are presumably senescent, has been observed in several autoimmune diseases including diabetes mellitus, RA, Wegener's granulomatosis, multiple sclerosis, and ankylosing spondylitis [60-64]. In general, these cells were clonally expanded and included autoreactive T cells, implicating them directly in the pathogenesis of these diseases. In RA, specifically, increased frequencies of CD4+CD28null T cells are associated with more severe disease, again providing evidence for a direct role of these cells in the disease manifestations. In early RA, the frequency of CD4+CD28null T cells is a predictor for erosive progression [65]. In the established disease, the frequency correlates with extra-articular manifestations [66]. Increased frequencies are seen in nodular disease, and the highest frequencies are found in patients with rheumatoid vasculitis. Also, the T-cell type of large granular lymphocytes seen in Felty-like conditions appears to be directly related to the senescent CD28null T cells [67].
At first sight, the loss of CD28 would suggest that these cells are functionally anergic and prone to apoptosis; however, the opposite is the case. These cells are very potent effector cells, and at least CD4+CD28null T cells are resistant to apoptosis (the data on CD8+ T cells are contradictory) [68-70]. Resistance to apoptosis-inducing signals cannot be attributed to a single mechanism but is acquired and multifactorial, consistent with the senescent phenotype of these cells. CD4+CD28null T cells express more bcl-2, which renders them less sensitive to growth-factor withdrawal [68]. CD4+CD28null T cells are also resistant to Fas-mediated apoptosis. These cells fail to degrade FLIP following T-cell activation and/or IL-2 stimulation. They, therefore, do not activate the death pathway upon Fas-ligand engagement [69]. The resistance to growth-factor withdrawal and Fas signaling may prevent the usual clonal downsizing in vivo after antigen-specific stimulation.
The accumulation of oligoclonal T-cell populations appears to be more the consequence of a prolonged survival than increased proliferation, again consistent with the concept of cellular senescence. Given the central role of T-cell apoptosis in T-cell homeostasis and peripheral tolerance, the prolonged survival of these cells may contribute to their role in inflammatory diseases. Specifically, overexpression of c-FLIP has been shown to induce autoimmunity [71].
In addition to resistance to apoptosis, other functional and phenotypic changes in senescent T cells in RA are of importance for their role in perpetuating chronic tissue inflammation. First, the shift in regulatory molecules, from the classic CD28-CD80/CD86 pathway to alternate immunoreceptors, changes the cellular context in which T-cell stimulation is facilitated. There is no longer a unique role for professional antigen-presenting cells that express CD80/CD86, but other cell types can be T-cell stimulatory. More importantly, CD4+CD28null T cells are very potent effector T cells and can cause tissue injury by virtue of their high cytotoxic activity and their excessive production of proinflammatory cytokines, including tumor necrosis factor alpha and IFN-γ. There is evidence that both dimensions are of functional importance in RA. Weissman and colleagues [72] were the first to postulate a role for perforin/granzyme-positive CD4+ T cells in the synovial inflammation of patients with RA, and also in one patient with ankylosing spondylitis. Namekawa and colleagues [54] demonstrated the presence of these cells in the synovial tissue of patients with RA, again postulating that the gain in cytotoxic function is of functional importance in maintaining chronic synovitis.
Regulatory genes of the KIR family have been identified as disease risk genes in RA and in psoriatic arthritis [73,74]. In patients with RA, in particular those who have extra-articular manifestations, oligoclonal T-cell populations were found to preferentially express the stimulatory KIR2DS2 gene, often in the absence of inhibitory KIRs or inhibitory receptors of the c-type lectin family, CD94/NKG2A [75]. Indeed, expression of KIR2DS2 had functional implications in that it sensitized the T cells to respond to subthreshold TCR stimulation. The KIR2DS2 gene, present in only 40% of a healthy Caucasian population, was found in association studies to be a risk factor for rheumatoid vasculitis [73]. Association studies also suggested a role for the stimulatory immune receptors, KIR2DS1 and KIR2DS2, in the risk of developing psoriatic arthritis [74].
Senescent T cells: shifting the balance from tissue homeostasis to tissue inflammation in coronary artery disease
Acquisition of new functions by senescent T cells appears not only to be important in autoimmune disease manifestations but also in more subtle inflammatory reactions that are associated with tissue homeostasis and repair. One characteristic example is coronary artery disease (CAD).
It is well established that activation of systemic inflammatory responses, as exemplified by elevated C-reactive protein levels, is a risk factor for adverse outcome in patients with CAD [76]. The atherosclerotic plaque is now understood to be an inflammatory lesion. Inflammation may lead to plaque rupture and subsequent thrombosis, and it may cause the clinical manifestations of acute coronary syndromes (ACS) such as myocardial infarction and unstable angina [77-79]. Patients with ACS have highly elevated frequencies of CD4+CD28null T cells, consistent with the notion that they have a pre-aged immune system [80]. CD4+CD28null T cells have been isolated from ruptured coronary plaques that have caused fatal myocardial infarction or have been isolated from plaque material that was harvested during angioplasty of unstable plaques [81]. CD4+CD28null T cells from patients with ACS produce large amounts of IFN-γ in vitro [82], and increased IFN-γ activity in vivo can be demonstrated. IFN-γ-inducible genes are upregulated in the peripheral blood of patients with ACS, and circulating monocytes show evidence of nuclear translocation of STAT-1 homodimers, indicative of IFN-γ receptor triggering. CD4+CD28null T cells are also cytotoxic towards endothelial cells, and this activity can be significantly enhanced by C-reactive protein [83].
Taking the data together, CD4+CD28null T cells appear to be instrumental in plaque rupture, either indirectly via IFN-γ-mediated activation of macrophages or directly via their cytotoxic activity. Again, as seen in patients with RA, the activity of CD4+CD28null T cells can be modulated by regulatory receptors of the KIR family [84]. CD4+ T cells frequently express KIRs, specifically stimulatory isoforms, in patients with ACS. Most interestingly, T cells in patients with ACS can also express the adaptor molecule, DAP12. The coexpression of DAP12 and the stimulatory receptor encoded by the KIR2DS2 gene is sufficient to form an independent antigen recognition unit that confers the ability to fully activate a T cell, even in the absence of TCR triggering. Such activation potential in T cells should have detrimental consequences for maintaining tolerance and tissue integrity, a characteristic example being the plaque rupture in a coronary artery lesion.
Sharing of immunosenescent mechanisms between ACS and RA provides a pathogenic framework for the recent clinical observations that the increased mortality of patients with RA can be attributed to coronary atherosclerosis and its complications [85]. In a case–control study, patients with RA were more likely to have multivessel coronary involvement at the first coronary angiogram compared with the general population (KJ Warrington, PD Kent, RL Frye, JF Lymp, SL Kopecky, JJ Goronzy, CM Weyand, manuscript submitted). The risk for accelerated CAD conferred by RA remained significant after adjustment for traditional risk factors. This example also illustrates how the distinction between the autoreactive response leading to autoimmune disease and the local inflammatory response of tissue repair can be blurred. The same mechanism, in this case immunosenescence, is responsible for the chronic destructive inflammatory disease itself as well as for its seemingly unrelated comorbidities.
Conclusion
RA is a disease that predominantly occurs in adults and has its highest incidence rates in the elderly [86]. This coincides with a period when the generation of new T cells is minimal and the ability to mount a naive T-cell response to new exogenous antigens starts to decline or is already severely compromised. Studies in patients with RA have shown that immune aging is accelerated, raising the question of whether the breakdown in tolerance can be truly explained within the classic models of an autoreactive T-cell response to a disease-inducing antigen or whether age-dependent changes of the immune system represent a critical factor.
The repertoire of naive T cells in RA is contracted and shows evidence of senescence, which may predispose the system to autoimmune responses that mirror the mechanisms in the lymphopenic mouse. In RA, presenescent memory T cells emerge that have acquired many functions of NK cells and are proinflammatory cells. We propose that the distinction between self and nonself requires a functional and competent immune system. Age-related degeneration of immunocompetence imposes an immediate risk on the complex processes of self-tolerance (Fig. 3). With premature immune aging in RA, failure of self-tolerance may occur more easily and earlier in life. Effector functions of presenescent T cells are critical for the autoimmune manifestations of RA, including some of the comorbidities of RA, such as CAD.
Figure 3.
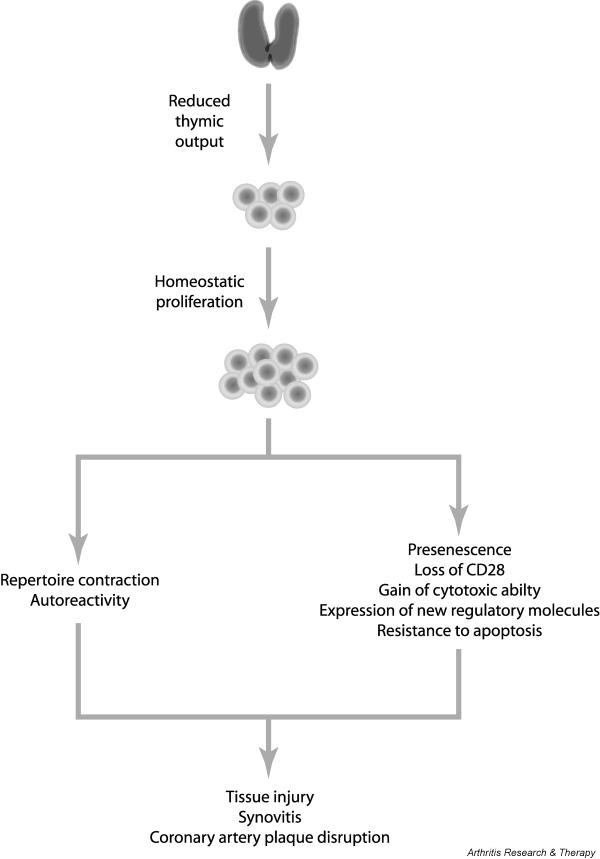
Pathomechanisms in rheumatoid arthritis. The diagram illustrates how aging, altered T-cell homeostasis, and cellular senescence may be involved in the pathogenic events leading to rheumatoid arthritis.
Competing Interests
None declared.
Abbreviations
ACS = acute coronary syndromes; CAD = coronary artery disease; IFN = interferon; IL = interleukin; KIR = killer immunoglobulin-like receptor; MHC = major histocompatibility complex; NK = natural killer; RA = rheumatoid arthritis; TCR = T-cell receptor; TREC = TCR excision circle.
Acknowledgments
Acknowledgements
Supported by grants from the National Institutes of Health (R01 AI44142, R01 AR42527, R01 EY11916, R01 HL 63919, R01 AG15043, and R01 AR41974) and by the Mayo Foundation. The authors thank James W Fulbright for assistance in manuscript preparation and for preparing the graphics and Linda H Arneson for secretarial support.
References
- Davis MM. T cell receptor gene diversity and selection. Annu Rev Biochem. 1990;59:475–496. doi: 10.1146/annurev.bi.59.070190.002355. [DOI] [PubMed] [Google Scholar]
- Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature. 1999;402:255–262. doi: 10.1038/46218. [DOI] [PubMed] [Google Scholar]
- Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- Wagner UG, Koetz K, Weyand CM, Goronzy JJ. Perturbation of the T cell repertoire in rheumatoid arthritis. Proc Natl Acad Sci USA. 1998;95:14447–14452. doi: 10.1073/pnas.95.24.14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, Polis MA, Haase AT, Feinberg MB, Sullivan JL, Jamieson BD, Zack JA, Picker LJ, Koup RA. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Douek DC, McFarland RD, Koup RA. Effects of exogenous interleukin-7 on human thymus function. Blood. 2002;99:2851–2858. doi: 10.1182/blood.V99.8.2851. [DOI] [PubMed] [Google Scholar]
- Schonland SO, Zimmer JK, Lopez-Benitez CM, Widmann T, Ramin KD, Goronzy JJ, Weyand CM. Homestatic control of T-cell generation in neonates. Blood. [DOI] [PubMed]
- Hazenberg MD, Otto SA, Cohen Stuart JW, Verschuren MC, Borl-effs JC, Boucher CA, Coutinho RA, Lange JM, Rinke de Wit TF, Tsegaye A, van Dongen JJ, Hamann D, de Boer RJ, Miedema F. Increased cell division but not thymic dysfunction rapidly affects the T-cell receptor excision circle content of the naive T cell population in HIV-1 infection. Nat Med. 2000;6:1036–1042. doi: 10.1038/79549. [DOI] [PubMed] [Google Scholar]
- Hazenberg MD, Borghans JA, de Boer RJ, Miedema F. Thymic output: a bad TREC record. Nat Immunol. 2003;4:97–99. doi: 10.1038/ni0203-97. [DOI] [PubMed] [Google Scholar]
- Evavold BD, Sloan-Lancaster J, Wilson KJ, Rothbard JB, Allen PM. Specific T cell recognition of minimally homologous peptides: evidence for multiple endogenous ligands. Immunity. 1995;2:655–663. doi: 10.1016/1074-7613(95)90010-1. [DOI] [PubMed] [Google Scholar]
- Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19:395–404. doi: 10.1016/S0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- Barthlott T, Kassiotis G, Stockinger B. T cell regulation as a side effect of homeostasis and competition. J Exp Med. 2003;197:451–460. doi: 10.1084/jem.20021387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, Weyand CM. T cell homeostasis and autoreactivity in rheumatoid arthritis. In: Goronzy JJ, Weyand CM, editor. In Rheumatoid Arthritis. Basel: Karger; 2001. pp. 112–132. [DOI] [PubMed] [Google Scholar]
- Freitas AA, Rocha B. Population biology of lymphocytes: the flight for survival. Annu Rev Immunol. 2000;18:83–111. doi: 10.1146/annurev.immunol.18.1.83. [DOI] [PubMed] [Google Scholar]
- Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- Groux H, Powrie F. Regulatory T cells and inflammatory bowel disease. Immunol Today. 1999;20:442–445. doi: 10.1016/S0167-5699(99)01510-8. [DOI] [PubMed] [Google Scholar]
- Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- Gleeson PA, Toh BH, van Driel IR. Organ-specific autoimmunity induced by lymphopenia. Immunol Rev. 1996;149:97–125. doi: 10.1111/j.1600-065x.1996.tb00901.x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101:455–458. doi: 10.1016/s0092-8674(00)80856-9. [DOI] [PubMed] [Google Scholar]
- Shevach EM. Suppressor T cells: rebirth, function and homeostasis. Curr Biol. 2000;10:R572–R575. doi: 10.1016/S0960-9822(00)00617-5. [DOI] [PubMed] [Google Scholar]
- Troy AE, Shen H. Cutting edge: homeostatic proliferation of peripheral T lymphocytes is regulated by clonal competition. J Immunol. 2003;170:672–676. doi: 10.4049/jimmunol.170.2.672. [DOI] [PubMed] [Google Scholar]
- Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, Nayak L, Moss PA. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol. 2002;169:1984–1992. doi: 10.4049/jimmunol.169.4.1984. [DOI] [PubMed] [Google Scholar]
- Haynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP. The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection. Annu Rev Immunol. 2000;18:529–560. doi: 10.1146/annurev.immunol.18.1.529. [DOI] [PubMed] [Google Scholar]
- Steinmann GG, Klaus B, Muller-Hermelink HK. The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand J Immunol. 1985;22:563–575. doi: 10.1111/j.1365-3083.1985.tb01916.x. [DOI] [PubMed] [Google Scholar]
- Neese RA, Misell LM, Turner S, Chu A, Kim J, Cesar D, Hoh R, Antelo F, Strawford A, McCune JM, Christiansen M, Hellerstein MK. Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc Natl Acad Sci USA. 2002;99:15345–15350. doi: 10.1073/pnas.232551499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 2000;97:9203–9208. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackall CL, Bare CV, Granger LA, Sharrow SO, Titus JA, Gress RE. Thymic-independent T cell regeneration occurs via antigen-driven expansion of peripheral T cells resulting in a repertoire that is limited in diversity and prone to skewing. J Immunol. 1996;156:4609–4616. [PubMed] [Google Scholar]
- Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity. 1999;11:183–190. doi: 10.1016/s1074-7613(00)80093-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viret C, Wong FS, Janeway CA., Jr Designing and maintaining the mature TCR repertoire: the continuum of self-peptide:self-MHC complex recognition. Immunity. 1999;10:559–568. doi: 10.1016/s1074-7613(00)80055-2. [DOI] [PubMed] [Google Scholar]
- Posnett DN, Sinha R, Kabak S, Russo C. Clonal populations of T cells in normal elderly humans: the T cell equivalent to 'benign monoclonal gammapathy'. J Exp Med. 1994;179:609–618. doi: 10.1084/jem.179.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain WD, Falta MT, Kotzin BL. Functional subsets within clonally expanded CD8(+) memory T cells in elderly humans. Clin Immunol. 2000;94:160–172. doi: 10.1006/clim.1999.4832. [DOI] [PubMed] [Google Scholar]
- Schwab R, Szabo P, Manavalan JS, Weksler ME, Posnett DN, Pannetier C, Kourilsky P, Even J. Expanded CD4+ and CD8+ T cell clones in elderly humans. J Immunol. 1997;158:4493–4499. [PubMed] [Google Scholar]
- Vallejo AN, Nestel AR, Schirmer M, Weyand CM, Goronzy JJ. Aging-related deficiency of CD28 expression in CD4+ T cells is associated with the loss of gene-specific nuclear factor binding activity. J Biol Chem. 1998;273:8119–8129. doi: 10.1074/jbc.273.14.8119. [DOI] [PubMed] [Google Scholar]
- Goronzy JJ, Bartz-Bazzanella P, Hu W, Jendro MC, Walser-Kuntz DR, Weyand CM. Dominant clonotypes in the repertoire of peripheral CD4+ T cells in rheumatoid arthritis. J Clin Invest. 1994;94:2068–2076. doi: 10.1172/JCI117561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Goronzy JJ, Weyand CM. CD4+CD7-CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. 1996;97:2027–2037. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JE, Ricalton NS, Meyer AC, West SG, Kaplan H, Behrendt C, Kotzin BL. Analysis of clonal CD8+ T cell expansions in normal individuals and patients with rheumatoid arthritis. J Immunol. 1995;154:3538–3547. [PubMed] [Google Scholar]
- Hingorani R, Monteiro J, Furie R, Chartash E, Navarrete C, Pergolizzi R, Gregersen PK. Oligoclonality of V beta 3 TCR chains in the CD8+ T cell population of rheumatoid arthritis patients. J Immunol. 1996;156:852–858. [PubMed] [Google Scholar]
- Waase I, Kayser C, Carlson PJ, Goronzy JJ, Weyand CM. Oligoclonal T cell proliferation in patients with rheumatoid arthritis and their unaffected siblings. Arthritis Rheum. 1996;39:904–913. doi: 10.1002/art.1780390606. [DOI] [PubMed] [Google Scholar]
- Rittner HL, Zettl A, Jendro MC, Bartz-Bazzanella P, Goronzy JJ, Weyand CM. Multiple mechanisms support oligoclonal T cell expansion in rheumatoid synovitis. Mol Med. 1997;3:452–465. [PMC free article] [PubMed] [Google Scholar]
- Ponchel F, Morgan AW, Bingham SJ, Quinn M, Buch M, Verburg RJ, Henwood J, Douglas SH, Masurel A, Conaghan P, Gesinde M, Taylor J, Markham AF, Emery P, van Laar JM, Isaacs JD. Dysregulated lymphocyte proliferation and differentiation in patients with rheumatoid arthritis. Blood. 2002;100:4550–4556. doi: 10.1182/blood-2002-03-0671. [DOI] [PubMed] [Google Scholar]
- Weng NP, Levine BL, June CH, Hodes RJ. Human naive and memory T lymphocytes differ in telomeric length and replicative potential. Proc Natl Acad Sci USA. 1995;92:11091–11094. doi: 10.1073/pnas.92.24.11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Replicative senescence: an old lives' tale? Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001;11:S27–S31. doi: 10.1016/S0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Fulbright JW, Goronzy JJ. Immunosenescence, autoimmunity, and rheumatoid arthritis. Exp Gerontol. 2003;38:833–841. doi: 10.1016/s0531-5565(03)00090-1. [DOI] [PubMed] [Google Scholar]
- Effros RB, Boucher N, Porter V, Zhu X, Spaulding C, Walford RL, Kronenberg M, Cohen D, Schachter F. Decline in CD28+ T cells in centenarians and in long-term T cell cultures: a possible cause for both in vivo and in vitro immunosenescence. Exp Gerontol. 1994;29:601–609. doi: 10.1016/0531-5565(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Effros RB. Loss of CD28 expression on T lymphocytes: a marker of replicative senescence. Dev Comp Immunol. 1997;21:471–478. doi: 10.1016/S0145-305X(97)00027-X. [DOI] [PubMed] [Google Scholar]
- Monteiro J, Batliwalla F, Ostrer H, Gregersen PK. Shortened telomeres in clonally expanded CD28-CD8+ T cells imply a replicative history that is distinct from their CD28+CD8+ counterparts. J Immunol. 1996;156:3587–3590. [PubMed] [Google Scholar]
- Vallejo AN, Weyand CM, Goronzy JJ. Functional disruption of the CD28 gene transcriptional initiator in senescent T cells. J Biol Chem. 2001;276:2565–2570. doi: 10.1074/jbc.M005503200. [DOI] [PubMed] [Google Scholar]
- Vallejo AN, Bryl E, Klarskov K, Naylor S, Weyand CM, Goronzy JJ. Molecular basis for the loss of CD28 expression in senescent T cells. J Biol Chem. 2002;277:46940–46949. doi: 10.1074/jbc.M207352200. [DOI] [PubMed] [Google Scholar]
- Bryl E, Vallejo AN, Weyand CM, Goronzy JJ. Down-regulation of CD28 expression by TNF-alpha. J Immunol. 2001;167:3231–3238. doi: 10.4049/jimmunol.167.6.3231. [DOI] [PubMed] [Google Scholar]
- Warrington KJ, Vallejo AN, Weyand CM, Goronzy JJ. CD28 loss in senescent CD4+ T cells: reversal by interleukin-12 stimulation. Blood. 2003;101:3543–3549. doi: 10.1182/blood-2002-08-2574. [DOI] [PubMed] [Google Scholar]
- Young NT, Uhrberg M. KIR expression shapes cytotoxic repertoires: a developmental program of survival. Trends Immunol. 2002;23:71–75. doi: 10.1016/S1471-4906(01)02113-5. [DOI] [PubMed] [Google Scholar]
- Namekawa T, Wagner UG, Goronzy JJ, Weyand CM. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998;41:2108–2116. doi: 10.1002/1529-0131(199812)41:12<2108::AID-ART5>3.3.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Appay V, Zaunders JJ, Papagno L, Sutton J, Jaramillo A, Waters A, Easterbrook P, Grey P, Smith D, McMichael AJ, Cooper DA, Rowland-Jones SL, Kelleher AD. Characterization of CD4(+) CTLs ex vivo. J Immunol. 2002;168:5954–5958. doi: 10.4049/jimmunol.168.11.5954. [DOI] [PubMed] [Google Scholar]
- Raulet DH, Vance RE, McMahon CW. Regulation of the natural killer cell receptor repertoire. Annu Rev Immunol. 2001;19:291–330. doi: 10.1146/annurev.immunol.19.1.291. [DOI] [PubMed] [Google Scholar]
- Moretta L, Biassoni R, Bottino C, Mingari MC, Moretta A. Human NK-cell receptors. Immunol Today. 2000;21:420–422. doi: 10.1016/S0167-5699(00)01673-X. [DOI] [PubMed] [Google Scholar]
- Warrington KJ, Takemura S, Goronzy JJ, Weyand CM. CD4+,CD28- T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum. 2001;44:13–20. doi: 10.1002/1529-0131(200101)44:1<13::AID-ANR3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Snyder MR, Lucas M, Vivier E, Weyand CM, Goronzy JJ. Selective activation of the c-Jun NH2-terminal protein kinase signaling pathway by stimulatory KIR in the absence of KARAP/DAP12 in CD4+ T cells. J Exp Med. 2003;197:437–449. doi: 10.1084/jem.20020383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosig F, Csernok E, Wang G, Gross WL. Costimulatory molecules in Wegener's granulomatosis (WG): lack of expression of CD28 and preferential up-regulation of its ligands B7-1 (CD80) and B7-2 (CD86) on T cells. Clin Exp Immunol. 1998;114:113–118. doi: 10.1046/j.1365-2249.1998.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R. CD4+CD28- costimulation-independent T cells in multiple sclerosis. J Clin Invest. 2001;108:1185–1194. doi: 10.1172/JCI200112516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M, Goldberger C, Wurzner R, Duftner C, Pfeiffer KP, Clausen J, Neumayr G, Falkenbach A. Circulating cytotoxic CD8(+) CD28(-) T cells in ankylosing spondylitis. Arthritis Res. 2002;4:71–76. doi: 10.1186/ar386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman A, Stewart SJ, Nepom GT, Green WF, Crowe D, Thomas JW, Miller GG. CD11b+CD28-CD4+ human T cells: activation requirements and association with HLA-DR alleles. J Immunol. 1996;157:4771–4780. [PubMed] [Google Scholar]
- Schmidt D, Martens PB, Weyand CM, Goronzy JJ. The repertoire of CD4+CD28- T cells in rheumatoid arthritis. Mol Med. 1996;2:608–618. [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, Matteson EL, Fulbright JW, Warrington KJ, Chang-Miller A, Hunder GG, Mason TG, Nelson AM, Valente RM, Crowson CS, Erlich HA, Reynolds RL, Swee RG, O'Fallon WM, Weyand CM. Prognostic markers for radiographic progression in early rheumatoid arthritis. Arthritis Rheum. [DOI] [PubMed]
- Martens PB, Goronzy JJ, Schaid D, Weyand CM. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40:1106–1114. doi: 10.1002/art.1780400615. [DOI] [PubMed] [Google Scholar]
- Starkebaum G. Chronic neutropenia associated with autoimmune disease. Semin Hematol. 2002;39:121–127. doi: 10.1053/shem.2002.31918. [DOI] [PubMed] [Google Scholar]
- Schirmer M, Vallejo AN, Weyand CM, Goronzy JJ. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28- T cells from rheumatoid arthritis patients. J Immunol. 1998;161:1018–1025. [PubMed] [Google Scholar]
- Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J Immunol. 2000;165:6301–6307. doi: 10.4049/jimmunol.165.11.6301. [DOI] [PubMed] [Google Scholar]
- Spaulding C, Guo W, Effros RB. Resistance to apoptosis in human CD8+ T cells that reach replicative senescence after multiple rounds of antigen-specific proliferation. Exp Gerontol. 1999;34:633–644. doi: 10.1016/S0531-5565(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Van Parijs L, Refaeli Y, Abbas AK, Baltimore D. Autoimmunity as a consequence of retrovirus-mediated expression of C-FLIP in lymphocytes. Immunity. 1999;11:763–770. doi: 10.1016/s1074-7613(00)80150-8. [DOI] [PubMed] [Google Scholar]
- Griffiths GM, Alpert S, Lambert E, McGuire J, Weissman IL. Perforin and granzyme A expression identifying cytolytic lymphocytes in rheumatoid arthritis. Proc Natl Acad Sci USA. 1992;89:549–553. doi: 10.1073/pnas.89.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen JH, Moore BE, Nakajima T, Scholl D, Schaid DJ, Weyand CM, Goronzy JJ. Major histocompatibility complex class I-recognizing receptors are disease risk genes in rheumatoid arthritis. J Exp Med. 2001;193:1159–1167. doi: 10.1084/jem.193.10.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MP, Nelson G, Lee JH, Pellett F, Gao X, Wade J, Wilson MJ, Trowsdale J, Gladman D, Carrington M. Cutting edge: susceptibility to psoriatic arthritis: influence of activating killer Ig-like receptor genes in the absence of specific HLA-C alleles. J Immunol. 2002;169:2818–2822. doi: 10.4049/jimmunol.169.6.2818. [DOI] [PubMed] [Google Scholar]
- Namekawa T, Snyder MR, Yen JH, Goehring BE, Leibson PJ, Weyand CM, Goronzy JJ. Killer cell activating receptors function as costimulatory molecules on CD4+CD28null T cells clonally expanded in rheumatoid arthritis. J Immunol. 2000;165:1138–1145. doi: 10.4049/jimmunol.165.2.1138. [DOI] [PubMed] [Google Scholar]
- Morrow DA, Ridker PM. C-reactive protein, inflammation, and coronary risk. Med Clin North Am. 2000;84:149–161. doi: 10.1016/s0025-7125(05)70211-x. [DOI] [PubMed] [Google Scholar]
- Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–671. doi: 10.1161/01.cir.92.3.657. [DOI] [PubMed] [Google Scholar]
- Arroyo LH, Lee RT. Mechanisms of plaque rupture: mechanical and biologic interactions. Cardiovasc Res. 1999;41:369–375. doi: 10.1016/S0008-6363(98)00308-3. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Goronzy JJ, Liuzzo G, Kopecky SL, Holmes DR, Jr, Frye RL. T-cell immunity in acute coronary syndromes. Mayo Clin Proc. 2001;76:1011–1020. doi: 10.4065/76.10.1011. [DOI] [PubMed] [Google Scholar]
- Liuzzo G, Kopecky SL, Frye RL, O'Fallon WM, Maseri A, Goronzy JJ, Weyand CM. Perturbation of the T-cell repertoire in patients with unstable angina. Circulation. 1999;100:2135–2139. doi: 10.1161/01.cir.100.21.2135. [DOI] [PubMed] [Google Scholar]
- Liuzzo G, Goronzy JJ, Yang H, Kopecky SL, Holmes DR, Frye RL, Weyand CM. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation. 2000;101:2883–2888. doi: 10.1161/01.cir.101.25.2883. [DOI] [PubMed] [Google Scholar]
- Liuzzo G, Vallejo AN, Kopecky SL, Frye RL, Holmes DR, Goronzy JJ, Weyand CM. Molecular fingerprint of interferon-gamma signaling in unstable angina. Circulation. 2001;103:1509–1514. doi: 10.1161/01.cir.103.11.1509. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Schulte S, Warrington KJ, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. 2002;105:570–575. doi: 10.1161/hc0502.103348. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Goek O, Zhang X, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM. De novo expression of killer immunoglobulin-like receptors and signaling proteins regulates the cytotoxic function of CD4 T cells in acute coronary syndromes. Circ Res. 2003;93:106–113. doi: 10.1161/01.RES.0000082333.58263.58. [DOI] [PubMed] [Google Scholar]
- Turesson C, Jacobsson L, Bergstrom U. Extra-articular rheumatoid arthritis: prevalence and mortality. Rheumatology (Oxford) 1999;38:668–674. doi: 10.1093/rheumatology/38.7.668. [DOI] [PubMed] [Google Scholar]
- Doran MF, Pond GR, Crowson CS, O'Fallon WM, Gabriel SE. Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum. 2002;46:625–631. doi: 10.1002/art.509. [DOI] [PubMed] [Google Scholar]
- Hellerstein M, Hanley MB, Cesar D, Siler S, Papageorgopoulos C, Wieder E, Schmidt D, Hoh R, Neese R, Macallan D, Deeks S, McCune JM. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat Med. 1999;5:83–89. doi: 10.1038/4772. [DOI] [PubMed] [Google Scholar]