

Abstract
Adapting metabolic enzymes of microorganisms to low temperature environments may require a difficult compromise between velocity and affinity. We have investigated catalytic efficiency in a key metabolic enzyme (dihydrofolate reductase) of Moritella profunda sp. nov., a strictly psychrophilic bacterium with a maximal growth rate at 2°C or less. The enzyme is monomeric (Mr = 18,291), 55% identical to its Escherichia coli counterpart, and displays Tm and denaturation enthalpy changes much lower than E. coli and Thermotoga maritima homologues. Its stability curve indicates a maximum stability above the temperature range of the organism, and predicts cold denaturation below 0°C. At mesophilic temperatures the apparent Km value for dihydrofolate is 50- to 80-fold higher than for E. coli, Lactobacillus casei, and T. maritima dihydrofolate reductases, whereas the apparent Km value for NADPH, though higher, remains in the same order of magnitude. At 5°C these values are not significantly modified. The enzyme is also much less sensitive than its E. coli counterpart to the inhibitors methotrexate and trimethoprim. The catalytic efficiency (kcat/Km) with respect to dihydrofolate is thus much lower than in the other three bacteria. The higher affinity for NADPH could have been maintained by selection since NADPH assists the release of the product tetrahydrofolate. Dihydrofolate reductase adaptation to low temperature thus appears to have entailed a pronounced trade-off between affinity and catalytic velocity. The kinetic features of this psychrophilic protein suggest that enzyme adaptation to low temperature may be constrained by natural limits to optimization of catalytic efficiency.
The temperature range compatible with procaryotic life extends from well below 0°C up to at least 113°C. The comparative analysis of series of homologous enzymes spanning this range suggested the molecular strategies which achieve the necessary compromises between structural flexibility and stability. Thermophilic enzymes appear to owe their stability to a variety of extrinsic or intrinsic factors (for a recent review, see reference 13). Moreover, oligomerization is the dominant stabilizing factor in several proteins (20, 22, 39); a basically similar situation is obtained when an enzyme is stabilized by physical association with another one (37).
At the other end of the temperature scale, psychrophilic (24) enzymes face the necessity of being efficient catalysts at a low energy cost (4, 9, 14). On the basis of a much more limited set of data than for thermophilic enzymes (see recent reviews in references 33, 35, and 45), two basic strategies for psychrophily were distinguished: either an overall increase of flexibility generally associated with a marked decrease of stability, or a more subtle strategy which keeps one domain rigid enough to provide enough affinity for the substrate while holding the catalytic domain in a more flexible state (2, 21, 45). In the former case thermodynamic and genetic data even suggest that some enzymes, such as the α-amylase from a psychrophilic bacterium (10), have achieved the maximum flexibility compatible with the maintenance of a correctly folded structure. This in turn indicates that the lowest temperature compatible with active life (by opposition to mere survival) may not be determined by the freezing point of the aqueous environment only. The challenge of stability at low temperature is however not the only one faced by cold-active enzymes. Kinetic optimization is another one, especially for intracellular biosynthetic enzymes since (i) improving kcat by decreasing the activation enthalpy may have a cost in terms of affinity due to a concomitant increase in conformational entropy (11, 21); (ii) such enzymes usually have to cope with low substrate concentrations. Investigations on the catalytic efficiency of cold-active enzymes acting at key metabolic steps are thus important.
In this work we report the characterization of dihydrofolate reductase (DHFR) (EC 1.5.1.3) from Moritella profunda (DHFRMp), a novel, strictly psychrophilic and moderately piezophilic species isolated from the deep sea (44). To our knowledge this is the first report on a cold-active DHFR.
DHFR catalyzes the NADPH-linked reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF). It is a metabolically and clinically important enzyme since it is involved in over 20 different one-carbon transfer reactions and is the target of several antibacterial, antiprotozoal and anticancer drugs. It is also an interesting model system because of its small size, thus amenable to nuclear magnetic resonance studies (with a molecular mass of about 19 kDa when monomeric).
MATERIALS AND METHODS
Culture conditions.
Escherichia coli strains were grown at 30 or 16°C in medium 853 (15) as a liquid medium or with the addition of 1.5% agar (Difco). For bacteria harboring recombinant plasmids, the media contained kanamycin and/or chloramphenicol at 50 μg ml−1. For genetic experiments M. profunda strain 2674T was grown in Difco marine broth and for enzyme assays in arginine- and uracil-free medium supplemented with artificial seawater (43).
Cloning of the DHFR gene.
Restriction enzymes and T4 ligase were purchased from Boehringer Mannheim and used according to the manufacturer's instructions. M. profunda genomic DNA partially digested with enzyme Sau3A was used to construct a genomic λZAP DNA library in pBK-CMV (Stratagene) vector (43). This genomic library was used for transformation of E. coli strain XL1-Blue MRF (Stratagene) and positive clones were selected for resistance to 5 mM trimethoprim (a DHFR inhibitor) at 30°C on plates of medium 853. The corresponding genomic DNA was retrieved by the polymerase-chain reaction and sequenced to check that no mutant gene had been isolated.
Overexpression and purification of recombinant DHFRMp.
The DHFR gene of M. profunda was amplified by PCR to produce NdeI and BamHI restriction sites at the ends of the fragment. The NdeI restriction site was designed to overlap the ATG start codon. The fragment was cloned into the pET24a vector (Novagen) which was used to transform E. coli strains and the nucleotide sequence was checked. Exponentially growing cells of E. coli BL21-codonPlus(DE3)-RIL (Stratagene) bearing the plasmid were induced with 1 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) for 20 h at 16°C, harvested by centrifugation and washed in 0.9% NaCl. Cells were disrupted with a high-pressure cell disruptor (basic Z model; Constant Systems Ltd.) at a pressure of 28,000 lb/in2 in 50 mM K phosphate buffer, pH 7.0. The extracts were freed of cell debris by centrifugation, first for 30 min at 12,000 × g and then for 1 h at 130,000 × g. The supernatant was applied to a methotrexate-agarose (Sigma) column (XK26/20; Pharmacia) equilibrated with buffer A (50 mM K phosphate buffer, 3 mM 2-mercaptoethanol, pH 7.5). The column was then washed three times until no more protein was detected at 280 nm in the effluent, first with the same buffer, then with buffer A containing 2.3 M KCl, and finally with buffer B (50 mM Tris HCl, 1 M KCl, 3 mM 2-mercaptoethanol, 5% glycerol [vol/vol], pH 8.5). The enzyme was then eluted using 15 ml buffer B containing 15 mM folate at pH 9.0 followed by buffer B until no absorbance at 280 nm was detected in the effluent. Fractions containing the eluted peak were combined and concentrated by ultrafiltration (YM-3 membrane; Amicon) until DHFR activity began to be detected in the flowthrough. Concentrated enzyme was applied to a Hiload 16/60 Superdex 75 prep-grade column equilibrated with 20 mM phosphate buffer, 0.15 M NaCl, 6 mM 2-mercaptoethanol, 5% glycerol (vol/vol), pH 6.5 and calibrated using the Pharmacia molecular weight standards. The activity of the eluted fractions was tested and the fractions pooled.
Fluorescence.
Fluorescence of DHFR samples was recorded using an SML-AMINCO Model 8100 spectrofluorimeter (Spectronic Instruments) at an excitation wavelength of 280 nm and at an emission wavelength of 330 nm (for heat denaturation) or 340 nm (for urea denaturation). In order to reduce photobleaching of the samples, the excitation band-pass was kept at 1 nm (4 nm for emission) and was closed between records. Samples (1 ml) in 20 mM K phosphate buffer at pH 7.0 were adjusted to an A280 of ∼0.1 and were heated in the fluorometer cell at a rate of 2°C/min using a programmed water bath Lauda Ecoline RE306. Temperature of the sample was recorded in the cell by a Cooper-Constantan thermocouple connected to a calibrated digital thermometer (Thermo Electric). Unfolding by urea was monitored at 15°C after 1 h of incubation of the samples at this temperature. Data were normalized using the pre- and posttransition baseline slopes as described (26). In the linear portion of the transition between the native and unfolded states, the equilibrium constant, K and the free-energy change, G can be calculated using the relations K = fraction unfolded/fraction folded and ΔG = −RT lnK. Least-squares analysis of G values as a function of urea concentrations allowed to estimate the conformational stability in the absence of denaturant, ΔGH2O according to the following equation: ΔG = ΔGH2O − m [urea].
DSC.
Differential scanning calorimetry (DSC) was performed using a MicroCal MCS-DSC instrument at a scan rate of 2°C/min and under a nitrogen pressure of 2 atm. Samples were desalted by gel filtration through a PD10 column (Pharmacia) equilibrated with 20 mM K phosphate buffer, pH 7.0. This buffer was used in the reference cell and for buffer baseline determination. Thermograms of DHFR were analyzed according to a single non-two-state transition model in which Tm, ΔHcal, and ΔHeff were fitted independently using the MicroCal Origin software (version 2.9).
Enzyme assays.
The standard reaction mixture contained 50 mM K phosphate buffer at pH 7.0, 1 mM 2-mercaptoethanol (or 10 mM 2-mercaptoethanol for saturation studies), 0.12 mM NADPH (Roche), 0.05 mM DHF (Sigma) and about 30 ng enzyme. Enzyme was incubated with NADPH, the reaction initiated by addition of DHF and monitored by recording absorption decrease at 340 nM using a PU8740 UV/VIS scanning spectrophotometer (Philips), in a temperature-controlled cuvette. A unit of enzyme is defined as the amount that will reduce 1 μmol of substrate per min, based on a molar extinction coefficient of 12,000 (17). Protein concentrations were determined by the method of Lowry.
In order to record thermal inactivation, DHFR (30 μg/ml) was incubated in 20 mM phosphate buffer (pH 7.0) in the absence of substrates for 15 min at different temperatures and then kept at 0°C. Residual activity was measured in the same buffer under standard conditions at 30°C.
Half-life of activity was determined by incubating enzyme (30 μg/ml) in 20 mM phosphate buffer at pH 7.0 with or without 0.5 mM 2-mercaptoethanol and 0.5% glycerol (vol/vol) at a fixed temperature. Residual activity was measured in the same buffer under standard conditions at 30°C.
The kinetic parameters kcat and Km determined by the initial velocity method using a nonlinear regression fit of the saturation curves on the Michaelis-Menten equation performed by SigmaPlot 4.0. The IC50s (inhibitor concentration required for 50% inhibition of the activity) were obtained by fitting the inhibition data on an nonexponential decay.
RESULTS
Cloning the DHFRMp gene and sequence analysis.
A λZAP library containing M. profunda genomic DNA was used to transform E. coli XL1-Blue recipient cells, selecting for resistance to trimethoprim as described in Materials and Methods. The copy number of the recombinant DHFR gene was expected to be sufficient to produce this phenotype since overexpressing (25- to 30-fold) the wild type E. coli DHFR gene in such a way confers trimethoprim resistance to the host (31). One such clone, found to contain an insert of about 2.7 kb, was analyzed further. Sequencing about 1,000 bp from this fragment revealed two open reading frames homologous to the E. coli DHFR and proline permease genes, respectively. We checked that the cloned DHFR gene was identical to the native one after retrieving the cognate genomic DNA by the polymerase chain-reaction. Enzymological studies (see “Kinetic parameters and temperature dependence”) showed that DHFRMp is actually less sensitive to trimethoprim than E. coli DHFR; this may have contributed to the success of the selection procedure. DHFR sequence data have been submitted to the EMBL database under accession number AJ487535.
The DHFRMp open reading frame encodes 162 amino acids and is clearly homologous to all known DHFRs despite a low general level of identity. It presents the highest score (more than 55% identity) with E. coli and Salmonella enterica serovar Typhimurium DHFRs and relatively high values are still obtained with other Proteobacteria belonging to the γ-3 group (Fig. 1). Codons with A in the third position, such as AGA, AUA, and CUA, are rare in E. coli but not in Moritella (43). This explains the necessity of using an ad hoc host strain for overexpression (see below).
FIG. 1.

Comparison of DHFR primary structures: alignment of amino acid sequences from M. profunda (MORPR), S. enterica serovar Typhimurium (SALTY), E. coli (ECOLI), Citrobacter freundii (CITFR), Enterobacter aerogenes (ENTAE), Klebsiella aerogenes (KLEAE), and Haemophilus influenzae (HAEIN) (all above 50% identity) and from T. maritima (TEMA). DHFRMp K20 (A19 in E. coli) and E27 (D26 in E. coli) are in bold and underlined. A dotted line above the alignment indicates the extent of the M20 loop (residues 10 to 24) as in E. coli DHFR. Secondary structures, as defined for E. coli DHFR (3) are indicated above the alignment (α-helices and β-strands). Asterisks indicate identical amino acids and dots indicate similar amino acids conserved in all eight proteins.
When comparing the derived amino acid sequence of DHFRMp with that of E. coli DHFR (Fig. 1) catalytically important residues were found to be extensively conserved. In E. coli DHFR, the M20 loop formed by residues 10 to 24 plays an important role in the catalytic mechanism (34). The psychrophilic DHFR shows high similarity in this region: all charged residues are either conserved or substituted in a conservative way. Moreover hydrophobic residues Gly15 and Pro21, both expected to play a structural role during the conformational changes of the M20 loop are also conserved. However, the replacement of Ala19 in E. coli by a Lys in DHFRMp is worth mentioning as Lys19 might thus interact with the substrates during the reaction. In the folate binding site, Asp27, which is close to the NA-2 group of folate in the E. coli enzyme, is replaced by a Glu in the psychrophilic DHFR, possibly affecting substrate binding. The other residues which appear important in defining the folate binding site in E. coli are conserved: Leu28, Phe31, Arg52, Met20, Trp22, Leu54, Ala7, Ile5, Lys32, and Arg57. Similarly, many residues of the E. coli NADPH binding site are strictly conserved: Gln102, Leu62, Thr124, Tyr100, Asn18, Arg44, Gln65, Thr46, Asp123, and Ile14 (12).
Expression, purification, and molecular mass of DHFRMp.
The DHFRMp gene was inserted into expression vector pET24a, which was used to transform E. coli strain BL21(DE3) (Novagen). However, the expression level of the recombinant protein was low. Since rare codons could have hampered expression (as the sequence analysis confirmed), BL21-codonPlus(DE3)-RIL (Stratagene) competent cells were used. After IPTG induction the expression level was so high that the affinity chromatography step followed by gel filtration yielded as much as 300 mg of pure DHFR (as shown by SDS and native PAGE) from 1 liter of culture. The enzyme was eluted as the major peak from the calibrated gel filtration column at about 19 kDa. Mass spectrometry indicated a molecular mass of 18,290 Da (courtesy of V. Villeret), thus in the expected range for a monomer and in excellent agreement with the value deduced from the amino acid sequence derived from the gene (18,291.8 Da).
Resistance to thermodenaturation.
As shown in Fig. 2, DHFRMp withstood incubation up to about 35°C for 15 min but became inactivated at higher temperatures, illustrating its psychrophilic character. The half-life of activity was 25 min at 45°C but in the presence of 0.5 mM 2-mercaptoethanol and 0.5% glycerol no activity loss was observed and the half-live at 50°C was 18 min. Heat-induced unfolding of DHFRMp recorded by fluorescence (Fig. 3) also demonstrated that addition of glycerol and 2-mercaptoethanol shifted the midpoint of the thermal transition upwards by about 25°C. This pronounced stabilization obtained by combining a solvophobic effect and reducing conditions allows proper storage and handling of this relatively unstable enzyme.
FIG. 2.

Effect of temperature on DHFRMp activity and stability. A: residual activity after 15 min incubation at the temperature indicated; assay carried out at 30°C. B: thermodependence of the activity; assay carried out for 5 min at the temperature indicated.
FIG. 3.

Stabilization of the heat-labile DHFRMp: heat-induced denaturation was performed in phosphate buffer (open symbols) and in phosphate buffer containing 0.1 mM. 2-mercaptoethanol and 0.1% glycerol (closed symbols).
Kinetic parameters and temperature dependence.
As shown in Fig. 2, the apparent optimal temperature of activity for DHFRMp is close to 34°C but about 19% of the maximal activity could still be recorded at 2°C, the temperature at which the highest growth rate of M. profunda has been observed (44). Almost identical results were obtained with the native enzyme in cell extracts of M. profunda. Table 1 compares the kinetic parameters of DHFRs from M. profunda, E. coli, Lactobacillus casei, and Thermotoga maritima. At mesophilic temperatures (30 and 20°C) DHFRMp is very active as indicated by the high kcat value, close to that of E. coli DHFR. Accordingly, at environmental temperature the enzyme still displayed a relatively high kcat. In sharp contrast, the apparent Km for both dihydrofolate and NADPH were much larger than those reported for other microbial DHFRs. Interestingly, Km values of MpDHFR were not drastically affected by temperature but as a result, the catalytic efficiency kcat/Km remained low at all temperatures. Km values for native enzyme were measured at the same temperatures in cell extracts and found to be very similar to those reported in Table 1 (not shown). A low binding constant for substrates was also suggested by the IC50s for two inhibitors: 14.5 ± 1.5 nM for methotrexate and 4.8 ± 0.5 μM for trimethoprim (similar values were obtained for native enzyme in cell extracts), whereas corresponding values for E. coli DHFR are about 1.0 nM for methotrexate and 0.05 μM for trimethoprim (18).
TABLE 1.
Kinetic parameters of psychrophilic, mesophilic, and thermophilic DHFRs
| Organism | Ta μmax (°C) | Tb (°C) | pH | Mean kcat ± SD (s−1) | Mean Km ± SD (μM)
|
kcat/Km
|
Reference | ||
|---|---|---|---|---|---|---|---|---|---|
| DHF | NADPH | DHF | NADPH | ||||||
| M. profunda | 2 | 30 | 7.0 | 21.2 ± 2.0 | 19.8 ± 2.2 | 9.6 ± 1.7 | 1.07 | 2.21 | This work |
| 20 | 7.0 | 13.8 ± 1.2 | 16.4 ± 1.5 | 12.3 ± 1.3 | 0.84 | 1.12 | This work | ||
| 5 | 7.0 | 4.9 ± 0.5 | 21.7 ± 1.3 | 9.9 ± 1.9 | 0.23 | 0.49 | This work | ||
| E. coli | 37 | 20 | 7.2 | 14.3 | 0.27 | 1.05 | 52.9 | 13.6 | 27 |
| NSd | 7.4 | 16.5 ± 0.3 | 0.47 ± 0.05 | 2.5 ± 0.1 | 35.1 | 6.6 | 36 | ||
| 23 | 7.2 | 10.0 ± 0.8 | 0.44 ± 0.05 | 6.45 ± 0.9 | 22.7 | 1.6 | 29 | ||
| L. casei | 37 | 25 | 7.5 | 4.0 ± 0.5 | 0.36 ± 0.08 | 0.78 ± 0.08 | 11.1 | 5.1 | 8 |
| T. maritima | 80 | 37 | 6.5 | 1.9 ± 0.2 | 0.3 ± 0.1 | 4.0 ± 1.0 | 6.3 | 0.5 | 41 |
| 77 | 6.5 | 15.8 ± 1.5 | 52.7c | 4.0c | 40 | ||||
Temperature for maximal growth rate.
Temperature of the assay.
Extrapolated values, taking the same Km as that at 37°C.
NS, not specified.
The optimal pH for catalysis of the reaction was around 7.0 in phosphate buffer at 30°C (data not shown). By contrast, E. coli DHFR exhibits two maxima, one at pH 7.0 and another at pH 4.5 (1). In the case of the heat-stable T. maritima DHFR, there is a single optimum at pH 6.5 (41).
Conformational stability.
In order to compare the structural stability of DHFRMp with that of its mesophilic and thermophilic homologues from E. coli and T. maritima, the reversible urea-induced unfolding curves were recorded by fluorescence at 15°C. As shown in Fig. 4 and Table 2, moderate concentrations of urea induced denaturation of the psychrophilic enzyme, with a concentration for half-denaturation of about 1.6 M. The m value of DHFRMp, which is a measure of the dependence of ΔG on denaturant concentration, is similar to that of E. coli DHFR. By contrast, linear extrapolation of the ΔG values for the main transition (Fig. 4) indicates that the conformational stability in the absence of denaturant (ΔGH2O) is much lower than that of both mesophilic and thermophilic homologs.
FIG. 4.

Urea-induced unfolding curves of DHFR. The unfolded fraction of DHFRMp is plotted for increasing urea concentrations (closed symbols). The sigmoidal transitions of E. coli DHFR (28) and of T. maritima DHFR (6) are included for comparison. The inset shows linear extrapolation of ΔG values in the main transition region of DHFRMp to zero denaturant concentration.
TABLE 2.
Thermodynamic parameters of urea-induced unfolding of DHFRs at 15°C
| Organism | C1/2c (M) | m (kcal mol−1 M−1) | ΔGH2O (kcal mol−1) |
|---|---|---|---|
| M. profunda | 1.59 | 2.0 | 3.2 |
| E. colia | 3.10 | 1.9 | 5.9 |
| T. maritimab | 5.45 | 4.7 | 34.5 |
Data from reference 28.
Data from reference 6.
C1/2, concentration for half-denaturation.
Differential scanning calorimetry was used to calculate the stability profile of DHFRMp which is described by the following modified Gibbs-Helmholtz equation (30):
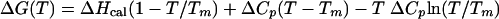 |
The calorimetric enthalpy ΔHcal, corresponding to the total amount of heat absorbed during unfolding, and the Tm value were obtained from thermograms of DHFRMp (Fig. 5). The ΔCp value, which determine the plot curvature, was taken from data for proteins of similar size (24). Figure 5 illustrates the resulting stability curve of DHFRMp with ΔCp values ranging from 1.5 to 2.0 kcal mol−1 K−1, as well as the profiles of the mesophilic E. coli DHFR (5, 19) and of the thermophilic T. maritima DHFR in the presence of 2.9 M guanidinium chloride (6). These stability curves represent the energy required to disrupt the native state (30). The increase in stability in the order (from most to least stable) psychrophile, mesophile, and thermophile is clearly illustrated by the progressively higher ΔGmax (top of the curve) and Tm (for ΔG = 0). Although ΔCp has not been determined experimentally for DHFRMp, this parameter is not prone to drastic variations between protein homologues (23) but mainly affects the predicted temperature of cold unfolding at subzero temperatures.
FIG. 5.

Stability profiles of DHFRs. The Gibbs free energy of denaturation for DHFRMp was calculated by equation 1 with ΔHcal = 50 kcal mol−1, Tm = 45.6°C, and ΔCp = 1.5 kcal mol−1 K−1(upper curve) or 2.0 kcal mol−1 K−1(lower curve). The stability curves for E. coli DHFR are from reference 19 for the lower curve and from reference 5 for the upper curve. Data for T. maritima DHFR in the presence of 2.9 M guanidinium chloride are from reference 6. The inset shows a differential scanning calorimetry thermogram of DHFRMp.
DISCUSSION
With a maximal growth rate (0.19 h−1) at 2°C (possibly less) and a maximal growth temperature of 12°C, M. profunda is one of the strictest psychrophiles described in the literature (16, 32, 44). This was the main reason to focus on DHFRMp as a model system. A second reason was that Moritella belongs to the family Vibrionaceae and is thus closely related to E. coli, whose DHFR was analyzed in detail. Actually the amino acid sequence of DHFRMp proved to be 55% identical to that of E. coli DHFR (12), whereas, in general, these enzymes are not very similar (3, 38, 41). DHFRs are usually monomeric enzymes. However, the thermophilic T. maritima DHFR was found to be a dimer (6, 41) and crystallographic analysis confirmed that the dimeric state plays an important role in the stabilization of this enzyme (7). Accordingly, DHFRMp was expected to be a monomeric protein, as confirmed here by gel filtration and mass spectrometry. The number of major hydrophobic residues (F, V, I, L, and M), conceivably involved in the formation of a hydrophobic core, is about the same in DHFRMp and E. coli DHFR but much less than in the dimeric T. maritima DHFR (Table 3) in which an increased proportion of Phe residues is localized in part within the hydrophobic core of the monomer but mainly at subunit interfaces (7). Thermolabile asparagine and glutamine residues are represented more often in DHFRMp and glutamine is even absent from T. maritima DHFR (Table 3). The total number of charged residues (K, R, E, and D), susceptible to provide stabilizing electrostatic interactions, is lower in DHFRMp than in E. coli DHFR and much lower than in T. maritima DHFR. Some of these differences are probably related to the psychrophilic nature of DHFRMp but direct three-dimensional studies are required to examine their significance.
TABLE 3.
Amino acid content in DHFRs from M. profunda, E. coli, and T. maritima
| Organism | % Amino acid content in DHFR
|
Σa (%)
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D | E | F | G | I | K | L | M | N | P | Q | R | V | LVIMF | KRED | |
| M. profunda | 5.6 | 8.0 | 2.5 | 6.2 | 6.7 | 3.1 | 10.5 | 3.1 | 5.6 | 5.9 | 3.7 | 5.6 | 6.7 | 29.6 | 22.3 |
| E. coli | 8.8 | 6.9 | 3.8 | 6.3 | 7.5 | 4.4 | 6.9 | 3.1 | 3.0 | 6.3 | 2.5 | 5.7 | 6.9 | 28.2 | 25.8 |
| T. maritima | 2.9 | 9.9 | 9.3 | 8.1 | 4.7 | 7.6 | 8.7 | 1.7 | 3.5 | 5.2 | 0.0 | 7.6 | 11.6 | 36.0 | 28.0 |
Sum of percentages for the cognate residues.
As expected for a cold-active enzyme, DHFRMp is less resistant to thermal denaturation than E. coli DHFR and much less than T. maritima DHFR. More than 80% of the activity was irreversibly inactivated after 15 min incubation at 45°C (Fig. 2) whereas DHFR from E. coli and T. maritima retained 100% activity after 15 min at 45°C and 75°C, respectively (40, 41). Regarding enzyme stability, unfolding experiments performed both by chemical means and high temperature pointed in the same direction. Figure 5 depicts the stability profile of DHFRs adapted to temperatures covering almost the whole biological range of temperatures. In principle, the low stability of DHFRMp could be reached in three ways (or a combination of them): by shifting the stability curve towards low temperatures (with similar maximal value of ΔG than E. coli DHFR but at much lower temperatures), by increasing the steepness of the stability curve (with similar maximal value of ΔG than E. coli DHFR at the same temperature but with a lower Tm), or by a global collapse of the stability curve. Each of these alternatives corresponds to a combination of alterations in the ΔHcal, Tm, or ΔCp values in equation 1. Obviously, the stability curve of DHFRMp follows the third alternative: the weak conformational energy arises from low values of both ΔΗcal and Tm. This demonstrates that the native state of DHFRMp is stabilized by a lower number of enthalpically driven interactions when compared with its mesophilic and thermophilic homologues. This is in agreement with the above-mentioned lower number of charged residues in DHFRMp. By contrast, the stability profile of T. maritima DHFR is shifted to higher temperatures with a sharply increased ΔGmax value (6). This indicates that the weak interactions responsible for the enhanced stability of the thermophilic enzyme are strengthened at high temperatures, suggesting the involvement of nonpolar groups (25). The relatively low number of hydrophobic amino acids in DHFRMp in comparison with T. maritima DHFR is also in agreement with this aspect.
Two further features revealed by previous work on a psychrophilic α-amylase (10) are also confirmed for DHFRMp. First the cold-adapted enzyme is not cold-stable as judged by the stabilization energy value at 0°C and by the extrapolated temperature of cold-unfolding at a subzero temperature. Secondly, the physiological temperature for DHFRMp lies on the left side of the bell-shaped stability curve, in sharp contrast with E. coli and T. maritima DHFRs. Accordingly, the thermal dissipative force is a main determinant of the structural mobility for the latter enzymes. By contrast, group hydration responsible for the decrease of ΔG values at low temperatures (23) significantly contributes to the conformational flexibility of DHFRMp which is required for enzyme activity around 0°C. It will be interesting to determine whether DHFRMp, like the above-mentioned α-amylase, approaches the maximal flexibility still compatible with stable folding. Indeed, the temperature of cold denaturation of some critical enzymes, and not the freezing point of the aqueous environment, may actually set the lower limit of bacterial development.
E. coli DHFR catalyzes reaction cycles through five detectable kinetic intermediates which have been depicted by crystallographic studies (3, 34). It was shown that the M20 loop (from residues 10 to 24) is predominantly closed in the E. coli holoenzyme. This loop moves over and between the reactants in the Michaelis and transition state complexes but during the remaining steps, the loop protrudes into the nicotinamide-ribose binding pocket. Thus, this loop is expected to assist the formation of the catalytic intermediates by driving the substrates into the active site. When compared to other DHFRs, the psychrophilic enzyme shows extensive conservation of catalytically important residues but differs by the A19K substitution in the M20 loop (Fig. 1). As a consequence, the basic side chain of Lys19 could interact with the oppositely charged folate during the reaction and could be responsible for the high kcat values. It should be mentioned that improved electrostatic fields at the active site of some psychrophilic enzymes seem involved in the adaptation to catalysis at low temperatures (35).
The apparent Km value for DHF of DHFRMp (Table 2) is more than 50 times the value currently reported by several authors for the cognate enzyme in E. coli (27, 29, 36) and is also considerably higher than for L. casei (8) and T. maritima (41). Detailed examination of published data does not suggest that assay conditions influenced actual values to an extent in anyway comparable to the differences we observe. Moreover, the sensitivity toward the inhibitors methotrexate and trimethoprim is much lower than for E. coli DHFR, indicating that the affinity of the enzyme has also been affected. In this respect, the specific replacement of Asp27 in the folate binding site by Glu in DHFRMp (Fig. 1) can possibly decrease the affinity as a result of steric hindrances arising from the larger acidic side chain. As regards NADPH, the Kmapp of DHFRMp is higher than in other homologues but remains in the same order of magnitude. Because these apparent Km values are not substantially different between 5°C and 30°C, the catalytic efficiency (kcat/Km) is definitely lower for DHFRMp than for E. coli DHFR, especially with respect to DHF. Therefore, DHFRMp appears to have adapted to low temperatures by maintaining a relatively high kcat at the expense of its affinity for one of the substrates of the reaction. The fact that this trade-off involves DHF rather than NADPH could be understood by comparison with E. coli DHFR since (i) efficient binding of NADPH appears necessary to promote the release of THF, product of the reaction, and (ii) the enzyme must keep a higher affinity for NADPH than for NADP because the latter is less efficient in THF release than NADPH and thus could slow down the reaction (34). Maintenance of a sufficient affinity for NADPH in DHFRMp was probably selected during the course of evolution.
From the kinetic point of view, DHFRMp thus appears to remain well below optimization, supporting the concept that complete metabolic adaptation to cold may remain out of reach. We have obtained similar data for ornithine carbamoyltransferase from a closely related species of Moritella (42). Since M. profunda is moderately piezophilic it will be interesting to determine whether increased hydrostatic pressure has any effect on the catalytic efficiency of the enzyme. Moreover, since the data presented in this work have been obtained with pure DHFR, one can wonder whether, in vivo, the relatively high Km is not partly alleviated by compartmentalization effects such as obtained in metabolic channeling.
Acknowledgments
This work was supported by the Belgian Foundation for Joint and Fundamental Research (FKFO), by the Flanders Actionprogramme Biotechnology, by the EEC programmes Coldzymes and Eurocold, and by a Concerted Action between the Belgian State and the Free University of Brussels (VUB).
We thank V. Villeret (Institut de Biologie de Lille, France) for helpful assistance in structural interpretation and for the molecular mass determination by mass spectrometry; we thank D. Georlette for her kind help.
REFERENCES
- 1.Baccanari, D. P., D. Stone, and L. Kuyper. 1981. Effect of a single amino acid substitution on Escherichia coli dihydrofolate reductase catalysis and ligand binding. J. Biol. Chem. 256:1738-1747. [PubMed] [Google Scholar]
- 2.Bentahir, M., G. Feller, M. Aittaleb, J. Lamotte-Brasseur, T. Himri, J. P. Chessa, and C. Gerday. 2000. Structural, kinetic and calorimetric characterization of the cold-active phosphoglycerate kinase from the antarctic Pseudomonas sp. TACII18. J. Biol. Chem. 275:11147-11153. [DOI] [PubMed] [Google Scholar]
- 3.Bolin, J. T., D. J. Filman, D. A. Matthews, R. C. Hamlin, and J. Kraut. 1982. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. I. General features and binding of methotrexate. J. Biol. Chem. 257:13650-13662. [PubMed] [Google Scholar]
- 4.Cavicchioli, R., K. S. Siddiqui, D. Andrews, and R. K. Sowers. 2002. Low-temperature extremophiles and their applications. Curr. Opin. Biotechnol. 13:253-261. [DOI] [PubMed] [Google Scholar]
- 5.Clark, A. C., and C. Frieden. 1999. Native Escherichia coli and murine dihydrofolate reductases contain late-folding non-native structures. J. Mol. Biol. 285:1765-1776. [DOI] [PubMed] [Google Scholar]
- 6.Dams, T., and R. Jaenicke. 1999. Stability and folding of dihydrofolate reductase from the hyperthermophilic bacterium Thermotoga maritima. Biochemistry 38:9169-9178. [DOI] [PubMed] [Google Scholar]
- 7.Dams, T., G. Auerbach, G. Bader, U. Jacob, T. Ploom, R. Huber, and R. Jaenicke R. 2000. The crystal structure of dihydrofolate reductase of Thermotoga maritima: molecular features of thermostability. J. Mol. Biol. 297:659-672. [DOI] [PubMed] [Google Scholar]
- 8.Dann, J. G., G. Ostler, R. A. Bjur, R. W. King, P. Scudder, P. C Turner, G. C. Roberts, and A. S. Burgen. 1976. Large-scale purification and characterization of dihydrofolate reductase from a methotrexate-resistant strain of Lactobacillus casei. Biochem. J. 157:559-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feller, G., and C. Gerday. 1997. Psychrophilic enzymes: molecular basis of cold adaptation. Cell. Mol. Life Sci. 53:830-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feller, G., D. d'Amico, and C. Gerday. 1999. Thermodynamic stability of a cold-active α-amylase from the Antarctic bacterium Alteromonas haloplanctis. Biochemistry 38:4613-4619. [DOI] [PubMed] [Google Scholar]
- 11.Fields, P. A., and G. N. Somero. 1998. Hot spots in cold adaptation: localized increases in conformational flexibility in lactate dehydrogenase A (4) orthologs of Antarctic notothenioid fishes. Proc. Natl. Acad. Sci. USA 95:11476-11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filman, D. J., J. T. Bolin, D. A. Matthews, and J. Kraut. 1982. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 Å resolution. II. Environment of bound NADPH and implications for catalysis. J. Biol. Chem. 257:13663-13672. [PubMed] [Google Scholar]
- 13.Gerday, C. Extremophiles. Basic concepts. In Unesco encyclopedia of life support systems, theme 6.73: extremophiles, in press.
- 14.Gerday, C., M. Aittaleb, J. L. Arpigny, E. Baise, J. P. Chessa, G. Garsoux, I. Petrescu, and G. Feller. 1997. Psychrophilic enzymes: a thermodynamic challenge. Biochim. Biophys. Acta 1342:119-131. [DOI] [PubMed] [Google Scholar]
- 15.Glansdorff, N. 1965. Topography of cotransductible arginine mutations in Escherichia coli K12. Genetics 51:167-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glansdorff, N., and Y. Xu. 2002. Microbial life at low temperatures: mechanisms of adaptation and extreme biotopes. Implications for exobiology and the origin of life. Recent Res. Dev. Microbiol. 6:1-21. [Google Scholar]
- 17.Hillcoat, B. L., P. F. Nixon, and R L. Blakley. 1967. Effect of substrate decomposition on the spectrophotometric assay of dihydrofolate reductase. Anal. Biochem. 21:178-189. [DOI] [PubMed] [Google Scholar]
- 18.Hitchings, G. H., Jr. 1989. Nobel lecture in physiology or medicine, 1988: selective inhibitors of dihydrofolate reductase. In Vitro Cell Dev. Biol. 25:303-310. [DOI] [PubMed] [Google Scholar]
- 19.Ionescu, R. M., V. F. Smith, J. C. O'Neill, Jr., and C. R. Matthews. 2000. Multistate equilibrium unfolding of Escherichia coli dihydrofolate reductase: thermodynamic and spectroscopic description of the native, intermediate, and unfolded ensembles. Biochemistry 39:9540-9550. [DOI] [PubMed] [Google Scholar]
- 20.Kohlhoff, M., A. Dahm, and R. Hensel. 1996. Tetrameric triosephosphate isomerase from hyperthermophilic Archaea. FEBS Lett. 383:245-250. [DOI] [PubMed] [Google Scholar]
- 21.Lonhienne, T., C. Gerday, and G. Feller. 2000. Psychrophilic enzymes: revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim. Biophys. Acta. 1543:1-10. [DOI] [PubMed] [Google Scholar]
- 22.Maes, D., J. P. Zeelen, N. Thanki, N. Beaucamp, M. Alvarez, M. H. Thi, J. Backmann, J. A. Martial, R. Jaenicke, R. K. Wierenga, and L. Wyns. 1999. The crystal structure of triosephosphate isomerase (TIM) from Thermotoga maritima: a comparative thermostability structural analysis of ten different TIM structures. Proteins 37:441-453. [PubMed] [Google Scholar]
- 23.Makhatadze, G. I., and P. L. Privalov. 1995. Energetics of protein structure. Adv. Protein Chem. 47:307-425. [DOI] [PubMed] [Google Scholar]
- 24.Morita, R. Y. 1975. Psychrophilic bacteria. Bacteriol. Rev. 39:144-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mozhaev, V. V., I. V. Berezin, and K. Martinek. 1988. Structure-stability relationship in proteins: fundamental tasks and strategy for the development of stabilized enzyme catalysts for biotechnology. Crit. Rev. Biochem. 23:235-281. [DOI] [PubMed] [Google Scholar]
- 26.Pace, C. N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131:266-280. [DOI] [PubMed] [Google Scholar]
- 27.Penner, M. H., and C. Frieden. 1987. Kinetic analysis of the mechanism of Escherichia coli dihydrofolate reductase. J. Biol. Chem. 262:15908-15914. [PubMed] [Google Scholar]
- 28.Perry, K. M., J. J. Onuffer, N. A. Touchette, C. S. Herndon, M. S. Gittelman, C. R. Matthews, J. T. Chen, R. J. Mayer, K. Taira, and S. J. Benkovic. 1987. Effect of single amino acid replacements on the folding and stability of dihydrofolate reductase from Escherichia coli. Biochemistry 26:2674-2682. [DOI] [PubMed] [Google Scholar]
- 29.Poe, M., N. J. Greenfield, J. M. Hirshfield, M. N. Williams, and K. Hoogsteen. 1972. Dihydrofolate reductase. Purification and characterization of the enzyme from an amethopterin-resistant mutant of Escherichia coli. Biochemistry 11:1023-1030. [DOI] [PubMed] [Google Scholar]
- 30.Privalov, P. L. 1979. Stability of proteins: small globular proteins. Adv. Protein Chem. 33:167-241. [DOI] [PubMed] [Google Scholar]
- 31.Rood, J. I., A. J. Laird, and J. M. Williams. 1980. Cloning of the Escherichia coli K12 dihydrofolate reductase gene following Mu-mediated transposition. Gene 8:255-265. [DOI] [PubMed] [Google Scholar]
- 32.Russell, N. J. 1992. Physiology and molecular biology of psychrophilic micro-organisms, p. 203-224. In R. A Herbert and R. J. Sharp (ed.), Molecular biology and bio/technology of extremophiles. Blackie & Son, Glasgow, Scotland.
- 33.Russell, N. J. 2000. Toward a molecular understanding of cold-activity of enzymes from psychrophiles. Extremophiles 4:83-90. [DOI] [PubMed] [Google Scholar]
- 34.Sawaya, M. R., and J. Kraut. 1997. Loop and subdomain momvements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry 36:586-603. [DOI] [PubMed] [Google Scholar]
- 35.Smalas, A. O., H. K. Leiros, V. Os, and N. P. Willassen. 2000. Cold adapted enzymes. Biotechnol. Annu. Rev. 6:1-57. [DOI] [PubMed] [Google Scholar]
- 36.Stone, S. R., and J. F. Morrison. 1982. Kinetic mechanism of the reaction catalyzed by dihydrofolate reductase from Escherichia coli. Biochemistry 21:3757-3765. [DOI] [PubMed] [Google Scholar]
- 37.Van de Casteele, M., P. Chen, M. Roovers, C. Legrain, and N. Glansdorff. 1997. Structure and expression of a pyrimidine gene cluster from the extreme thermophile Thermus strain ZO5. J. Bacteriol. 179:3470-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van de Casteele, M., C. Legrain, V. Wilquet, and N. Glansdorff. 1995. The dihydrofolate reductase-encoding gene dyrA of the hyperthermophilic bacterium Thermotoga maritima. Gene 158:101-105. [DOI] [PubMed] [Google Scholar]
- 39.Villeret, V., B. Clantin, C. Tricot, C. Legrain, M. Roovers, V. Stalon, N. Glansdorff, and J. Van Beeumen. 1998. The crystal structure of Pyrococcus furiosus ornithine carbamoyltransferase reveals a key role for oligomerization in enzyme stability at extremely high temperatures. Proc. Natl. Acad. Sci. USA 95:2801-2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilquet, V. 2000. NAD(P)H-dependent enzymes from extremophilic microorganisms: the eubacterial thermophilic dihydrofolate and dihydropteridine reductases and a cold-adapted glutamate dehydrogenase. Ph.D. thesis. Université Libre de Bruxelles, Brussels, Belgium.
- 41.Wilquet, V., J. A. Gaspar, M. Van de Lande, M. Van de Casteele, C. Legrain, E. M. Meiering, and N. Glansdorff. 1998. Purification and characterization of recombinant Thermotoga maritima dihydrofolate reductase Eur. J. Biochem. 255:628-637. [DOI] [PubMed] [Google Scholar]
- 42.Xu, Y., G. Feller, C. Gerday, and N. Glansdorff. 2003. Metabolic enzymes from psychrophilic bacteria: challenge of adaptation to low temperatures in ornithine carbamoyltransferase from Moritella abyssi. J. Bacteriol. 185:2161-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu, Y., Z. Liang, C. Legrain, H. J. Ruger, and N. Glansdorff. 2000. Evolution of arginine biosynthesis in the bacterial domain: novel gene-enzyme relationships from psychrophilic Moritella strains (Vibrionaceae) and evolutionary significance of N-α-acetyl ornithinase. J. Bacteriol. 182:1609-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu, Y., Y. Nogi, C. Kato, Z. Liang, H. J. Rüger, D. De Kegel, and N. Glansdorff. 2003. Moritella profunda sp. nov. and Moritella abyssi sp. nov., two psychropiezophilic organisms isolated from deep Atlantic sediments. Int. J. Syst. E vol. Microbiol. 53:533-538. [DOI] [PubMed] [Google Scholar]
- 45.Zecchinon, L., P. Claverie, T. Collins, S. D'Amico, D. Delille, G. Feller, D. Georlette, E. Gratia, A. Hoyoux, M. A. Meuwis, G. Sonan, and C. Gerday. 2001. Did psychrophilic enzymes really win the challenge? Extremophiles 5:313-321. [DOI] [PubMed] [Google Scholar]