

Abstract
The genetic and biochemical properties of three human MutS homologues, hMSH2, hMSH3, and hMSH6, have been examined. The full-length hMSH6 cDNA and genomic locus were isolated and characterized, and it was demonstrated that the hMSH6 gene consisted of 10 exons and mapped to chromosome 2p15-16. The hMSH3 cDNA was in some cases found to contain a 27-bp deletion resulting in a loss of nine amino acids, depending on the individual from which the cDNA was isolated. hMSH2, hMSH3, and hMSH6 all showed similar tissue-specific expression patterns. hMSH2 protein formed a complex with both hMSH3 and hMSH6 proteins, similar to protein complexes demonstrated by studies of the Saccharomyces cerevisiae MSH2, MSH3, and MSH6. hMSH2 was also found to form a homomultimer complex, but neither hMSH3 nor hMSH6 appear to interact with themselves or each other. Analysis of the mismatched nucleotide-binding specificity of the hMSH2–hMSH3 and hMSH2–hMSH6 protein complexes showed that they have overlapping but not identical binding specificity. These results help to explain the distribution of mutations in different mismatch-repair genes seen in hereditary nonpolyposis colon cancer.
Elevated rates of spontaneous mutations are a hallmark of defects in genes associated with postreplicative mismatch repair and led to their original designation as mutator (mut) genes in bacteria. Many of these mutator genes have been found to be conserved throughout evolution. At present there have been six homologues, MSH1–MSH6, of the bacterial MutS protein identified in the budding yeast Saccharomyces cerevisiae (1, 2, 3, 4, 5). MSH1 and MSH2 were found to be nuclear-encoded genes in which mutations resulted in elevated mutation rates in mitochondrial and nuclear DNA, respectively (6). Although its function was not immediately recognized, the first eukaryotic homologue of MutS was found as a divergently transcribed gene adjacent to the dihydrofolate reductase gene (DUT/DUG in humans and REP-3 in mice; now called MSH3) (7, 8). This gene was later found to be identical to a third yeast MutS homologue, MSH3 (2), a gene that has recently been implicated in mismatch repair (5). MSH4 and MSH5 were identified as genes involved in regulation of meiotic crossing-over in yeast, but not likely having a role in mismatch repair (3, 4). MSH6 was identified in the S. cerevisiae genome database and determined to play a role in mismatch repair (5); homology relationships and genetic analysis indicate that MSH6 is the homologue of human “G/T-binding protein” (GTBP or p160) (5, 9).
The connection of mutator genes to frequently occurring human cancers was first recognized when inherited mutations in the hMSH2 gene were found to underlie a significant proportion of hereditary nonpolyposis colon cancer (HNPCC) and subsequently acquired mutations in hMSH2 were found in sporadic tumors and tumor cell lines that display microsatellite instability characteristic of mismatch-repair defects in eukaryotes (10, 11, 12, 13, 14, 15, 16, 17, 18). The purified yeast and human MSH2 proteins have been shown to bind mismatched nucleotides and insertion/deletion loop-type (IDL) mismatched substrates, suggesting that it has a function similar to that of MutS, albeit for eukaryotic mismatch recognition (19, 20, 21). Purification of an activity that complements the biochemical mismatch-repair deficiency exhibited by Lovo cells, which contain a deletion of hMSH2 exons 3 through 8 (17), identified a heterodimeric complex containing hMSH2 and a 160-kDa polypeptide (22). This heterodimer was suggested to be the mismatch-recognition component of human mismatch repair. The latter protein was found to be identical to the GTBP (or p160) that was mapped to a region of chromosome 2 (2p15-16) approximately 1 megabase away from hMSH2 (23, 24). Because there is at present no evidence that the G/T-binding protein by itself actually binds mismatched nucleotides, and because functional and evolutionary relatedness studies indicate that GTBP is the homologue of S. cerevisiae MSH6, we will refer to the human MutS homologues with respect to their homology to yeast genes (e.g., hMSH2, hMSH3, and hMSH6) to simplify the nomenclature of MutS homologue genes.
The involvement of MSH3 and MSH6 in MSH2-dependent mismatch repair has been detailed in genetic studies of the mutator phenotype caused by mutation of these genes in yeast (5, 9, 25, 26). msh2 mutations were found to cause a general mutator phenotype consistent with defects in repair of single-base-pair substitution, single-nucleotide insertion, and multiple-nucleotide insertion mispairs. msh3 mutations caused a limited mutator phenotype, suggesting that it might primarily be involved in the repair of multiple-nucleotide insertion mispairs, whereas msh6 mutations caused strong defects in the repair of single-base-pair substitution mispairs and partial defects in the repair of single-nucleotide insertion mispairs. The msh3 msh6 double mutant combination appeared to cause the same repair defect as the msh2 single mutation, indicating that MSH3 and MSH6 had overlapping functions in MSH2-dependent mismatch repair in yeast. The demonstration that MSH2 could form complexes with both MSH3 and MSH6 suggested that eukaryotic mismatch repair might involve two different heterodimeric mismatch-recognition complexes, each having a different mispair-recognition specificity. If similar functions were identified with respect to the human homologues, these data would have important implications for the distribution of germ-line mutations in hMSH2, hMSH3, and hMSH6 that would be expected to be found in HNPCC patients (5, 9).
Here we report that hMSH3 and hMSH6 interact with hMSH2 and that these two complexes contain different mismatch-recognition specificity similar to that predicted by the genetic analysis of the corresponding S. cerevisiae genes. We have also characterized the complete cDNA and genomic locus of hMSH6 and the expression of hMSH6 in human tissues, and have further characterized the hMSH3 coding sequence.
METHODS
Chemicals and Enzymes.
[35S]Methionine was purchased from New England Nuclear. Protein A-agarose beads were purchased from Santa Cruz Biotechnology. hMSH2 polyclonal antibody (OS118) was obtained from Oncogene Science. All fine chemicals used were purchased from Amresco (Euclid, OH). In Vitro Transcription-Translation kits were purchased from Promega. Prestained markers were from Bio-Rad. Oligonucleotides were synthesized using solid-phase cyanoethyl phosphoramidite chemistry and were obtained from the Dana-Farber Cancer Institute Core Molecular Biology Facility or from the Kimmel Cancer Institute Nucleic Acids Facility. The human multiple-tissue Northern blot IV was purchased from CLONTECH.
Isolation and Characterization of cDNA and Genomic DNA Clones.
The hMSH6 genomic locus was cloned from a human P1 library screened by Genome Systems (St. Louis). One round of screening was performed with primers 22980 5′-GTA TGA AGA AAC TAC ATA CAG C and 23045 5′-AAG TCC AGT CTT TCG AGC C (23) to obtain three P1 phages. One additional P1 phage containing the N terminus of the gene was obtained by screening with primers 24141 5′-CAG AAG GGA GGT CAT TTT TAC AG and 23444 5′-GAA GGC TCA TCA CAC ACT GCC. The resulting phages were transduced into the CRE-negative Escherichia coli strain NS3516 using a protocol provided by Genome Systems. DNA preparations were made using the alkaline lysis protocol described by Genome Systems and then spot-dialyzed on a VSWP 02500 Millipore filter against H2O for 30 min. The hMSH6 coding sequence and flanking intron sequences were then determined on an Applied Biosystems 373 DNA sequencer using Taq DNA polymerase and dye terminators and protocols supplied by the manufacturer (Perkin–Elmer), and sequencing primers derived from the cDNA sequence essentially as previously described (12, 27). The sizes of the hMSH6 introns were estimated by PCR essentially as previously described (12, 27) or by determining the sequence of the entire intron. All of the genomic sequences have been submitted to GenBank.
An initial ≈4-kb hMSH6 cDNA clone was isolated by screening a λ ZAP II (Stratagene) library constructed from fibroblast 1262 poly(A) mRNA using conventional methods (28). However, this cDNA clone appeared to be ≈200 to 500 bp shorter than the full-length hMSH6 mRNA as determined by Northern blotting, suggesting that a portion of the 5′ end was missing from the cDNA clone.
To isolate an intact hMSH6 cDNA clone, the 5′ end of the cDNA was first cloned by 5′ rapid amplification of cDNA ends (RACE) (29). Single-strand, random-primed cDNA was prepared from human testis poly(A) mRNA (CLONTECH) using a Promega cDNA synthesis kit. The cDNA was then treated with RNase H, extracted with phenol, and precipitated with ethanol. The single-strand cDNA was used directly in a RACE reaction. Four different oligonucleotides were used in this experiment: RACE anchor, 5′-CA CGG ATC CAC TAT CGA TTC TGG AAC; RACE primer, 5′-CCA GAA TCG ATA GTG GAT CCG T; hMSH6-A, 5′-ACG TTG CAT TGC TCT CAG TAT TTC; and hMSH6-B, 5′-CCA AAC CAA ATC TCC TGG TGA. Dideoxythymine (Perkin–Elmer) was added to the RACE anchor primer using terminal transferase (Pharmacia). The RACE anchor was then ligated to the single-strand cDNA for 6 hr at 37°C using RNA ligase (New England Biolabs) in 50 mM Tris, pH 7.8/10 mM magnesium chloride/1 mM hexamine cobalt chloride/20 μM ATP/10 μg/ml BSA/50% PEG 8000, and this cDNA was used directly for PCR. The first round of PCR was carried out for 35 cycles using the RACE primer and hMSH6-A primer. One microliter of the resulting PCR product was then used as a template for a second round of PCR using the RACE primer and hMSH6-B primer for 25 cycles. PCR was performed in 75 mM Tris, pH 9.0/20 mM ammonium sulfate/2 mM magnesium chloride/0.01% Tween 20/10% glycerol/0.2 mM each of the four dNTPs/0.4 μM of each primer. Cycling temperatures consisted of 30 sec at 94°C, 1 min at 50°C, and 1 min at 72°C, and was performed in a Perkin–Elmer 2400. Amplified fragments were cloned using a TA Cloning Kit (Invitrogen) and sequenced using a Sequenase 3.0 sequencing kit (United States Biochemical) or a ABI 373 sequencer as described above.
A 350-bp fragment containing the 5′ RACE-derived hMSH6 sequence was used to probe a λ ZAP II (Stratagene) library constructed from fibroblast 1262 poly(A) mRNA (28). The probe was labeled using a Nick Translation Kit (GIBCO) and [32P]dCTP (New England Nuclear). Library screening and phagemid rescue was performed using protocols provided by the manufacturer (Stratagene). One cDNA clone (M6-1) was then sequenced and used for all experiments in which the hMSH6 protein was analyzed. The sequence of this clone has been submitted to GenBank (accession no. U54777U54777). A hMSH3 cDNA (M3-12), beginning at nucleotide 54 of the published hMSH3 sequence (7, 30) and extending 899 nucleotides 3′, to include a poly(A), was similarly isolated by screening a HeLa λ ZAP cDNA library (Stratagene) and used for all experiments in which the hMSH3 protein was analyzed (GenBank accession number U61981U61981).
In Vitro Transcription and Translation.
hMSH2, hMSH3, and hMSH6, subcloned into the pET vectors pET 3d, pET 24d, and pET 29a (Novagen), respectively, were used for in vitro transcription and translation studies. DNA was prepared and purified with the Qiagen (Chatsworth, CA) kit. In vitro transcription and translation of the different genes was performed in 50-μl volumes, according to the standard procedures provided by the manufacturer (Promega). The amount of each template DNA added to the in vitro transcription and translation was optimized to obtain the maximum possible translation for the respective gene. From 1 to 2 μg of DNA was used as template in typical reactions. [35S]Methionine-labeled rabbit reticulocyte translation products of comparable translation efficiencies were mixed and further incubated at 37°C for 30 min or at 4°C for 6 hr. This was then processed for either immunoprecipitation directly or cross-linking followed by immunoprecipitation. For mismatch binding experiments, unlabeled proteins were synthesized by omitting the [35S]methionine from the reactions.
Immunoprecipitation.
For immunoprecipitation, 5 μl of in vitro transcription and translation mixture was diluted in 100 μl of ice-cold buffer containing 0.1 M potassium phosphate, pH 7.5, and 100 mM Tris, pH 7.5. Eighty microliters of this was added to 250 μl of ice-cold IP buffer (20 mM Tris, pH 7.5/10% glycerol/150 mM NaCl/5 mM EDTA/1 mM DTT/0.1% Tween 20/0.75 mg/ml BSA/0.5 mM phenylmethylsulfonyl fluoride/0.8 μg/ml leupeptin) containing 5–10 μg/ml hMSH2 polyclonal antibody. The samples were incubated at 4°C on a rocking platform for 6–8 hr. Protein A-agarose beads were washed in IP buffer, a 1:1 suspension of Protein A-agarose beads was prepared in IP buffer and incubated for 30 min at 4°C, and then 50 μl of this suspension was added to the samples. Incubation was continued for 12–15 hr at 4°C on the rocking platform. The beads were harvested by centrifugation at 2000 rpm for 30 sec and washed three times with 500 μl of ice-cold IP buffer at 4°C. Thirty microliters of 2× SDS sample loading buffer (0.25 M Tris, pH 6.8/20% glycerol/4% SDS/10% 2-mercaptoethanol/0.25% bromophenol blue) was added to the beads, the samples were incubated at 95°C for 5 min and centrifuged for 2 min in a microfuge at room temperature, and equivalent amounts of protein were analyzed by PAGE on 6% SDS gels. The gels were fixed and dried, and the proteins were visualized with a Molecular Dynamics PhosphorImager.
Protein Cross-Linking.
For cross-linking, 5 μl of the protein samples were diluted in 100 μl of 0.1 M potassium phosphate buffer, pH 7.5, containing 0.002% glutaraldehyde and incubated at 25°C for 30 min. The cross-linking reaction was terminated by the addition of 1 M Tris, pH 7.5, to a final concentration of 100 mM. Eighty microliters of the reaction was processed for immunoprecipitation and analyzed by 5% SDS/PAGE as described above.
Mismatch Binding Assay.
The hMSH2, hMSH3, and hMSH6 constructs were in vitro transcribed and translated as described above. A sample without DNA was processed as a control. Five microliters of the hMSH2 sample was mixed with 5 μl of hMSH3 or hMSH6 sample in separate reactions and incubated at room temperature for 15 min. 32P-labeled DNA duplexes containing either a mismatch (G/T) or IDL [+T, +CA, +(CA)5] were prepared as previously described (21). The sequences of the oligonucleotides used to construct these substrates are as follows: G/T, top: 5′-CGGCGAATTCCACCAAGCTTGATCGCTCGAGGTACCAGG-3′, bottom: 5′-CCTGGTACCTCGAGCGATCGAGCTTGGTGGAATTCGCCG-3′; +T, top: 5′-CGGCGAATTCCACCCAGCTTGATCTCTCGAGGTACCAGG-3′; +G, top: 5′-CGGCGAATTCCACCCAGCTGGATCTCTCGAGGTACCAGG-3′; +CA, top: 5′-CGGCGAATTCCACCCAGCTCAGATCTCTCGAGGTACCAGG-3′; +(CA)5, top: 5′-CGGCGAATTCCACCCAGCTCACACACACAGATCTCTCGAGGTACCAGG-3′; bottom strand for +T, +G, +CA, and +(CA)5: 5′-CCTGGTACCTCGAGAGATCAGCTGGGTGGAATTCGCCG-3′. Mismatched DNA binding assays were carried out in 30-μl reactions containing 10 μl of the protein mixtures described above, DNA binding buffer [50 mM potassium phosphate, pH 7.5/50 mM NaCl/0.1 mM EDTA/1 mM DTT/5 mM AMP/10% glycerol/2 μg poly(dI·dC)/100 ng of single-strand DNA oligomer (39 bp)/2 ng of 32P-labeled DNA duplex], and incubated at 37°C for 10 min. The binding reactions were loaded onto a 1.5-mm-thick, 22-cm-long by 14-cm-wide 4% polyacrylamide gel containing 2.5% glycerol that was run at 35 mA for 3 hr as previously described (21). The gels were then dried and analyzed on a Molecular Dynamics PhosphorImager.
RESULTS
Characterization of the hMSH3 and hMSH6 Genes.
The hMSH6 cDNA was isolated by a combination of cDNA library screening and 5′ RACE and the hMSH6 genomic locus was isolated by screening a human P1 library. hMSH6 was found to be transcribed as a 4245-bp mRNA encoding a 153-kDa protein with 1360 amino acids. We found one apparent nucleotide polymorphism (C185A) from the published partial cDNA sequence (24) that did not alter the coding amino acid at position 62. In addition, both cDNA and genomic sequencing revealed a single cytosine deletion 36 nucleotides downstream from the stop codon compared with the original cDNA sequence (24). The genomic locus was found to have 10 exons that cover 20 kb of chromosomal DNA; a diagram of the genomic organization and the DNA sequences defining the intron-exon junctions are shown in Fig. 1. The map location of hMSH6 was determined by analyzing a Genebridge-4 radiation hybrid panel (Research Genetics, Huntsville, AL) and found to be 2p15-16 (data not shown). The expression of hMSH6 was analyzed by probing a tissue-specific Northern blot (Fig. 2). The hMSH6 mRNA (≈4.5 kb) was expressed in all tissues examined, but appeared to be more highly expressed in testis, thymus, and uterus. This expression distribution is similar to that previously observed for hMSH2 and hMSH3 (30, 31). The hMSH6 cDNA probe also hybridized weakly to an ≈7.3-kb RNA species the identity of which is unknown at present.
Figure 1.
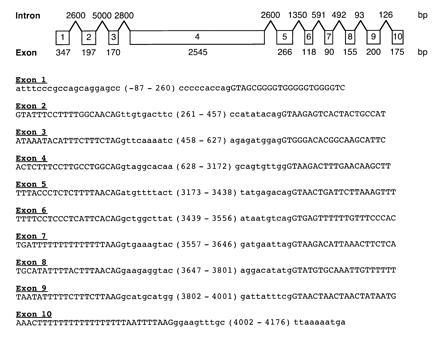
Organization of hMSH6 genomic locus and sequence of the intron region flanking each MSH6 exon. Boxes containing numbers 1 through 10 indicate the individual MSH6 exons and their relative sizes. The size of each exon is given below each exon and the size of each intron is given above the region between each pair of exons. The sizes of exons 1 and 10 are the sizes of the mRNA sequence upstream and downstream of the first and last introns, respectively; these sizes were calculated from the longest MSH6 cDNA sequence available. The first 20 nucleotides of each intron sequence up to the intron-exon junction is given in uppercase letters except for the 3′ side of the last intron, where additional sequence is given. The first 10 nucleotides of each exon sequence up to the intron-exon junction is given in lowercase letters. In addition, the sequence of the 5′ and 3′ ends of the cDNA, minus the poly(A) sequence, is also given in lowercase letters. The numbers in parentheses between intron sequences are the nucleotide coordinates of the exon sequences or cDNA sequences, assuming the A of the ATG is nucleotide 1. (Additional intron sequence including the complete sequence of some introns has been determined in all cases and is available on request from M.F.K. and R.K.)
Figure 2.
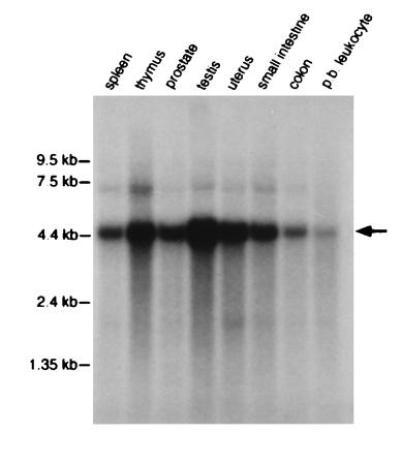
Tissue expression of hMSH6 mRNA. A human multiple-tissue Northern blot (CLONTECH) was probed with the 32P-labeled complete cDNA clone of hMSH6 according to the manufacturer’s protocol. Colon refers to mucosal lining; p.b. leukocyte refers to peripheral blood leukocyte. The hMSH6 transcript is indicated by the arrow and corresponds to ≈4.5 kb. The amount of RNA loaded in each lane was adjusted to comparable levels as judged spectrophotometrically and by the levels of actin present (CLONTECH).
We isolated a complete cDNA coding for the human hMSH3 gene (MS3-12) by screening a HeLa cDNA library essentially as described for hMSH6. Sequence analysis of this clone indicated that it contained a 27-bp deletion from nucleotides 229 to 255, which eliminated codons 57 to 65 compared with the published sequence (GenBank accession no. J04810J04810) (7). This alteration appears to result from the deletion of 3 of 10 9-bp imperfect direct-repeat sequences (consensus: GSCYSCAGC) spanning nucleotides 184 to 273 of the original published hMSH3 cDNA sequence and appears similar to a previously noted deletion in a HeLaR genomic clone (7, 32). This appeared to result from either recombination or replication slippage between repeats five and seven. A cDNA PCR assay was devised in which this region was amplified and the resulting product was separated based on its size (Fig. 3). Using this assay, a similar deletion was found in cDNA from the HeLa and SW480 tumor cell lines. In contrast, the originally reported HL60R cDNA clone kindly provided by Shimada and coworkers (Nippon Medical School, Tokyo) did not contain the deletion, nor did cDNA derived from the liver of a 40-year-old Caucasian male and testis cDNA pooled from seven Caucasian males aged 10–37. Interestingly, small intestine cDNA from a 22-year-old male and kidney cDNA from an unidentified individual as well as pooled cDNAs from bone marrow and placenta contained both the deletion and the wild-type sizes. At present we have no evidence that this deletion has any functional consequences, and its widespread dissemination throughout the cDNAs that we have tested appears to indicate that the 27-bp deletion is a sequence polymorphism. In addition, we found two other nucleotide differences with the published sequence that resulted in silent changes (T162C and T204G), and two nucleotide differences that resulted in amino acid changes (G1865A/G622E and A3133G/T1045A); these sequence changes could also be the result of silent polymorphisms in the population.
Figure 3.
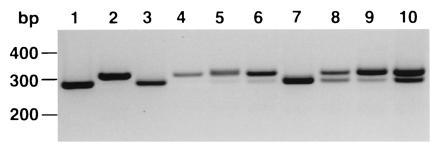
Analysis of hMSH3 27-bp deletion in various cDNAs. Tissue cDNAs from individuals or combined groups (CLONTECH) were used in a PCR reaction with primers flanking the region of the 27-bp deletion. The expected sizes of the products are 307 bp for the wild type and 280 bp for the deletion. Lane 1, MS3-12 HeLa hMSH3 cDNA clone; lane 2, HL60R hMSH3 cDNA clone; lane 3, HeLa cDNA; lane 4, liver cDNA from 40-year-old Caucasian male; lane 5, testis cDNA pooled from seven Caucasians aged 10–37; lane 6, placenta cDNA; lane 7, SW480 colorectal carcinoma cell line cDNA; lane 8, small intestine cDNA from a 22-year-old male; lane 9, bone marrow cDNA pooled from 24 Caucasian males and females aged 16–70; lane 10, kidney cDNA (unknown origin).
Interactions Between hMSH2, hMSH3, and hMSH6.
Two approaches were used to study possible interactions between hMSH2, hMSH3, and hMSH6. The first approach involved in vitro transcription and translation followed by co-immunoprecipitation of interacting proteins using a polyclonal antibody directed against hMSH2. The second approach involved chemical cross-linking of the in vitro translated products with glutaraldehyde followed by immunoprecipitation with the hMSH2-specific polyclonal antibody or direct examination of the cross-linked products. The results of these experiments are shown in Figs. 4 and 5. In some co-immunoprecipitation experiments, hMSH2 was labeled with [35S]methionine to monitor its synthesis and immunoprecipitation, whereas in other experiments hMSH2 was not labeled and only the co-immunoprecipitated proteins were monitored. We found that hMSH3 was immunoprecipitated by the hMSH2-specific antibody only in the presence of hMSH2 protein (Fig. 4, lanes 6, 9, and 12). Likewise, hMSH6 was immunoprecipitated by the hMSH2-specific antibody only when present in the context of hMSH2 (Fig. 4, lanes 7, 10, and 13). In vitro translated luciferase protein could not be immunoprecipitated by the hMSH2-specific antibody either by itself or in the presence of hMSH2 under the conditions in which hMSH3 and/or hMSH6 co-immunoprecipitate with hMSH2, indicating the interactions between hMSH2 and hMSH3 or hMSH6 were specific. We employed highly stringent wash conditions for the immunoprecipitates shown in Fig. 4, which appeared to reduce the expected 1:1 stoichiometry of hMSH2 interaction with hMSH6 (22) or hMSH3; similarly, it also eliminated background nonspecific interactions.
Figure 4.
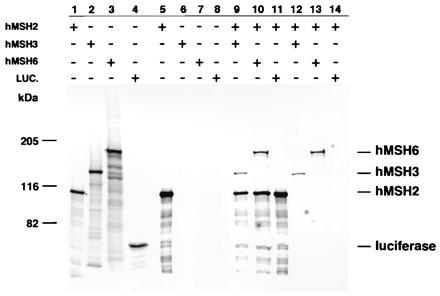
Immunoprecipitation of hMSH2, hMSH3, and hMSH6 protein complexes. In vitro transcribed and translated hMSH2, hMSH3, and hMSH6 were mixed and processed for immunoprecipitation (see Materials and Methods). Lanes 1–4, translations of hMSH2, hMSH3, hMSH6, and luciferase, respectively; lanes 5–8, immunoprecipitates of individual translated hMSH2, hMSH3, hMSH6, and luciferase, respectively; lane 9, co-immunoprecipitate of hMSH2 and hMSH3; lane 10, co-immunoprecipitate of hMSH2 and hMSH6; lane 11, co-immunoprecipitate of hMSH2 and luciferase; lane 12, co-immunoprecipitate of hMSH2 (unlabeled) and hMSH3; lane 13, co-immunoprecipitate of hMSH2 (unlabeled) and hMSH6; lane 14, co-immunoprecipitate of hMSH2 (unlabeled) and luciferase.
Figure 5.
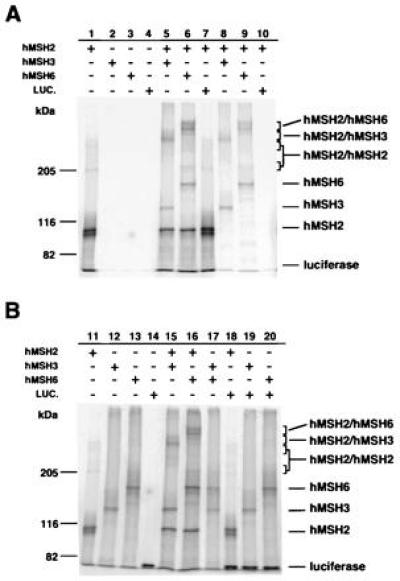
Chemical cross-link analysis of hMSH2, hMSH3, and hMSH6 protein complexes. The in vitro transcribed and translated proteins were mixed, cross-linked, and processed for immunoprecipitation (see Materials and Methods). (A) Lanes 1–4, immunoprecipitates of individually cross-linked hMSH2 (lane 1), hMSH3 (lane 2), hMSH6 (lane 3), and luciferase (lane 4); lanes 5–10, immunoprecipitates of the cross-linked complexes hMSH2 and hMSH3 (lane 5), hMSH2 and hMSH6 (lane 6), hMSH2 and luciferase (lane 7), hMSH2 (unlabeled) and hMSH3 (lane 8), hMSH2 (unlabeled) and hMSH6 (lane 9), hMSH2 (unlabeled) and luciferase (lane 10). (B) Lanes 11–18, cross-linked complexes before immunoprecipitation including hMSH2 (lane 11), hMSH3 (lane 12), hMSH6 (lane 13), luciferase (lane 14), hMSH2 and hMSH3 (lane 15), hMSH2 and hMSH6 (lane 16), hMSH3 and hMSH6 (lane 17), hMSH2 and luciferase (lane 18), hMSH3 and luciferase (lane 19), hMSH6 and luciferase (lane 20).
To further investigate these interactions and determine whether these proteins interact with themselves, chemical cross-linking using glutaraldehyde was performed and the complexes were immunoprecipitated using the hMSH2-specific polyclonal antibody. In the presence of hMSH2 and hMSH3, a high molecular mass complex consistent with the formation of a heteromultimer was observed (Fig. 5A, lanes 5 and 8). Similarly, in the presence of hMSH2 and hMSH6, a high molecular mass complex consistent with the formation of a heteromultimer was observed (Fig. 5A, lanes 6 and 9). These specific complexes were not due to interaction of hMSH2, hMSH3, or hMSH6 with themselves (see below). The higher-order hMSH2–hMSH3 and hMSH2–hMSH6 complexes do not appear to correspond exactly to calculated heterodimer sizes, although it is possible that the cross-linking procedure or cross-linking to alternative translation products present at low levels results in a complex that runs anomolously.
hMSH2 alone was also found to form a stable cross-linked complex having an approximate molecular mass of 210 kDa, consistent with the formation of a homodimer as well as a larger multimer (Fig. 5A, lane 1). The hMSH2 complex was formed only when hMSH2 alone was present and was either not observed or largely reduced in the presence of hMSH3 or hMSH6 (Fig. 5A, compare lanes 1, 5, and 6). These multimeric complexes could also be detected prior to immunoprecipitation (Fig. 5B, lanes 11, 15, and 16). A heteromultimeric complex was not formed when a nonspecific protein, luciferase, was mixed with hMSH2 before chemical cross-linking (Fig. 5A, lanes 7 and 10). No higher-order complexes were observed in cross-linked samples containing hMSH3 or hMSH6 alone or when hMSH3 and hMSH6 were mixed (Fig. 5B, lanes 12, 13, and 15), indicating that these two proteins do not appear to interact with each other or form multimers.
Mismatch Binding Specificity of hMSH2–hMSH3 and hMSH2–hMSH6 Complexes.
A gel-shift assay was utilized to examine mismatch binding by hMSH2–hMSH3 and hMSH2–hMSH6 complexes (Fig. 6). DNA heteroduplexes containing either a single G/T mismatch (Fig. 6B), a +1 thymine (+T) nucleotide insertion (Fig. 6C), a +1 guanine (+G) nucleotide insertion (Fig. 6D), a +2 cytosine-adenine (+CA) dinucleotide insertion (Fig. 6E) or a +10 cytosine-adenine (+CA)5 dinucleotide repeat insertion (Fig. 6F) were tested. A homoduplex DNA oligonucleotide was used as a control (Fig. 6A). The results of this assay indicated that under these binding conditions, hMSH2, hMSH3, and hMSH6 alone are not capable of binding to the heteroduplex DNA oligonucleotides, nor do any of the individual proteins or mixtures bind to the homoduplex substrate. However, when mixed, hMSH2 and hMSH3 formed a complex with the +G, +CA, and +(CA)5 substrates but not with the substrate containing a G/T mispair or the +T insertion mispair. In contrast, the mixed hMSH2 and hMSH6 bound to a G/T mismatch and the +T and +G insertion mismatches, but did not form complexes with the other IDL substrates. The lack of binding to homoduplex substrate DNA indicates that these complexes bind specifically to mismatch-containing substrate DNA. These results indicate that functional hMSH2–hMSH3 and hMSH2–hMSH6 heteromultimeric complexes are formed and that both of these complexes can recognize mispaired bases in DNA. Faster-migrating species were observed in all lanes in Fig. 6. These complexes are likely to be formed by endogenous DNA binding proteins present in the reticulocyte lysate because these complexes are also present in lane 6 (control, no DNA added).
Figure 6.
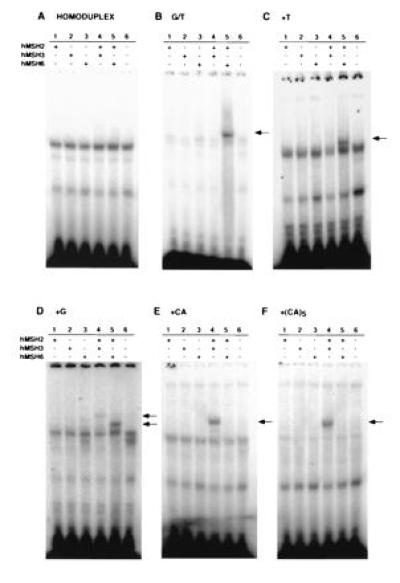
Mismatch-binding specificity of hMSH2, hMSH3, and hMSH6 protein complexes. In vitro transcribed and translated hMSH2, hMSH3, hMSH6, hMSH2–hMSH3, and hMSH2–hMSH6 complexes and a control without DNA were incubated with different mispair-containing, 32P-labeled DNA oligoduplexes and the resulting protein-DNA complexes were analyzed using a gel-shift assay (see Materials and Methods). The DNA oligoduplexes were: (A) homoduplex; (B) G/T mismatch; (C) +T IDL; (D) +G IDL; (E) +CA; (F) +(CA)5. All gels were loaded as follows: lane 1, hMSH2; lane 2, hMSH3; lane 3, hMSH6; lane 4, hMSH2–hMSH3; lane 5, hMSH2–hMSH6; lane 6, control (no DNA).
DISCUSSION
The present study has demonstrated the physical and functional interactions between the human MutS homologues hMSH2, hMSH3, and hMSH6. Immunoprecipitation and cross-linking experiments show that hMSH2 forms a stable complex with both hMSH3 and hMSH6 in vitro. Additionally, the cross-linking experiments indicated that hMSH2 is capable of associating with itself in vitro, although at a lower efficiency. This observation is important because hMSH2 appears to be in excess of hMSH3 and hMSH6 in most proliferating human cells (S.A., T.W., and R.F., unpublished observations). It is possible that most if not all of the hMSH3 and hMSH6 are bound in complexes with hMSH2 and that the remaining free hMSH2 may then form a hMSH2 complex capable of substrate recognition and/or other cellular functions. In this regard, it has been shown that both purified hMSH2 and S. cerevisiae MSH2 alone are capable of binding both single-base-pair and IDL mismatches with affinities similar to those of the bacterial MutS protein (19, 20, 21).
The studies reported here demonstrate that hMSH2 functionally interacts with hMSH3 and hMSH6 to form complexes that recognize mispaired bases in DNA. The hMSH2–hMSH3 complex appeared to recognize some +1 nucleotide IDL (+G) and larger IDL (+CA and +CA5) substrates but did not recognize a G/T mismatch or a +T nucleotide IDL. In contrast, the hMSH2–hMSH6 complex recognized the G/T mismatch and some of the +1 nucleotide IDL (+T and +G) substrates, but did not appear to recognize any of the larger IDL substrates. These results suggest that the hMSH2–hMSH3 and hMSH2–hMSH6 complexes have both different and overlapping mispair-recognition specificity. Previous studies have shown that an in vitro transcribed and translated truncated version of GTBP obtained using an incomplete cDNA clone (GTBP/p160 = hMSH6) also binds to G/T mismatches in the presence of hMSH2 (24). Furthermore, a “hMutSα complex” (hMSH2 plus hMSH6/GTBP/p160) purified from HeLa cells has been demonstrated to bind to a single-base mismatch (G/T), a +T insertion, and a +(T)3 insertion (22). The binding of the +(T)3 substrate is in contrast to our observations that the hMSH2–hMSH6 complex did not bind to larger IDL substrates. In addition, unlike purified hMSH2 protein (20, 21), the in vitro transcribed and translated hMSH2 alone was incapable of binding the mismatched heteroduplexes used in the present gel-shift assay system. A possible explanation for these apparent discrepancies is that the assays performed here involve the analysis of limiting amounts of in vitro transcribed and translated repair proteins (substrate excess) and thus may detect only the highest-binding affinity interactions, precluding our ability to detect functionally significant, lower-affinity binding complexes under the reaction conditions used. Indeed, analysis of S. cerevisiae suggests that the MSH2-MSH6 complex should recognize larger IDL mismatches, albeit at a lower affinity than the MSH2-MSH3 complex (9), and preliminary studies with purified S. cerevisiae MSH2-MSH6 complex have demonstrated that this is the case (G.T.M. and R.K., unpublished results). It is likely that a much more detailed study of the mispair-binding properties of these complexes using purified proteins, combined with a detailed analysis of the in vivo repair defects caused by mutations in the genes encoding these proteins, will be required to fully understand the functional mispair-recognition properties of these proteins.
Recent genetic studies of the corresponding yeast homologues have implicated the formation of MSH2–MSH3 and MSH2–MSH6 complexes and presented a detailed analysis of the mismatch-repair defects caused by mutations in these genes (5, 9, 25, 26). These studies have suggested that repair of single-base substitution mispairs requires the MSH2–MSH6 complex, that repair of single-base insertion/deletion mispairs can utilize both the MSH2–MSH6 or MSH2–MSH3 complexes, and that repair of larger insertion/deletion mispairs likely utilizes the MSH2–MSH3 complex more frequently than the MSH2-MSH6 complex (5, 9). The data presented here, combined with previously reported data on the mispair-binding properties on hMSH2–hMSH6 (GTBP) complexes, are in general agreement with studies of the yeast MutS homologues.
The functional relationships between hMSH2, hMSH3, and hMSH6 appear to at least partially explain the distribution of mutations in mismatch-repair genes associated with inherited and acquired cancer susceptibility (17, 18). Our results indicate that hMSH2 is central to all mismatch recognition, thereby providing an explanation for the high prevalence of hMSH2 mutations observed in HNPCC patients compared with hMSH6 (GTBP) mutations (MSH3 has not yet been tested) (18). Single mutations in hMSH3 and hMSH6 are unlikely to cause the same types of mismatch-repair defects as mutations in hMSH2 because they have partially redundant functions. This suggests that mutations in hMSH3 and hMSH6 may account for a very small subset of the HNPCC patients, if any, or alternatively that they may account for different types of inherited cancer susceptibility typified by the nature of the mismatch-repair defects caused by mutations in these genes. However, mutation of both hMSH3 and hMSH6 may mimic the mutation of hMSH2 in a manner similar to what has been found in yeast (5, 9). More detailed studies to determine the relative binding affinities for the different complexes are in progress and should provide greater insight into both the mechanism of mismatch recognition and the possible involvement of defects in hMSH3 and hMSH6 in cancer susceptibility.
Acknowledgments
We thank Mon-Li Chu for fibroblast 1262 cDNA library and T. Shimada for providing hMSH3 cDNA. We thank Paul Morrison, Marissa Dovido, and Christene Earabino for oligonucleotides and sequence analysis of hMSH3 and hMSH6 cDNAs and the hMSH6 genomic locus. This work was supported by National Institutes of Health Grants GM50006, CA44704 (R.K.), CA06516 (Dana–Farber Cancer Institute), and CA56542 and CA67007 (R.F.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: GTBP, G/T-binding protein; HNPCC, hereditary nonpolyposis colon cancer; IDL, insertion/deletion loop-type; RACE, rapid amplification of cDNA ends.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base [accession nos. U61981U61981 (hMSH3), U54777U54777 (hMSH6), cDNAs, and U73732-7 (hMSH6) genomic locus].
References
- 1.Reenan R A G, Kolodner R D. Genetics. 1992;132:963–973. doi: 10.1093/genetics/132.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.New L, Liu K, Crouse G F. Mol Gen Genet. 1993;239:97–108. doi: 10.1007/BF00281607. [DOI] [PubMed] [Google Scholar]
- 3.Ross–Macdonald P, Roeder G S. Cell. 1994;79:1069–1080. doi: 10.1016/0092-8674(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 4.Hollingsworth N M, Ponte L, Halsey C. Genes Dev. 1995;9:1728–1739. doi: 10.1101/gad.9.14.1728. [DOI] [PubMed] [Google Scholar]
- 5.Marsischky G T, Filosi N, Kane M F, Kolodner R. Genes Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 6.Reenan R A G, Kolodner R D. Genetics. 1992;132:975–985. doi: 10.1093/genetics/132.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujii H, Shimada T. J Biol Chem. 1989;264:10057–10064. [PubMed] [Google Scholar]
- 8.Linton J P, Yen J Y, Selby E, Chen Z, Chinsky J M, Liu K, Kellems R E, Crouse G F. Mol Cell Biol. 1989;9:3058–3072. doi: 10.1128/mcb.9.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolodner R. Genes Dev. 1996;10:1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- 10.Fishel R A, Lescoe M K, Rao M R S, Copland N, Jenkins N, Garber J, Kane M, Kolodner R. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 11.Leach F S, Nicolaides N C, Papadopoulos N, Liu B, Jen J, et al. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 12.Kolodner R D, Hall N R, Lipford J, Kane M F, Rao M R S, Morrison P, Wirth L, Finan P J, Burn J, Chapman P, Earabino C, Merchant E, Bishop T. Genomics. 1994;24:516–526. doi: 10.1006/geno.1994.1661. [DOI] [PubMed] [Google Scholar]
- 13.Liu B, Parsons R E, Hamilton S R, Petersen G M, Lynch H T, Watson P, Markowitz S, Willson J K V, Green J, de la Chapelle A, Kinzler K W, Vogelstein B. Cancer Res. 1994;54:4590–4594. [PubMed] [Google Scholar]
- 14.Borresen A L, Lothe R A, Meling G I, Lystad S, Morrison P, Lipford J, Kane M F, Rognum T O, Kolodner R D. Hum Mol Genet. 1995;4:2065–2072. doi: 10.1093/hmg/4.11.2065. [DOI] [PubMed] [Google Scholar]
- 15.Liu B, Nicolaides M C, Markowitz S, Willson J K V, Parsons R E, Jen J, Papadopolous N, Peltomaki P, de la Chapelle A, Hamilton S R, Kinzler A W, Vogelstein B. Nat Genet. 1995;9:48–55. doi: 10.1038/ng0195-48. [DOI] [PubMed] [Google Scholar]
- 16.Umar A, Boyer J C, Thomas D C, Nguyen D C, Risinger J I, Boyd J, Ionov Y, Perucho M, Kunkel T A. J Biol Chem. 1994;259:1–4. [PubMed] [Google Scholar]
- 17.Boyer J C, Umar A, Risinger J I, Lipford J R, Kane M, Yin S, Barrett J C, Kolodner R D, Kunkel T A. Cancer Res. 1995;55:6063–6070. [PubMed] [Google Scholar]
- 18.Liu B, Parsons R, Papadopoulos N, Nicolaides N C, Lynch H T, Watson P, Jass J R, Dunlop M, Wyllie A, Peltomaki P, de la Chapelle A, Hamilton S R, Vogelstein B, Kinzler K W. Nat Med. 1996;2:169–174. doi: 10.1038/nm0296-169. [DOI] [PubMed] [Google Scholar]
- 19.Alani E, Chi N W, Kolodner R. Genes Dev. 1995;9:234–247. doi: 10.1101/gad.9.2.234. [DOI] [PubMed] [Google Scholar]
- 20.Fishel R, Ewel A, Lescoe M K. Cancer Res. 1994;54:5539–5542. [PubMed] [Google Scholar]
- 21.Fishel R, Ewel A, Lee S, Lescoe M K, Griffith J. Science. 1994;266:1403–1405. doi: 10.1126/science.7973733. [DOI] [PubMed] [Google Scholar]
- 22.Drummond J T, Li G-M, Longley M J, Modrich P. Science. 1995;268:1909–1912. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- 23.Papadopoulos N, Nicolaides N C, Liu B, Parsons R, Lengauer C, Palombo F, D’Arrigo A, Markowitz S, Willson J K V, Kinzler K W, Jiricny J, Vogelstein B. Science. 1995;268:1915–1917. doi: 10.1126/science.7604266. [DOI] [PubMed] [Google Scholar]
- 24.Palombo F, Gallinari P, Iaccarino I, Lettieri T, Hughes M, D’Arrigo A, Truong O, Hsuan J J, Jiricny J. Science. 1995;268:1912–1914. doi: 10.1126/science.7604265. [DOI] [PubMed] [Google Scholar]
- 25.Johnson R E, Kovvali G K, Prakash L, Prakash S. J Biol Chem. 1996;271:7285–7288. doi: 10.1074/jbc.271.13.7285. [DOI] [PubMed] [Google Scholar]
- 26.Strand M, Earley M C, Crouse G F, Petes T D. Proc Natl Acad Sci USA. 1995;92:10418–10421. doi: 10.1073/pnas.92.22.10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolodner R D, Hall N R, Lipford J, Kane M F, Rao M R S, Morrison P, Wirth L, Finan P J, Burn J, Chapman P, Earabino C, Merchant E, Bishop D T. Genomics. 1995;28:613. doi: 10.1006/geno.1994.1661. [DOI] [PubMed] [Google Scholar]
- 28.Pan T-C, Zhang R-Z, Mattei M-G, Timpl R, Chu M-L. Proc Natl Acad Sci USA. 1992;89:6565–6569. doi: 10.1073/pnas.89.14.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Apte A N, Siebert P D. BioTechniques. 1993;15:890–893. [PubMed] [Google Scholar]
- 30.Watanabe A, Ikejima M, Suzuki N, Shimada T. Genomics. 1996;31:311–318. doi: 10.1006/geno.1996.0053. [DOI] [PubMed] [Google Scholar]
- 31.Wilson T M, Ewel A, Duguid J R, Eble J N, Lescoe M K, Fishel R, Kelley M R. Cancer Res. 1995;55:5146–5150. [PubMed] [Google Scholar]
- 32.Nakajima E, Orimo H, Ikejima M, Shimada T. Jpn J Human Genet. 1995;40:343–345. doi: 10.1007/BF01900603. [DOI] [PubMed] [Google Scholar]