

Abstract
MAP kinase is associated with delta-opioid receptor (DOR) signaling and plays a role in cell survival/death. Since anisomycin may alter MAP kinase activity and affect neuronal survival, we investigated whether anisomycin alters neuronal response to hypoxic stress and DOR inhibition. The experiments were performed in cultured cortical neurons. MAP kinase activities were determined by immunoblotting and neuronal viability was assessed by LDH leakage and live/dead morphological study. DOR inhibition with naltrindole (10 μM) led to significant injury in normoxic neurons after 24 hours of treatment and exacerbated hypoxia-induced injury. Along with the injury, either by hypoxia or naltrindole, phosphorylated p38 increased in a major way, while phosphorylated ERK and JNK had no significant change or slightly decreased. Anisomycin (50 ng/ml) prevented the increase in phosphorylated p38 immunoreactivity induced by naltrindole and reduced the neuronal injury. The results suggest that 1) MAP kinases are differentially involved in neuronal response to hypoxia and DOR inhibition in cortical neurons with phosphorylated p38 immunoreactivity being up-regulated and 2) Anisomycin attenuates the increase in phosphorylated p38 immunoreactivity and reduces neuronal injury induced by hypoxia- and DOR inhibition.
Keywords: Anisomycin, ERK, p38, JNK, Hypoxia, Delta-opioid receptor
1. Introduction
Mitogen-activated protein kinases (MAP kinases) transduce extracellular signals from tyrosine-kinase receptors and G protein-coupled receptors to cytoplasmic and nuclear effectors (Chang and Karin, 2001; Schaeffer and Weber, 1999). Three distinct but interlinked MAP kinases have been identified in mammalian cells, including neurons (Sweatt, 2001; Obata et al., 2004), i.e, the extracellular-signal regulated kinase (ERK), the c-Jun -terminal kinase (JNK) and p38. Several lines of evidence have suggested that ERK cascades mediate cell development, growth and survival (Skaper and Walsch, 1998; Seger and Krebs, 1995), while p38 and JNK respond to inflammatory cytokines and cellular stress and promote inflammation and cell death (Schaeffer and Weber, 1999). This has been seen in neurons in response to stress. For instance, we have shown that activation of delta-opioid receptors (DOR) protects cortical neurons from hypoxic stress with a down-regulation of p38 and up-regulation of ERK (Ma et al., 2005), while DOR inhibition is associated with worsening hypoxic injury and an increase in phosphorylated p38 immunoreactivity (Zhang et al., 2002; Ma et al., 2005).
However, controversy continues regarding the role of MAP kinases in cellular survival and death, especially with regard to p38 (Zheng and Zuo, 2004; Harada and Sugimoto, 1999; Horstmann et al., 1998; Maroney et al., 1998). For example, Zheng et al. (2004) suggest that p38 signals are neuroprotective in the brain, while others shows that neuroprotection from apoptosis is dependent on the inhibition of p38 kinase (Pi et al., 2004; Serbest et al., 2006). The role of p38 kinases in cardiac injury/protection is also controversial (Steenbergen 2002). Several studies (Marais et al., 2001; Schneiders et al., 2001) show that the attenuated activation of p38 plays an important role in protection from ischemic necrosis. There is, however, evidence to the contrary showing that the activation of p38 protects against ischemic insults to the heart (Steenbergen 2002; Yellon et al., 2003).
One of the reasons for this controversy may be attributed to the complex effects of non-specific p38 activators and inhibitors. Among these chemical tools, anisomycin, an antibiotic, has been used as an activator of JNK/p38 (Shifrin and Anderson, 1999). There is evidence that in addition to its role in inhibition of protein synthesis at the translational step (Torocsik and Szeberenyi, 2000; Luft et al., 2004), anisomycin may affect not only p38, but also influence multiple MAP kinases in certain cell types. For instance, anisomycin activates all three MAP kinases in HepG2 cells (Dhawan et al., 1999). Therefore, it is important to clarify the consequences of neuronal exposure to anisomycin with regard to MAP kinase activities, which may provide clues to reconcile the contradictory data in the past.
Anisomycin has been shown to protect cerebellar neurons in culture from 24-hour hypoxia (Dessi et al., 1992). The anisomycin-induced protection is also seen in other cells under stress, e.g., PC 12 cells with KCl deprivation (Kharlamov et al., 1995), and cardiac myocytes under hypoxia (Mackay and Mochly-Rosen, 1999). Such protection, we believe, is less likely mediated by an increase in p38 activity although anisomycin is thought to be a p38 activator (Shifrin and Anderson, 1999). Our argument is based on our recent observation that phosphorylated p38 immunoreactivity increases with hypoxic injury and decreases with neuroprotection induced by neuronal preconditioning (Ma et al., 2005). However, the cellular responses to anisomycin could be different between cortical neurons and other cells. It is essential to clarify this important issue since there has been no data available regarding effect of anisomycin on MAP kinases in cortical neurons under hypoxia or other stress.
In this work, therefore, we asked three fundamental questions regarding effects of anisomycin on hypoxic injury and p38 in cortical neurons. First, does anisomycin protect cortical neurons from hypoxic insults and other stress, e.g., DOR inhibition that causes neuronal injury (Zhang et al., 2002)? Second, is the effect of anisomycin dependent on neuronal maturity or anisomycin concentration? This is important because there is evidence showing that effects of anisomycin vary with age (Kharlamov et al., 1995) and are dose-dependent (Dessi et al., 1992). Third and most important, if anisomycin is protective against stress in cortical neurons, is this protection associate with changes in MAP kinase activities which are critically involved in neuronal injury/survival during stress? This will be done by measuring activities of three MAP kinases from the same samples to provide comparative information on anisomycin-mediated regulation of MAP kinases.
2. Results
This work was mainly carried out in cortical neurons at day 8 because the neurons are sensitive to hypoxic stress and DOR inhibition at this age as shown in our previous work (Hong et al., 2004; Zhang et al., 2000, 2002). To compare neuronal responses to different stresses, we exposed cortical neurons to either hypoxia, naltrindole or both since MAP kinases are involved in DOR signaling and DOR inhibition can leads to neuronal injury (Zhang et al., 2002). LDH leakage from the neurons to culture medium was measured to determine neuronal viability since it is a very reliable index of neuronal injury in our model of cortical neurons (Zhang et al., 2000 and 2006).
2.1 Anisomycin-induced neuroprotection from hypoxic stress and/or DOR inhibition
First, we tested whether anisomycin protects cortical neurons from stress. Because a concentration of 50 ng/ml is sufficient to cause significant changes in MAP kinase immunoreactivity in-vitro (Dhawan et al., 1999), we applied anisomycin to our in-vitro neurons at this concentration.
As shown in Figure 1, 24-hour hypoxia induced significant injury as indicated by an 35.0±8.1% increase in LDH leakage (P< 0.05) and decrease in live neurons in live/dead staining Fig. 1B). DOR inhibition with NTI (10 μM) caused similar injury to the cortical neurons with a 40.8±5.4% increase in LDH leakage (P < 0.05, n= 3). The combination of hypoxia with naltrindole increased LDH leakage by 105.0±5.4% (P < 0.05, n= 3).
Fig. 1. Effect of low-dose anisomycin on neuronal injury during hypoxia.

Day-8 neurons were exposed to hypoxia (1% O2), hypoxia + naltrindole (NTI), or hypoxia + naltrindole (NTI) + anisomycin (AN, 50 ng/ml) for 24 hrs. (A) Cell viability was measured by lactate dehydrogenase assay. Each group represents mean ± SE (n=3). ★: significant difference between the compared groups (p < 0.05). (B) Culture dishes were subjected to live/dead staining (Molecular Probes) and viewed under a fluorescence microscope system. Left, phase-contrast photomicrograph. Middle, intracellular calcein fluorescence signifying viable neurons. Right, ethidium homodimer fluorescence signifying injured and nonviable neurons. The three images of each row were taken from the same field. Note that low dose of anisomycin attenuated neuronal injury induced by hypoxia- and/or naltrindole.
Anisomycin (50 ng/ml) almost completely abolished the hypoxia-induced injury and significantly reduced the increased LDH leakage (105.0±8.1% vs 100% in normoxia control, P <0.05, n=3). The same treatment with anisomycin markedly reduced the neuronal injury due to naltrindole with a 50% reduction in LDH leakage (Fig. 1A). Although the combination of hypoxia and naltrindole caused remarkable injury, anisomycin largely reduced the neuronal injury and led to a >70% reduction of LDH leakage (from 105.0±.8.1% to 27.5±6.0% over the control). The morphological studies confirmed the results of LDH leakage, i.e., hypoxia or naltrindole decreased viable cellular density, but anisomycin increased the viability of neurons in the culture exposed to hypoxia and naltrindole (Fig. 1B).
2.2 Neurotoxicity of high-dose Anisomycin
Since the effect of pharmacological agents may vary with concentrations, we performed dose-response experiments and tested the differential effects of anisomycin at higher concentrations on cortical neurons. We started the experiments with 10 μg/ml of anisomycin. This high concentration was based on the work of Dessi et al. (1992) who found that no protective effect of anisomycin was apparent at <10 μg/ml whereas 50–100 μg/ml anisomycin offered strong protection to cerebellar neurons in culture against injury from 24h-hypoxia. Since in our model 50 ng/ml of anisomycin was sufficient to induce significant protection from 24h-hypoxia, we asked whether a higher concentration could offer a more benefit. In the same neuronal cultures, day-8 neurons were treated with anisomycin at 10 μg/ml and then exposed to either normoxia or hypoxia for 24 hours with or without naltrindole. As shown in Figure 2, anisomycin at the high concentration was not neuroprotective in any of the groups. Instead, it exacerbated the neuronal injury induced by hypoxia and/or naltrindole. Even in the normoxic neurons without naltrindole, 10 μg/ml of anisomycin caused major injury with a ~200% increase in LDH leakage. In the DOR inhibition group, this concentration of anisomycin caused a 6-fold increase in LDH leakage (from 34.8 ± 8.1% in DOR inhibition only to 292.2 ± 26.2% in DOR inhibition plus anisomycin.
Fig. 2. Effect of high-dose anisomycin on the cortical neurons treated with naltrindole.

The cortical neurons at day 8 were treated with anisomycin (AN, 10 μg/ml) and naltrindole (NTI, 10 μM) in normoxia or hypoxia. Cell viability was assessed by lactate dehydogenase assay after 24 hrs of the treatment. Each group represents mean ± SE (n = 3).
★: significant difference between compared groups (p < 0.05). Note that high dose of anisomycin cause significant injury in the cortical neurons.
2.3 Effect of anisomycin on immature neurons exposed to hypoxia and/or naltrindole
Since there is evidence showing that anisomycin had differential effects on immature versus more mature neurons (Kharlamov et al., 1995), we asked whether the neuronal responses to anisomycin was different between ages by applying 50 ng/ml of anisomycin in immature day-4 in culture neurons exposed to hypoxia and/or naltrindole (Fig. 3). In the immature neurons, the injury was much less than that of day-8 neurons after 24-hour treatment with hypoxia and/or naltrindole. In the hypoxia group, 24-hour stress did not induce any appreciable change in LDH leakage, suggesting that the immature neurons were more resistant to hypoxic stress than day-8 neurons. However, naltrindole caused a significant 40.8 ± 8.2 % increase in LDH leakage (P <0.05, n=10), which was similar to that seen in the day-8 neurons. In the presence of naltrindole, hypoxia could not further increase LDH leakage over that resulting from DOR inhibition (140.8 ± 8.1 % in DOR inhibition (NTI) vs 128.7 ± 9.3 % in DOR inhibition plus hypoxia (H+NTI), n=10) (Fig. 3A). In the immature neurons with 24-hour stress, anisomycin did not significantly influence the level of LDH leakage except for a slight reduction in the normoxic group with naltrindole (from 40.8 ± 8.2 % to 23.8 ± 9.7 % over the control, P>0.05).
Fig. 3. Effect of low-dose anisomycin on immature neurons with DOR inhibition.
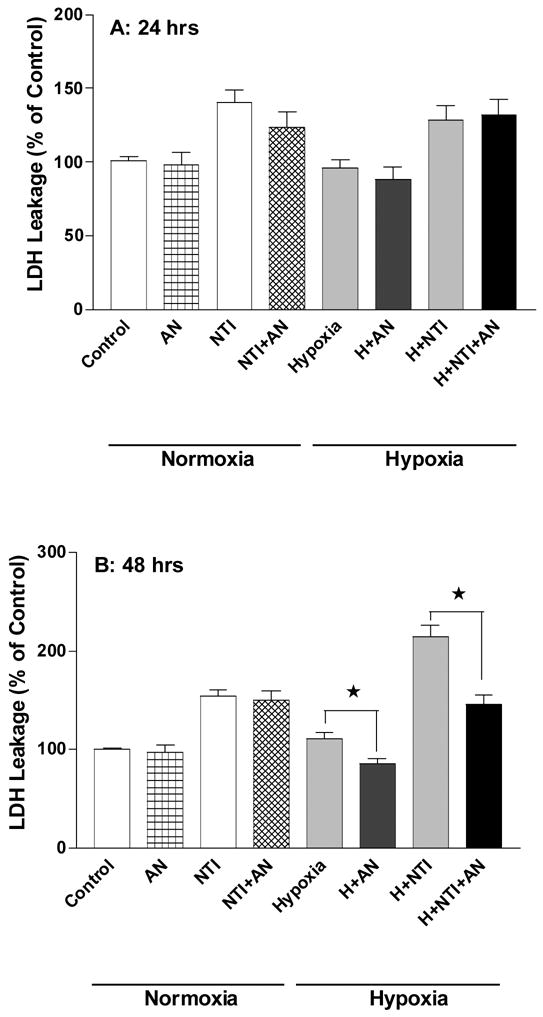
The cortical neurons at day 4 were treated with naltrindole (NTI,10 μM) with or without anisomycin (AN, 50 ng/ml) in normoxia or hypoxia. Cell viability was determined by lactate dehydogenase assay after 24hrs or 48 hrs of the treatment. Each group represents mean ± SE (n = 10). ★: significant difference between compared groups (p < 0.05). Note that as compared to day-8 neurons (Figures 1 and 2), immature neurons had much less injury after exposure to hypoxia or naltrindole for 24 hours. With extended exposure (48 hours), the immature neurons showed a significant LDH leakage which could be attenuated by the treatment with low dose of anisomycin.
Extending hypoxic exposure to 48 hrs resulted in slightly more evident injury in the immature neurons. On the other hand, naltrindole greatly increased neuronal injury during extended hypoxia. As shown in Figure 3B, LDH leakage was 113.8 ± 12.5 % over the control in the group of hypoxia plus naltrindole, which was similar to that of day-8 neurons. Although anisomycin had no significant effect on the injury induced by naltrindole, it significantly protected the neurons from hypoxia-induced injury, especially the hypoxic injury enhanced by naltrindole. After application of anisomycin, LDH leakage in the neurons treated with hypoxia plus naltrindole was similar to that of the neurons treated with naltrindole alone, suggesting that anisomycin completely abolished the hypoxic effect in theses neurons.
2.4 Anisomycin-increased p38 down-regulation and ERK upregulation in hypoxic neurons
To determine whether anisomycin-induced neuroprotection was associated with specific changes in MAP kinases, we applied 50 ng/ml of anisomycin to day-8 cortical neurons and measured phosphorylated p38, JNK and ERK in the same samples exposed to hypoxia, naltrindole or both stresses.
In the naltrindole-treated neurons, MAP kinase activities were changed in a type-specific manner (Fig. 4B). In both normoxic and hypoxic neurons, naltrindole largely increased phosphorylated p38 immunoreactivity. In contrast, phosphorylated JNK was slightly decreased in the same samples. These results suggest that phosphorylated p38 immunoreactivity was specifically up-regulated in response to hypoxic stress and naltrindole.
Fig. 4. Effect of DOR inhibition on phosphorylated MAP kinase immunoreactivity.

The cortical neurons at day 8 were treated with naltrindole (NTI, 10 μM) for DOR inhibition. (A) Cell viability was determined by lactate dehydrogenase assay. (B) Phospho-MAP kinases in cell lysates (30 μg of protein) were measured by immunoblotting using antibodies against phospho-ERK, phospho-JNK, and phospho-p38. Actin signal was measured as an internal standard. Each group represents mean ± SE (n=3). ★: significant difference between the compared groups (p < 0.05). Note that naltrindole caused major injury in the cortical neurons, as evident by a large increase in LDH leakage. In the injured neurons, MAP kinases were differentially affected with an up-regulation of phosphorylated p38 immunoreactivity.
In the cortical neurons treated with anisomycin (Fig. 5), the quantitative analysis with normalization to β-actin immunoreactivity showed that three types of MAP kinases were differentially regulated 24 hours after the stress. In normoxia, anisomycin decreased phosphorylated p38 and ERK immunoreactivity with no significant changes in phosphorylated JNK immunoreactivity. In the naltrindole-treated neurons, anisomycin reduced phosphorylated p38 immunoreactivity by 95.0 ± 1.0% and increased phosphorylated ERK immunoreactivity by 160.0±30% with no change in phosphorylated JNK immunoreactivity. In hypoxic neurons, anisomycin attenuated phosphorylated p38 immunoreactivity by 44.5±10.7% and increased phosphorylated ERK immunoreactivity by 52.3±48.0% as compared to those of hypoxic neurons without anisomycin. In the neurons exposed to both hypoxia and naltrindole, anisomycin attenuated phosphorylated p38 immunoreactivity by 47.8±12.6% and phosphorylated JNK immunoreactivity by 51.1±7.7% with a tendency to increase phosphorylated ERK immunoreactivity. These data indicate that 24 hours after application of anisomycin, all groups had a major decrease in phosphorylated p38 immunoreactivity with an increase in phosphorylated ERK immunoreactivity. In contrast, JNK kinase was relatively insensitive to anisomycin. Since three MAP kinases were simultaneously studied from the same samples, the differential changes in their activities suggest that the regulation of p38 differs from other MAP kinases. The lack of any effect on actin immunoreactivity further supports a specific change in phosphorylated p38 immunoreactivity.
Fig. 5. Effect of low-dose anisomycin on phosphorylated MAP kinase immunoreactivity.


Cortical day-8 neurons were treated with naltrindole (ΝΤΙ, 10 μM) with/without anisomycin (AN, 50 ng/ml) in normoxia (N) or hypoxia (H) for 24 hrs. Western blots were used for measurements of phospho-MAP kinases. Cell lysates (30 μg of protein) were loaded on 7.5% SDS PAGE and analyzed by immunoblotting using antibodies against phospho-ERK, phospho-JNK, and phospho-p38. The upper panel shows representative blots and the lower panel, mean ± SE from at least 4 independent experiments per group. ★: significant difference between compared groups (p < 0.05). Note that low dose of anisomycin differentially regulated MAP kinases in the cortical neurons exposed to hypoxia or naltrindole with phosphorylated p38 immunoreactivity being significantly decreased.
To confirm a direct role of ERK in the neuronal response to the stress, we inhibited ERK with a specific ERK inhibitor, U0126 (5 μM). In both normoxic and hypoxic conditions, the ERK inhibition led to increased LDH leakage induced by naltrindole (Fig. 6), suggesting that ERK inhibition caused neuronal injury in normoxia and worsened hypoxic injury.
Fig. 6. Effect of ERK inhibitor on naltrindole-induced neuronal injury.

U0126, an ERK inhibitor, was applied to the naltrindole (NTI) treated neurons in normoxia or hypoxia for 24 hrs. Cell viability was determined by lactate dehydrogenase assay. Each group represents mean ± SE. ★: significant difference between the compared groups (p < 0.05). Note that U0126 increased naltrindole-induced injury in the cortical neurons under either normoxia or hypoxia, suggesting that ERK signals may be protective against neuronal injury induced by DOR inhibition.
3. Discussion
The major findings of the present study are that 1) anisomycin in a low concentration protects cortical neurons from hypoxia and naltrindole induced injury and the protective effect varies with neuronal maturation; 2) hypoxia and/or DOR inhibition induced neuronal injury with an increase in phosphorylated p38 immunoreactivity, while anisomycin attenuated the injury and the increase in phosphorylated p38 immunoreactivity; and 3) anisomycin activity is associated with an increase in phosphorylated ERK immunoreactivity, the inhibition of which worsened neuronal injury in hypoxia and DOR inhibition by naltrindole.
This is the first study to show that anisomycin provides neuroprotection from hypoxia and DOR inhibition with naltrindole in cortical neurons. Our data suggest that 50 ng/ml of anisomycin is sufficient to induce significant neuroprotection, whereas a high concentration (10 μg/ml) causes severe neuronal injury. These results are different from those of cerebellar neurons in culture. Dessi et al. (1992) show that in cultured cerebellar neurons, anisomycin at 10 μg/ml had no protective effect against neuronal injury caused by 24-hour hypoxia, whereas the concentrations between 50 and 100 μg/ml offered a strong neuroprotection. Kharlamov et al. (1995) observed that in primary cultures of cerebellar granule neurons, 10 μg/ml of anisomycin significantly reduced neuronal injury induced by potassium deficiency. Our data may reflect a major difference between neuronal types in response to anisomycin with cortical neurons being much more sensitive to anisomycin than cerebellar neurons, with only low concentrations (e.g., 50 ng/ml in this work) offering a protective effect. Our findings are supported by data from PC12 cells that show that anisomycin at 1 μg/ml caused internucleosomal DNA fragmentation characteristic of apoptosis (Torocsik and Szeberenyi, 2000). The opposing effects of anisomycin at low vs. high concentrations (Torocsik and Szeberenyi, 2000; Luft et al., 2004) suggest that its actions may involve several intracellular processes at multiple levels.
There is evidence showing that in primary cultures of rat cerebellar granule neurons, the effect of anisomycin varies with neuronal age (culture day 1–7), i.e., protective in the mature but toxic in the immature (Kharlamov et al., 1995). In the present study, however, we did not observe any neurotoxicity in the immature cortical neurons at the concentration that induced significant neuroprotection in the mature. Interestingly, the efficacy of anisomycin was largely decreased or delayed in the immature neurons. Although anisomycin significantly reduced LDH leakage in the immature neurons exposed to an extended duration of hypoxia, it had no significant effect on neuronal injury induced by naltrindole neither at 24 or 48 hours after application of the DOR antagonist. These data indicate that the immature cortical neurons are less sensitive to anisomycin-induced protection as compared to mature cortical neurons. The mechanism underlying the age-difference is unclear at this point. It may be partially related to the differential expression of membrane proteins (e.g., NMDA receptors and DOR) and intracellular components of signal transduction during neuronal development (Xia and Haddad, 1991; Zhang et al., 2006).
It has been well documented that DOR activation is neuroprotective (Borlongan et al., 2004; Lim et al., 2004; Zhang J, 2002, Zhang J, 2005). The mechanism of DOR-inhibition induced injury is partially related to the disturbance of MAP kinase signaling, and appears to be specific in nature, affecting p38 and ERK but not JNK. A wealth of evidence suggests that MAP kinases may play a role in intracellular signaling of DOR-mediated neuroprotection. First, activation of DOR alters MAP kinase activity (Kramer and Simon, 2000; Shahabi et al., 1999; Zhang et al., 1999; Polakiewicz et al., 1998; Fukuda et al., 1996). Second, hypoxia modulates MAP kinase activity in a variety of cells (Jin et al., 2002; Dougherty et al. 2002; Jin et al., 2000). Third, MAP kinases are involved in control of cellular survival and death (Schaeffer and Weber, 1999; Skaper and Walsch, 1998; Seger and Krebs, 1995). However, the precise role of p38 in neuronal death remains unclear. Some studies have shown that p38 is neuroprotective (Zheng and Zuo, 2004; Barancik et al., 1999). In sharp contrast, other studies suggest that up-regulation of p38 kinase causes neuronal apoptosis/death (Harada and Sugimoto, 1999; Horstmann et al., 1998; Maroney et al., 1998; Skaper and Walsh, 1998; Yang et al., 1997), and that p38 inhibitors promote the survival of a variety of neurons (Harada and Sugimoto, 1999; Horstmann et al., 1998; Skaper and Walsh, 1998; Xia et al., 1995). In NG108-15 cells, activation of DOR up-regulates p38 kinase (Zhang et al., 1999). However, our recent work shows that DOR upregulation is associated with a down-regulation of p38 during neuroprotection induced by hypoxic preconditioning (Ma et al., 2005). The difference between our data and those from NG108-15 cells may stem from the difference in cellular types (cortical neurons vs. NG108-15 cells). Since hypoxia induced similar change in p38 kinase in injured neurons, we believe that 1) the role of p38 in the control of neuronal survival/death is complex and dependent on cellular type and conditions and 2) p38 plays an important role in neuronal injury in cortical neurons during stress.
If p38 upregulation is associated with neuronal injury, one may predict that anisomycin would exacerbate neuronal injury during hypoxia and/or DOR inhibition with naltrindole because anisomycin is thought to be a p38 activator (Hazzalin et al., 1998, Harada and Sugimoto, 1999; Horstmann et al., 1998). Interestingly, we observed that anisomycin (50 ng/ml) significantly attenuated hypoxia or naltrindole-induced injury in the cortical neurons, as was evident by decreased LDH leakage and confirmed by live/dead morphological studies. These data suggest that anisomycin at a low concentration is neuroprotective against conditions of hypoxia and DOR inhibition via a down-regulation of p38. The anisomycin induced protection is consistent with that seen in other cells under stress, e.g., PC 12 cells with KCl deprivation (Kharlamov et al., 1995), and cardiac myocytes under hypoxia (Mackay and Mochly-Rosen, 1999) in which anisomycin protects cells from environmental stress. This however raises several intriguing questions regarding how anisomycin, a putative p38 activator, could induce neuroprotection against stress. Is the neuroprotection associated with an increase in phosphorylated p38? A direct approach to answer this question is to determine whether anisomycin increased phosphorylated p38 in the cortical neurons under hypoxia and/or DOR inhibition. Surprisingly, our data show that anisomycin (50 ng/ml) did not increase phosphorylated p38 immunoreactivity in all experimental groups. On the contrary, it significantly decreased the immunoreactivity. On the other hand, anisomycin increased phosphorylated ERK immunoreactivity (Figure 6). Because anisomycin consistently down-regulated p38 in both naive and hypoxia/naltrindole treated neurons, we believe that the long-term effect of anisomycin on the cortical neurons is a down-regulation rather than an up-regulation of p38. Indeed, there is evidence showing that anisomycin at 50 ng/ml can activate both p38 and ERK with p38 up-regulation being short-lasting and ERK upregulation being long-lasting (Dhawan et al., 1999). Since we have previously observed that the crosstalk between ERK and p38 MAP kinases displays a “yin-yang” antagonism (Ma et al., 2005), it is possible that the long-lasting activation of ERK suppresses p38 in the long-term (e.g., 24 hours) after application of anisomycin. Further studies are needed to verify this possibility.
In summary, the present data suggest that hypoxic insult and DOR inhibition with naltrindole increase p38 immunoreactivity and cause injury in cortical neurons. Anisomycin at 50 ng/ml, though known as a p38 activator, protects the neurons from hypoxic insult and DOR inhibition with a decrease in phosphorylated p38 immunoreactivity and an increase in phosphorylated ERK immunoreactivity. This protective effect is greatly influenced by neuronal age and concentration of anisomycin. Our data suggests that cortical neurons are different from other cells in terms of cellular response to anisomycin during stress.
4. Experimental Procedure
4.1 Animals
Sprague-Dawley pregnant (embryonic day 17–18) rats were purchased from Charles River Laboratories (Wilmington, MA) and sacrificed under Halothane anesthesia (Sigma, St Louis, MO). For all procedures, the guidelines of the Animal Care Committee of Yale University School of Medicine which is accredited by the American Association of Laboratory Animal Care were followed.
4.2 Cortical neuron culture
Cortical neuron cultures were prepared from embryonic day 17–18 rat fetuses, as we have previously described (Zhang et al., 2002). In brief, fetuses were decapitated and brains cleaned of meninges. Cortical tissue was collected under sterile conditions and then placed into 50 ml of Dulbecco’s phosphate-buffer saline solution (Gibco-BRL, Life Technologies, Rockville, MD) containing 0.6% glucose. The supernatant was carefully removed and 1 ml of trypsin-EDTA solution with 10 μl of HEPES buffer solution was added to dissociate tissue connections for 15 minutes at 37°C. Subsequently, a trypsin inhibitor solution (1 ml of Hanks’ balanced salt solution, 110 μl of fetal bovine serum, 100 μl of trypsin inhibitor and 10 μl of HEPES buffer solution; all purchased from Gibco-BRL) was added and incubated for 5 minutes at 37°C. The mixture was triturated 10 times using a 1 ml pipette and then centrifuged at 14,000 rpm for 5 minutes at 4°C. After removal of the supernatant, the cells were resuspended in serum-free Neurobasal Medium (Gibco-BRL), supplemented with B-27, glutamine (0.5 mM), glutamate (25 μM) and a combination of penicillin (100 U/ml) and streptomycin (100 μg/ml). The cells were plated onto poly-D-lysine (100 μg/ml, Sigma, St Louis, MO) coated 35-mm culture dishes at 1 × 106 cells/ml and maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
4.3 Neuronal injury induced by hypoxia
Culture dishes were randomly divided into normoxic and hypoxic groups. At either day 4 or day 8, dishes selected for hypoxic treatment were moved to a separate incubator maintained at 1% O2, 5% CO2 and 94% N2. Neurons were exposed to hypoxia beginning at day 4 or 8 for 24–48 hours.
4.4 Neuronal injury induced by naltrindole
Cells were treated with 10 μM of naltrindole, a DOR antagonist, at day 4 or day 8 in culture under normoxic and hypoxic conditions. After 24–48 hours, neuronal viability was determined by measurement of LDH leakage or live/dead staining. We have previously shown that DOR activation is protective in cortical neurons, while DOR inhibition with naltrindole at 10 μM for 24 hours induces neuronal injury under normoxia and exacerbates hypoxic injury (Zhang et al., 2000 and 2002).
4.5 Drug application
Cells were treated with anisomycin or U0126 (an ERK inhibitor) at final concentrations of 50 ng/ml (10 μg/ml for a high dose) and 5 μM, respectively. All chemicals were purchased from Calbiochem (San Diego, CA). For DOR inhibition, Naltrindole hydrochloride (NTI; DOR-antagonist, Sigma Co., St. Louis, MO) was used at 10 μM. Note that anisomycin and U0126 treatments contained 0.025% dimethyl sulfoxide (DMSO) upon final dilution. After 24 or 48 hr of exposure, neurons were assessed for neuronal injury via lactate dehydrogenase release and live/dead assay.
4.6 Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) activity in culture medium was assessed using Sigma Diagnostics’ Lactate Dehydrogenase Kit (Procedure No. 228-UV) with a Beckman DU-640 spectrophotometer system (Beckman Instruments, Inc., Fullerton, CA). Culture medium was collected and centrifuged to remove cellular debris from the supernatant. Subsequently, 100 μl of the sample was added to a polystyrene cuvet containing 1 ml of LDH reagent (50 mmol/L lactate, 7 mmol/L NAD+ in 0.05% sodium azide buffer, pH 8.9). The cuvet was placed immediately into the spectrophotometer, maintained at 30°C. After stabilization for 30 seconds, absorbance at 340 nm was recorded at 30-second intervals for 2 minutes. The change in absorbance was then expressed as a concentration in units (U) per liter as described in the kit.
4.7 Live/dead assay
After photomicrographs of cultured cells were taken using a phase-contrast microscope to establish baseline viability, neuronal survival was quantified at the same field using a live/dead viability/cytotoxicity kit (L-3224) from Molecular Probes. In accordance with the manufacturer’s protocol, neurons were exposed to cell permeant calcein AM (3 μM), which is hydrolyzed by intracellular esterases, and to ethidium homodimer-1 (4 μM), which binds to nucleic acids. (The cleavage product of calcein AM, calcein, produces a green fluorescence when exposed to 494-nm light and is used to identify live cells. Bound ethidium homodimer-1 produces a red fluorescence when exposed to 528-nm light, allowing the identification of dead cells.) Culture dishes were dually stained and neuronal viability was examined under a fluorescence microscope system (Zeiss Axiovert 25; Sony Progressive 3CCD and Camera Adapter CMA-D2; blue excitation filter: 488/515 nm, green excitation filter: 514/550 nm).
4.8 SDS-PAGE and immunoblotting
After removing the culture medium, the neuronal cells were washed twice with PBS and harvested with cell lysis buffer (Cell Signaling Technology # 9803: 20 mM Tris (pH7.5), 150 mM NaCl, 1 mM ethylene diaminetetraacetate (EDTA), 1 mM ethylene glycolbis (2-aminoethyl)-N,N,N′,N′-tetraacetic acid (EGTA), 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4, 1μg/ml leupeptin). All procedures were performed on ice. The mixture was centrifuged at 620 × g for 5 min to remove cellular debris, and samples of the supernatant were subjected to electrophoresis. The protein concentration was determined using a Bio-Rad Dc protein assay (Hercules, CA). Protein samples (100 μg) were separated on 12 % SDS-polyacrylamide gels and transferred to polyvinyldifluoridine membranes. The nonspecific binding was blocked for 1 hr at room temperature with 5% milk in TBS-0.1% Tween 20 and then incubated overnight at 4°C in the primary antibody diluted in 5% milk in TBS-0.1% Tween 20. The primary antibodies used were as follows: phospho-specific ERK (1:4000) from Cell Signaling Technology (Beverly, MA) and phospho-specific JNK (1:4000) and phospho-specific p38 (1:200) from Santa Cruz Biotechnology (Santa Cruz, CA). Membranes were washed with TBS-0.1% Tween 20, incubated at room temperature for 1 hr with horseradish peroxidase-goat anti-rabbit (Bio-Rad, Hercules, CA) or goat anti-mouse antibody (1:5000; Santa Cruz Biotechnology, Santa Cruz, CA), and washed. Immunoblots were developed with the Super Signal reagent (Pierce, Rockford, IL). After immunoblots of MAP kinases, membranes were stripped and then reprobed with a goat polyclonal antibody to β-actin (1:500; Santa Cruz, CA) and secondary anti-goat antibody (Santa Cruz, CA) at a dilution of 1:2000. The band density was determined by densitometry using an image analysis system (Molecular Dynamics, Sunnyvale, CA).
4.9 Data Analysis
All data were expressed as mean values ± standard error, and were subjected to statistical analysis via non-paired, two-tailed Student’s t-test using Graphpad Prism 3.0 software (GraphPad Software, Inc., San Diego, CA). The level of statistical significance was set at P < 0.05.
Acknowledgments
This work was supported by NIH (HD-34852 and AT01094) grants to YX and BK21 project of Advanced Medical Education, Inha University School of Medicine (Korea) to SSH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barancik M, Htun P, Schaper W. Okadaic acid and anisomycin are protective and stimulate the SAPK/JNK pathway. J Cardiovasc Pharmacol. 1999;34:182–190. doi: 10.1097/00005344-199908000-00002. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Wang Y, Su TP. Delta opioid peptide (D-Ala 2, D-Leu 5) enkephalin: linking hibernation and neuroprotection. Front Biosci. 2004;9:3392–3398. doi: 10.2741/1490. [DOI] [PubMed] [Google Scholar]
- Boutin H, Dauphin F, MacKenzie ET, Jauzac P. Differential time-course decreases in nonselective, mu-, delta-, and kappa-opioid receptors after focal cerebral ischemia in mice. Stroke. 1999;30:1271–1277. doi: 10.1161/01.str.30.6.1271. [DOI] [PubMed] [Google Scholar]
- Cao J, Semenova MM, Solovyan VT, Han J, Coffey ET, Courtney MJ. Distinct requirements for p38alpha and c-Jun N-terminal kinase stress-activated protein kinases in different forms of apoptotic neuronal death. J Biol Chem. 2004;279:35903–35913. doi: 10.1074/jbc.M402353200. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Dessi F, Charriaut-Marlangue C, Ben-Ari Y. Anisomycin and cycloheximide protect cerebellar neurons in culture from anoxia. Brain Res. 1992;581:323–326. doi: 10.1016/0006-8993(92)90726-p. [DOI] [PubMed] [Google Scholar]
- Dhawan P, Bell A, Kumar A, Golden C, Mehta KD. Critical role of p42/44(MAPK) activation in anisomycin and hepatocyte growth factor-induced LDL receptor expression: activation of Raf-1/Mek-1/p42/44(MAPK) cascade alone is sufficient to induce LDL receptor expression. J Lipid Res. 1999;40:1911–1919. [PubMed] [Google Scholar]
- Dougherty CJ, Kubasiak LA, Prentice H, Andreka P, Bishopric NH, Webster KA. Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress. Biochem J. 2002;362:561–571. doi: 10.1042/0264-6021:3620561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Kato S, Morikawa H, Shoda T, Mori K. Functional coupling of the delta-, mu-, and kappa-opioid receptors to mitogen-activated protein kinase and arachidonate release in Chinese hamster ovary cells. J Neurochem. 1996;67:1309–1316. doi: 10.1046/j.1471-4159.1996.67031309.x. [DOI] [PubMed] [Google Scholar]
- Harada J, Sugimoto M. An inhibitor of p38 and JNK MAP kinases prevents activation of caspase and apoptosis of cultures cerebellar granule neurons. Jpn J Pharmacol. 1999;79:369–378. doi: 10.1254/jjp.79.369. [DOI] [PubMed] [Google Scholar]
- Hazzalin CA, Le Panse R, Cano E, Mahadevan LC. Anisomycin selectively desensitizes signaling components involved in stress kinase activation and fos and jun induction. Mol Cell Biol. 1998;18:1844–1854. doi: 10.1128/mcb.18.4.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SS, Gibney GT, Esquilin M, Yu J, Xia Y. Effect of protein kinases on lactate dehydrogenase activity in cortical neurons during hypoxia. Brain Res. 2004;1009:195–202. doi: 10.1016/j.brainres.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Kahle PJ, Borasio GD. Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J Neurosci Res. 1998;52:483–490. doi: 10.1002/(SICI)1097-4547(19980515)52:4<483::AID-JNR12>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Jin N, Hatton N, Swartz DR, Xia X, Harrington MA, Larsen SH, Rhoades RA. Hypoxia activates jun-N-terminal kinase, extracellular signal-regulated protein kinase, and p38 kinase in pulmonary arteries. Am J Respir Cell Mol Biol. 2000;23:593–601. doi: 10.1165/ajrcmb.23.5.3921. [DOI] [PubMed] [Google Scholar]
- Jin K, Mao XO, Zhu Y, Greenberg DA. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem. 2002;80:119–125. doi: 10.1046/j.0022-3042.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- Kharlamov E, Cagnoli CM, Atabay C, Ikonomovic S, Grayson DR, Manev H. Opposite effect of protein synthesis inhibitors on potassium deficiencyinduced apoptotic cell death in immature and mature neuronal cultures. J Neurochem. 1995;65:1395–1398. doi: 10.1046/j.1471-4159.1995.65031395.x. [DOI] [PubMed] [Google Scholar]
- Kramer HK, Simon EJ. mu and delta-opioid receptor agonists induce mitogen-activated protein kinase (MAPK) activation in the absence of receptor internalization. Neuropharmacology. 2000;39:1707–1719. doi: 10.1016/s0028-3908(99)00243-9. [DOI] [PubMed] [Google Scholar]
- Lim YJ, Zheng S, Zuo Z. Morphine preconditions purkinje cells against cell death under in vitro simulated ischemia-reperfusion conditions. Anesthesiology. 2004;100:562–568. doi: 10.1097/00000542-200403000-00015. [DOI] [PubMed] [Google Scholar]
- Luft AR, Buitrago MM, Ringer T, Dichgans J, Schulz JB. Motor skill learning depends on protein synthesis in motor cortex after training. J Neurosci. 2004;24:6515–6520. doi: 10.1523/JNEUROSCI.1034-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MC, Qian H, Ghassemi F, Zhao P, Xia Y. Oxygen sensitive d-opioid receptor-regulated survival and death signals: Novel insights into neuronal preconditioning and protection. J Biol Chem. 2005;280:16208–16218. doi: 10.1074/jbc.M408055200. [DOI] [PubMed] [Google Scholar]
- Mackay K, Mochly-Rosen D. An inhibitor of p38 mitogen-activated protein kinase protects neonatal cardiac myocytes from ischemia. J Biol Chem. 1999;274:6272–6279. doi: 10.1074/jbc.274.10.6272. [DOI] [PubMed] [Google Scholar]
- Marais E, Genade S, Huisamen B, Strijdom JG, Moolman JA. Activation of p38 MAPK induced by a multi-cycle ischaemic reconditioning protocol is associated with attenuated p38 MAPK activity during sustained ischaemia and reperfusion. Journal of Mol Cell Cardiol. 2001;33:769–778. doi: 10.1006/jmcc.2001.1347. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Glicksman MA, Basma AN, et al. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro A, Ciccarelli C, De Cesaris P, Scoglio A, Bouche M, Molinaro M, Aquino A, Zani BM. PKCalpha-mediated ERK, JNK and p38 activation regulates the myogenic program in human rhabdomyosarcoma cells. J Cell Sci. 2002;115:3587–3599. doi: 10.1242/jcs.00037. [DOI] [PubMed] [Google Scholar]
- Mayfield KP, Kozak W, Malvin GM, Porreca F. Hypoxia decreases opioid delta receptor expression in mouse brain. Neuroscience. 1996;72:785–789. doi: 10.1016/0306-4522(95)00585-4. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi R, Li W, Lee NT, Chan HH, Pu Y, Chan LN, Sucher NJ, Chang DC, Li M, Han Y. Minocycline prevents glutamate-induced apoptosis of cerebellar granule neurons by differential regulation of p38 and Akt pathways. J Neurochem. 2004;91:1219–1230. doi: 10.1111/j.1471-4159.2004.02796.x. [DOI] [PubMed] [Google Scholar]
- Polakiewicz RD, Schieferl SM, Gingras AC, Sonenberg N, Comb MJ. mu-Opioid receptor activates signaling pathways implicated in cell survival and translational control. J Biol Chem. 1998;273:23534–23541. doi: 10.1074/jbc.273.36.23534. [DOI] [PubMed] [Google Scholar]
- Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiders S, Chen W, Hou J, Steenbergen C, Murphy E. Inhibition of p38 MAPK a/b reduces ischemic injury and does not block the protective effects of preconditioning. Am J Physiol. 2001;280:H499–H580. doi: 10.1152/ajpheart.2001.280.2.H499. [DOI] [PubMed] [Google Scholar]
- Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- Serbest G, Horwitz J, Jost M, Barbee K. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. 2006;20:308–310. doi: 10.1096/fj.05-4024fje. [DOI] [PubMed] [Google Scholar]
- Shahabi NA, Daaka Y, McAllen K, Sharp BM. Delta opioid receptors expressed by stably transfected jurkat cells signal through the map kinase pathway in a ras-independent manner. J Neuroimmunol. 1999;94:48–57. doi: 10.1016/s0165-5728(98)00211-2. [DOI] [PubMed] [Google Scholar]
- Shifrin VI, Anderson P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J Biol Chem. 1999;274:13985–13992. doi: 10.1074/jbc.274.20.13985. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Walsh FS. Neurotrophic molecules: Strategies for designing effective therapeutic molecules in neurodegeneration. Mol Cell Neurosci. 1998;12:179–193. doi: 10.1006/mcne.1998.0714. [DOI] [PubMed] [Google Scholar]
- Steenbergen C. The role of p38 mitogen-activated protein kinase in myocardial ischemia/reperfusion injury; relationship to ischemic preconditioning. Basic Res Cardiol. 2002;97:276–285. doi: 10.1007/s00395-002-0364-9. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Torocsik B, Szeberenyi J. Anisomycin affects both pro- and antiapoptotic mechanisms in PC12 cells. Biochem Biophys Res Commun. 2000;278:550–556. doi: 10.1006/bbrc.2000.3836. [DOI] [PubMed] [Google Scholar]
- Xia Y, Haddad GG. Ontogeny and distribution of opioid receptors in the rat brainstem. Brain Res. 1991;549:181–193. doi: 10.1016/0006-8993(91)90457-7. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Downey JM. Preconditioning the myocardium: From cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gibney GT, Zhao P, Xia Y. Neuroprotective role of delta-opioid receptors in cortical neurons. Am J Physiol - Cell Physiol. 2002;282:C1225–C1234. doi: 10.1152/ajpcell.00226.2001. [DOI] [PubMed] [Google Scholar]
- Zhang J, Haddad GG, Xia Y. δ-, not μ- and κ-, opioid receptor activation protects neocotical neurons from glutamate-induced excitotoxic injury. Brain Res. 2000;885:143–153. doi: 10.1016/s0006-8993(00)02906-1. [DOI] [PubMed] [Google Scholar]
- Zhang J, Qian H, Zhao P, Hong SS, Xia Y. Rapid hypoxia preconditioning protects cortical neurons from glutamate Toxicity Through δ-Opioid Receptor. Stroke. 2006;37:1094–1099. doi: 10.1161/01.STR.0000206444.29930.18. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Xin SM, Wu GX, Zhang WB, Ma L, Pei G. Endogenous deltaopioid and ORL1 receptors couple to phosphorylation and activation of p38 MAPK in NG108-15 cells and this is regulated by protein kinase A and protein kinase C. J Neurochem. 1999;73:1502–1509. doi: 10.1046/j.1471-4159.1999.0731502.x. [DOI] [PubMed] [Google Scholar]
- Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of P38 mitogen-activated protein kinases. Mol Pharmacol. 2004;65:1172–1180. doi: 10.1124/mol.65.5.1172. [DOI] [PubMed] [Google Scholar]