

Abstract
In this study, we tested the hypothesis that factor XI (FXI) activation occurs in plasma following activation of the extrinsic pathway by thrombin-mediated feedback activation. We used two different assays: (i) a direct measurement of activated FXI by ELISA and (ii) a functional assay that follows the activation of the coagulation cascade in the presence or absence of a FXI inhibiting antibody by monitoring thrombin activity. We failed to detect any FXI activation or functional contribution to the activation of the coagulation cascade in platelet poor or platelet-rich plasma, when activation was initiated by thrombin or tissue factor. Additionally, we found that, in the absence of a contact system inhibitor during blood draw, contact activation of FXI can mistakenly appear as thrombin- or tissue-factor-dependent activation. Thus, activation of FXI by thrombin in solution or on the surface of activated platelets does not appear to play a significant role in a plasma environment. These results call for reevaluation of the physiological role of the contact activation system in blood coagulation.
Keywords: coagulation, platelets, contact activation
Blood coagulation is mediated by a cascade of proteolytic enzymes termed the coagulation factors. The cascade of coagulation factors endoproteolytic cleavage events culminates in the cleavage of fibrinogen, by thrombin, and the formation of fibrin fibers which form blood clots. During in vitro plasma clotting assays, factor XI (FXI) is activated by factor XII (FXII). This process was termed the contact activation system as it depends on the binding of FXII to negatively charged artificial surfaces. Binding of FXII to these surfaces leads to its autoactivation, which is further augmented by the other two components of the contact activation system, plasma prekallikrein and high molecular weight kininogen (HK). Activated FXI (FXIa) ties into the intrinsic pathway of blood coagulation by activating factor IX (FIX), which together with activated factor VIII, initiates the common pathway by activating factor X leading to prothrombin activation and clot formation (see refs. 1–4 for recent reviews of the contact activation system).
The physiological function of the contact system in hemostasis is not clear because deficiencies in components of the contact system, plasma prekallikrein, HK, and FXII are not associated with a bleeding diathesis. On the other hand, mutations in the downstream intrinsic pathway components, FVIII and FIX, lead to hemophilia A and hemophilia B, respectively. Interestingly, FXI deficiency leads to a mild bleeding disorder, hemophilia C. The existence of bleeding in FXI-deficient individuals raised the possibility that FXI can be activated independently of the contact system.
Attempts to identify alternative mechanisms for FXI activation, using purified coagulation proteins, revealed that FXI can be activated by thrombin as well as being autoactivated by FXIa. Both thrombin-mediated activation and autoactivation, however, occur at a very slow rate in solution. The rate of autoactivation and thrombin-mediated activation is greatly enhanced by negatively charged polymers such as dextran sulfate (5, 6). It was suggested that FXI activation by thrombin can serve as a positive feedback mechanism to augment the common pathway. In accordance with this hypothesis, several studies have demonstrated that FXI contributes to the activation of the coagulation cascade in plasma by low concentrations of tissue factor (TF) (7–9). This observation is particularly significant because TF, which is exposed to blood during injury, initiates hemostatic blood clot formation through the extrinsic pathway. Furthermore, FXI was also found to inhibit fibrinolysis by promoting activation of thrombin-activatable fibrinolysis inhibitor (9–11). The involvement of thrombin mediated FXI activation in these processes was questioned, however, because no FXI activation was detected in plasma even when high concentrations of TF and thrombin were added (12). Furthermore, it was found that physiological concentrations of plasma proteins such as fibrinogen and HK (which is complexed with FXI under physiological conditions) interfere with FXI activation by thrombin (5, 13).
An apparent solution to the discrepancy was offered by the observation that FXI activation by thrombin is enhanced several orders of magnitude by activated platelets in systems reconstituted from purified coagulation proteins (14–16). This process depended on the presence of HK or prothrombin (14, 15), suggesting that FXI activation on activated platelets could also occur in plasma. In support of this model, binding of FXI to activated platelets has been demonstrated (17) and the platelet binding domain in FXI has been identified (18). Nevertheless, platelet-dependent FXI activation by thrombin has not yet been directly demonstrated in a plasma environment.
In this work, we tested whether FXI can be activated independently of the contact system, in a plasma environment when platelets are present as suggested by the above studies. To this end, we measured FXI activation when the coagulation cascade was activated by thrombin or TF using a sensitive ELISA. In addition, we followed the contribution of FXI to the activation of the coagulation cascade by thrombin and TF using a functional assay in the presence or absence of a FXIa inhibiting antibody. The results of this study suggest that, in a plasma environment, thrombin or TF do not activate FXI, even in the presence of platelets. These results, along with the recent identification of an antithrombotic phenotype in FXII knockout mice (19), suggest that FXI activation by the contact system may be physiologically relevant, after all.
Results
FXI Activation in Plasma Environment.
To study the mechanism of FXI activation in a plasma environment, we monitored FXIa formation when the coagulation cascade was initiated by different procoagulants. In plasma, FXIa is rapidly inactivated by covalent complex formation with plasma serine protease inhibitors (serpins). C1 inhibitor (C1inh) is the major inactivator of FXIa in plasma (20). Therefore, we used a FXIa–C1inh sandwich ELISA to monitor FXI activation. This ELISA (Fig. 1A) was found to be very sensitive in detecting FXIa added to citrated platelet-poor plasma (PPP) and platelet-rich plasma (PRP). The specificity of the assay was confirmed by the reduction of the signal with the addition of FXI, which competes with FXIa–C1inh complex, for binding to the anti-FXI coating antibody or by inclusion of FXIa active site-directed inhibitor, which prevents the interaction with C1inh (not shown). When added in the absence of calcium, to avoid potential FXI feedback activation by thrombin, 10–40 pM of FXIa was sufficient to give a signal that was twice the background signal observed in the absence of exogenously added FXIa. Because FXI plasma concentration is 30 nM, the FXIa–C1inh ELISA can detect as little as 0.03–0.12% FXI activation in plasma. To study the effect of platelets and different procoagulants on FXI activation, PRP or PPP were recalcified and incubated with the indicated procoagulants for 1 h at 37°C (Fig. 1B). FXIa formation was determined by FXIa–C1inh ELISA and quantified using a standard curve formed in the same plasma sample with exogenously added FXIa. Initially, plasmas from different donors were stimulated by low concentrations of thrombin, TF, and the contact system activator, Alexin (50 pM, 1/80,000 and 1/320, respectively). These concentrations were chosen because they similarly reduce the activation time of the coagulation cascade in the functional assay described below (≈60%, not shown). Activation of FXI was only detected with Alexin stimulation under these conditions, with or without platelets (Fig. 1B). In subsequent experiments, higher levels of thrombin and TF were tested. Even at a 1000-fold higher thrombin or a 200-fold higher TF concentration, no FXIa generation was detected in the presence or absence of platelets in any of the three donors that were tested (Fig. 1B). In sharp contrast, stimulation with increasing concentrations of Alexin, gave a corresponding increase in the concentrations of FXIa detected by FXIa–C1inh ELISA (Fig. 1B).
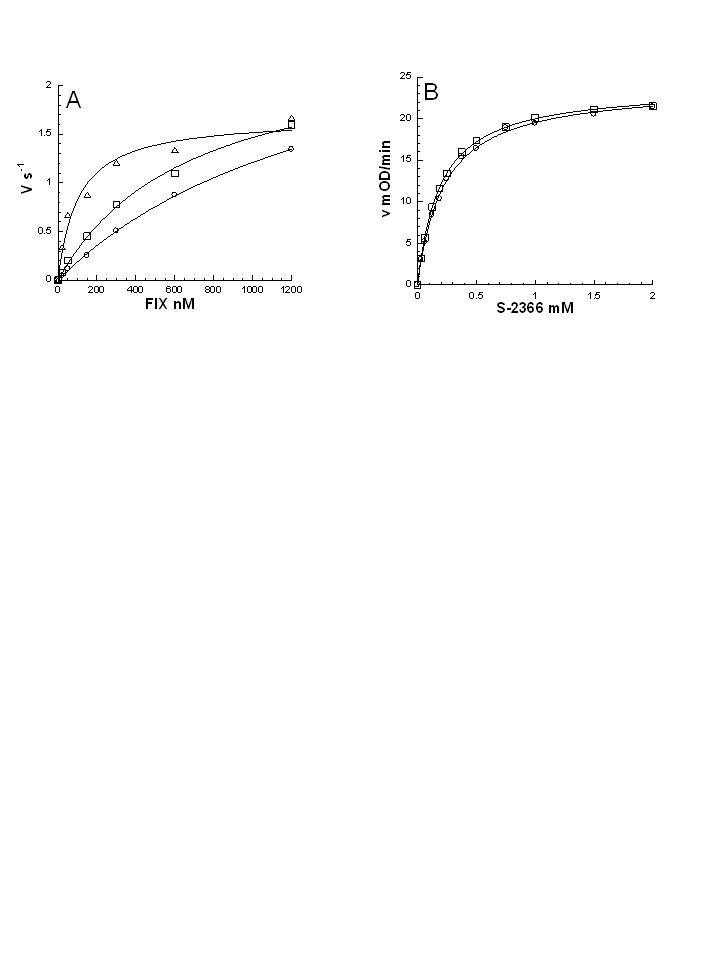
Fig. 1.

Detection of FXIa in plasma. (A) Standard curves for the FXIa–C1inh ELISA. PPP or PRP prepared from citrated blood was incubated with the indicated concentrations of FXIa. Diluted samples containing 10% or 0.2% plasma were analyzed (FXIa concentration reflects the concentration in the undiluted plasma). Each data point is run in duplicates with standard deviation typically below 10% of the signal. A representative experiment out of >10 experiments conducted with blood from four different donors is shown. (B) FXIa levels in PPP, PRP, and PPP reconstituted with washed activated platelets, stimulated with different procoagulants. Plasma samples from healthy donors were stimulated by the indicated procoagulants and the resulting FXIa-C1 inhibitor complexes were determined by ELISA. The average ± standard deviation for all donors tested (in pM) is shown, with the number of donors in parenthesis. The average lower limit of detection defined as the concentration of FXIa which gives a signal 2-fold higher than the background was 26 ± 11 pM for PPP, 19 ± 9 pM for PRP, and 26 ± 7 pM for PRP with washed activated platelets. BQL, below quantification limit.
The lack of FXI activation by thrombin or TF in these experiments was in apparent conflict with the observation that FXI is activated by thrombin on the surface of activated platelets in a reconstituted system (14, 15). Therefore, we performed a number of control experiments to confirm our results. To exclude the possibility that platelets, when activated, interfere with the detection of FXIa by secreting serpins that compete with C1inh for FXIa binding [e.g., protease nexin II (21, 22)], we repeated the experiments with PRP that was reconstituted from PPP and washed activated platelets. This procedure ensures the removal of serpins that might have been released during platelet activation. As in the case of the untreated platelet-rich plasma, no FXIa was detected after stimulation by high concentrations of TF or thrombin, whereas FXIa was readily detectable after stimulation of the contact system (Fig. 1B, last column). This observation, along with the lack of platelet effect on the FXIa standard curve (Fig. 1A) and on the detection of exogenously added FXIa (Fig. 1B) suggest that platelets do not interfere with the detection of FXIa in our system. The reduced concentration of FXIa after stimulation of PRP with Alexin (Fig. 1B) might have been caused by platelet interference with the contact activation system.
Despite failing to detect FXI activation when TF and thrombin were added, it is possible that low levels of FXIa, below our detection limit, were generated. We have attempted several strategies to reduce our detection limit. We were able to significantly reduce our detection limit only when corn trypsin inhibitor [CTI, a selective inhibitor of FXIIa (23, 24)] was added to the citrate tubes used for the blood draw (Fig. 2A). This observation suggested that most of the background in the FXIa–C1inh ELISA results from activation of FXI via the contact system during the blood draw and/or plasma preparation. Therefore, we drew plasma from two donors into CTI-containing citrate tubes and stimulated it with high concentrations of thrombin or TF. Despite reduction of the FXIa detection limit from 19–26 pM without CTI to 5.1–5.2 pM with CTI, FXIa levels were still below our detection limit (Fig. 2B). CTI had no effect on the detection of exogenously added FXIa in recalcified plasma (Fig. 2B).
Fig. 2.

Detection of FXIa in plasma prepared from blood drawn into CTI. (A) Standard curves prepared from blood drawn into citrate or citrate + CTI as described in the legend for Fig. 1. A representative experiment out of two experiments where samples with or without CTI were compared directly is shown. (B) FXIa levels in PPP and PRP prepared from blood drawn into citrate + CTI, stimulated with thrombin or TF. Plasma samples from two healthy donors were stimulated by the indicated procoagulants, and FXIa concentration was determined using the FXIa–C1inh ELISA. The average lower limit of detection, defined as the concentration of FXIa that gives a signal 2-fold higher than the background, was 5.1 ± 0.1 pM for PPP and 5.2 ± 0.2 pM for PRP. BQL, below quantification limit.
Inhibition of FXIa in Plasma Environment.
To complement the direct measurements of FXI activation, we used a functional assay to study the contribution of FXI activation by thrombin and TF to the activation of the coagulation cascade in a plasma environment. Using our previously developed FIXa detection assay (25), we have identified a monoclonal antibody that inhibited FIX activation by FXIa [supporting information (SI) Fig. 4 and SI Materials and Methods]. We used a promiscuous chromogenic substrate to monitor the progression of coagulation cascade activation in plasma with or without the FXIa inhibiting antibody and expressed the results as time to reach an OD405 increase of 1.0 (SI Fig. 5, see also ref. 26 for an example of a similar assay). The FXI antibody elicited a concentration dependent inhibition of the activation of the coagulation cascade by recalcification and by low concentration of tissue factor as was evident from the prolongation of the time required to reach an OD405 increase of 1.0 (Fig. 3A). The antibody also inhibited the activation of the coagulation cascade by the contact system activator, Alexin, in normal plasma but not in FXI deficient plasma (Fig. 3B), thus confirming the FXI-specificity of the antibody effect.
Fig. 3.

The effect of FXI antibody 5061 on the activation of the coagulation cascade in a plasma environment. (A) FXI antibody concentration response. Normal plasma was stimulated by recalcification (open symbols) or by recalcification and TF (1/106, filled circles) in the presence of the indicated concentration of FXI antibody 5061. Total IgG concentration (specific and nonspecific) was adjusted to 50 μg/ml. Each concentration was tested in triplicates. (B) The specificity of antibody 5061. Normal pooled plasma or FXI deficient plasma were stimulated with Alexin (1/80) in the presence of 10 μg/ml antibody 5061 or mouse IgG. Each sample was tested in triplicates.
Effect of Platelets, Thrombin, and TF on the Contribution of FXI to the Activation of the Coagulation Cascade.
The experiments shown in Fig. 3 were carried out in the absence of an inhibitor of the contact activation system. Based on our observation that the contact system activates FXI during blood draw (Fig. 2A) and the potential for further activation by the assay plate surface, we carried out additional experiments using PPP and PRP that were drawn into CTI containing tubes. PPP and PRP from three donors were stimulated by activating the extrinsic and the common pathways with several concentrations of TF and thrombin as well as by recalcification alone (Table 1, first four columns). In all cases, procoagulant concentration-dependent reductions in coagulation cascade activation time were observed. Platelets significantly reduced the activation time of the coagulation cascade as expected from the role of platelet phospholipids in promoting the assembly of the Xase and prothrombinase complexes. Nevertheless, no significant effect of the FXI inhibiting antibody was observed when the coagulation cascade was activated by thrombin or TF in either PPP or PRP. This lack of effect was in apparent conflict with earlier studies reporting a contribution of FXI to the activation of the coagulation cascade activation by TF even in the presence of CTI (7). One important difference between the two studies was the inclusion of CTI in the blood draw in this study, whereas it was added only before assay initiation in the previous study. To test whether this could account for the different results, we repeated our experiments drawing blood into citrate alone but preincubating the plasma with the same CTI concentration before initiation of the coagulation cascade (Table 1, last two columns). When CTI was added after plasma isolation, the FXI antibody generated statistically significant prolongation of the activation lag time in all cases. In line with the previous studies, the maximal effect of FXIa inhibition was observed with the mildest stimulus (recalcification alone). Moreover, the coagulation activation lag time was significantly shorter when CTI was added only after plasma isolation (Table 1, compare the two middle columns to the last two columns), indicating that contact system activation during blood draw or plasma preparation contributes significantly to the activation of the coagulation cascade in citrated plasma.
Table 1.
The effect of FXI inhibition on the activation of the coagulation cascade in plasma
| CTI added to blood draw tubes |
CTI added to PPP after plasma preparation | |||||
|---|---|---|---|---|---|---|
| PRP | PPP | |||||
| FXI antibody | - | + | - | + | - | + |
| Recalcification | 65.1 ± 4.5 | 71.0 ± 12.5 | 90 ± 0 | 90 ± 0 | 15.5 ± 2.3 | 25.5 ± 5.6* |
| TF 10−6 | 37.1 ± 6.8 | 34.4 ± 4.2 | 81.8 ± 14.2 | 86.6 ± 5.8 | 14.5 ± 2.6 | 20.6 ± 3.9* |
| TF 2.5 × 10−6 | 24.9 ± 1.9 | 25.3 ± 1.4 | 66.9 ± 15.4 | 68.8 ± 19.7 | 13.2 ± 2.2 | 17.9 ± 3.3* |
| TF 1.25 × 10−5 | 13.4 ± 0.5 | 15.1 ± 1.8 | 19.5 ± 4.2 | 19.3 ± 3.9 | 10.1 ± 1.5 | 12.2 ± 1.9* |
| Thrombin 50 pM | 24.1 ± 6.7 | 23.2 ± 5.3 | 58.6 ± 4.8 | 68.0 ± 3.9 | 10.8 ± 0.2 | 13.9 ± 0.5* |
| Thrombin 150 pM | 22.3 ± 3.7 | 21.0 ± 4.0 | 37.8 ± 4.0 | 41.9 ± 1.7 | 8.6 ± 0.7 | 10.8 ± 0.9* |
PRP or PPP from three healthy donors prepared from blood drawn into citrate or citrate + CTI was preincubated with 10 μg/ml of FXI antibody 5061 or mouse IgG and CTI (when it was not included in the drawing tubes). After preincubation, plasma was stimulated with the indicated procoagulant. The average time (in min) for the three donors is shown. Conditions where the P value of a paired t test for reactions with or without the FXI antibody is ≤ 0.05 are denoted with *.
Discussion
The aim of this study was to investigate the mechanism of FXI activation in a plasma environment. Despite our sensitive detection method (5 pM FXIa, corresponding to activation of 0.017% of plasma FXI), no FXI activation by the extrinsic pathway or the common pathway was detected in plasma with or without platelets. Furthermore, no contribution of FXI to the activation of the coagulation cascade by the extrinsic pathway or the common pathway was observed after elimination of the contact system. These observations suggest that activation of the extrinsic and/or the common pathway is insufficient to promote FXI activation in plasma.
How can the results of this study be reconciled with the previous reports, which used similar plasma assays and linked FXI activation to the extrinsic and/or the common pathway? The most important reason for the differences is the lack of complete elimination of FXI activation through the contact system in the previous studies. Most earlier studies used plasma samples obtained from blood drawn into citrate in the absence of any inactivator of the contact system (7, 8, 27). Although these studies attempted to eliminate the contribution of the contact system to FXI activation by adding CTI or a FXII antibody to the isolated plasma, these additions did not eliminate the FXIa that was already formed via contact system activation during the blood draw and plasma isolation (ref. 28 and Fig. 2 and Table 1). Thus, these studies have mistakenly attributed the FXI dependency of their assays to the added procoagulant rather than to contact system activation during blood draw and plasma isolation. The study by von dem Borne et al. (9) appears to be better controlled for inadvertent activation of FXI by the contact system because it used plasma drawn from a FXII-deficient patient. An important caveat of this study, however, is the use of FXII deficient plasma that was immunodepleted for endogenous FXI and reconstituted with purified FXI. As reported in ref. 9, the purified FXI contained traces of FXIa and these traces could have been mistakenly taken for FXI activated by the extrinsic pathway. Furthermore, the exogenously added FXI may have not been appropriately bound by its numerous reported partners (e.g., HK, prothrombin and platelets), thereby facilitating non physiologically relevant activation. Finally, the inverse correlation between the concentration of TF and the FXI dependency in plasma clotting assays (refs. 7 and 9, and in this study when CTI is absent from the blood draw tubes) further confirms that contact activation rather than thrombin formed by the extrinsic pathway is the real cause for the FXI dependency in these assays.
A number of studies using purified coagulation proteins have reported activation of FXI by thrombin (29) with several orders of magnitude enhancement by activated platelets (14–16). In this study, however, we did not observe a platelet effect on FXI activation using the FXIa–C1inh ELISA and the coagulation cascade activation assay. Theoretically, FXIa generated on platelets may have been sequestered from binding to C1inh and therefore undetectable in our assay. The observation that clinically relevant FXIa was found to associate with C1inh (30, 31) and the rapid disassociation of FXIa from platelets (t1/2 off <5 min; ref. 32) relative to the duration of our experiments (1 h) makes this possibility unlikely. Another potential cause for our inability to replicate the platelet effects observed in the reconstituted systems may have been the difficulty to restore sufficient concentration of free Zn++ ions to citrated plasma because of the high affinity of citrate to Zn++. Although it was reported that Zn++ ions are important for binding of FXI to platelets via HK, Ca2+, which was restored to the citrated plasma in all assays performed in this study, can substitute for Zn++ using prothrombin as a cofactor for platelet binding (14, 15). In preliminary experiments, we were unable to detect FXIa formation in whole blood drawn into citrate-free tubes containing CTI and TF or thrombin, thus suggesting that Zn++ depletion was not the cause for the discrepancy between this study and the reconstituted systems studies.
It is also possible that platelets did promote FXI activation in our experiments, below the detection limit of 5 pM (Fig. 2B). If this was the case, then the rate of FXI activation in plasma environment is at least 7 orders of magnitude lower than the rate reported for the reconstituted system, in the absence of plasma (<5 pM/h with 50 nM thrombin vs. 25 nM/min with 1.25 nM thrombin; ref. 14). The inability of platelets to promote detectable FXI activation in plasma is probably the result of interference by plasma component(s). Accordingly, several plasma components were shown to interfere with FXI activation by thrombin (5, 13, 33). Thus, the observations, made in regard to FXI activation in reconstituted systems, may not apply in a plasma environment.
How is FXI activated in vivo during hemostasis? The results of this study question the present hypothesis that FXI activation by thrombin on activated platelets augments the coagulation cascade during hemostasis. An alternative mechanism could involve the contact activation system which was shown to mediate efficient activation of FXI in plasma. This possibility has been dismissed in the past because patients with FXII, HK or prekallikrein deficiency do not bleed excessively. However, a number of recent observations argue that the contact activation system is the main pathway for FXI activation in vivo. FXI and FXII knockout mice have very similar phenotypes. They show minimal or no excessive bleeding while displaying reduced capacity for thrombus formation (19, 34, 35), thus suggesting similar function for FXI and the contact system during hemostasis and thrombosis. The mouse phenotype may be more reliable than the human phenotype because it involves subjects with much more uniform genetic backgrounds. The identity of the surface that mediates contact system activation during hemostasis or thrombosis is still unknown as the negatively charged polymers that are used in in vitro assays are artificial. The observation that RNA can activate the contact system and contribute to thrombus formation in vivo provides a physiologically relevant template for contact system activation during hemostasis (36).
The diversion between the phenotypes of FXI deficient individuals and individuals with deficiency in other components of the contact system can also be explained by the numerous thrombin-inhibiting and profibrinolytic activities of the contact system and its products (1, 2). These activities are mediated by plasma prekallikrein, FXII, HK, and its degradation products, and are largely independent of FXI. Thus, the antihemostatic effect of the lack of FXI activation in individuals deficient in prekallikrein FXII, HK is compensated by the reduced thrombin-inhibitory and profibrinolytic activity of the contact system. FXI-deficient individuals, on the other hand, maintain the thrombin-inhibitory and profibrinolytic effects of the contact system and are therefore more prone to excessive bleeding.
Yet another alternative is that the contact activation system might activate FXI under conditions that promote pathological thrombosis, whereas contact system-independent mechanism(s) could activate FXI during functional hemostasis (37). In this respect, it is important to note that none of the in vitro plasma assays that were used to dissect the mechanism of FXI activation in the present or past studies can replicate the contribution of the endothelium, injured tissue or blood flow and resulting shear stress to the activation of FXI. Each of them could further promote a contact system-dependent or independent activation of FXI.
Materials and Methods
Plasma Samples and Procoagulants.
Citrated pooled normal plasma and congenital FXI-deficient plasma were obtained from George King Bio-medical (Overland Park, KS). Samples from healthy donors were collected by venous puncture into buffered sodium citrate containing tubes [3.2%, Becton Dickinson (Franklin Lakes, NJ)]. In some cases the tubes contained CTI (Haematologic Technologies, Essex Junction, VT), final concentration 100 μg/ml) to minimize the activation of the contact system during blood draw. PRP and PPP were isolated after 10 min centrifugation at room temperature at 175 × g and 3,100 × g, respectively. Procoagulants, thrombin, Innovin (TF, the concentration of tissue factor is expressed as dilution factor because the concentration of tissue factor in the Innovin preparation is not disclosed by the manufacturer) and Alexin (containing purified rabbit brain cephalin and 0.1 mM ellagic acid) were obtained from Haematologic Technologies, Dade Behring (Deerfield, IL), and Sigma Diagnostics (St. Louis, MO), respectively.
Detection of FXIa–C1inh Complex.
Citrated PPP or PRP (90 μl) were combined with 10 mM CaCl2, 25 μM ZnCl2, and the procoagulant of choice in a final reaction volume of 100 μl. For the generation of standard curves, FXIa was added and CaCl2 and ZnCl2 were omitted to avoid potential feedback activation of FXI. The reactions were incubated at 37°C for 60 min and were quenched by the addition of 900 μl of PBSTB (PBS, 0.05% Tween 20, 0.1% BSA) containing 2 mM EDTA. After quenching, clots were dissolved by brief sonication or by homogenization using a Polytron (Brinkman, Westbury, NY). The samples were spun down using an Eppendorf centrifuge (6 min, 16,000 × g), and the supernatant was recovered. FXIa–C1inh complexes in these samples were detected as follows: A Microlite 2 plate (Thermo Labsystems, Waltham, MA) was coated with 100 μl of 10 μg/ml of mouse anti human FXI antibody 5061 (Haematologic Technologies). After blocking with 5% BSA in PBS (200 μl), appropriately diluted plasma samples (100 μl) were added and incubated for 2 h. After sample removal and wash, 100 μl of PBSTB containing biotinylated affinity-purified chicken antibody, which we have generated against human C1inh (The Binding Site, San Diego, CA), was added and incubated for additional 1 h. The complex was detected by incubation with streptavidin horseradish peroxidase (HRP) conjugate (Zymed, Carlsbad, CA) in PBSTB, followed by an HRP chemiluminesence detection reagent (SuperSignal ELISA Pico, Pierce, Rockford, IL). The concentrations of FXIa in samples were determined by using a linear regression of the log of FXIa concentration versus the log of background-subtracted chemiluminesence counts on the corresponding standard curve. Typically, 10% plasma samples were used for FXIa concentrations of up to 300 pM, whereas 0.2% plasma dilutions were used for FXIa concentrations ranging from 300 pM to 10 nM.
Preparation of Washed Activated Platelets.
Blood was drawn into buffered citrate for PPP and into 85 mM trisodium citrate, 71 mM citric acid, and 111 mM dextrose (pH 4.5) for PRP. The platelets were isolated from PRP by centrifugation (20 min, 365 × g) and resuspended in a similar volume of 137 mM NaCl, 2.7 mM KCl, 12 mM NaHCO3, 0.4 mM NaH2PO4, 1 mM MgCl2, 5 mM CaCl2 5 mM Hepes (pH 7.4), 100 nM Roxifiban (a GpIIb/IIIa antagonist; ref. 38), and 25 μM of the protease activated receptor 1 agonist peptide SFLLRN (Bachem, King of Prussia, PA). After incubation for 30 min at 37°C, the platelets were pelleted and resuspended in a similar volume of PPP to generate PRP reconstituted with activated platelets. The combination of SFLLRN and Roxifiban allows platelet activation without aggregation (it should be noted that GPIIb/IIIa antagonists do not prevent platelet activation and secretion by PAR1 agonist peptides even though outside-in signaling via GPIIb/IIIa is abolished).
Activation of the Coagulation Cascade in Plasma.
Plasma (100 μl per well, 50% final concentration) was preincubated for 30 min at room temperature with the indicated concentration of FXI antibody 5061 or mouse IgG control (Sigma) and, when indicated, with CTI (100 μg/ml). The reactions were initiated by the addition of the plasma-antibody mixture to a 96-well clear polypropylene plate (Corning, Lowell, MA) containing 200 μM S-2366 (Diapharma, West Chester, OH), 10 mM CaCl2, 25 μM ZnCl2 and the indicated procoagulant in FXIa assay buffer. The reactions (200 μl total volume) were carried out at 37°C and OD405 was monitored for up to 90 min using a Spectramax Plus plate reader (Molecular Devices, Sunnyvale, CA). The efficiency of activation of the coagulation cascade is expressed as the time to reach an OD405 change of 1.0. This value was chosen to ensure a robust signal within the dynamic range of the spectrophotometer. Using OD values of 0.2 or 0.5 did not have a significant effect on the results.
Supplementary Material
Abbreviations
- FXI
factor XI
- FXIa
activated FXI
- HK
migh molecular weight kininogen
- TF
tissue factor; PPP platelet-poor plasma
- PRP
platelet-rich plasma
- CTI
corn trypsin inhibitor
- serpin
serine protease inhibitor
- C1inh
C1 inhibitor
- FXII
factor XII.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705566104/DC1.
References
- 1.Colman RW. In: Hemostasis and thrombosis. Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN, editors. Philadelphia: Lippincott Williams and Wilkins; 2001. pp. 104–121. [Google Scholar]
- 2.Joseph K, Kaplan AP. Adv Immunol. 2005;86:159–208. doi: 10.1016/S0065-2776(04)86005-X. [DOI] [PubMed] [Google Scholar]
- 3.Shariat-Madar Z, Schmaier AH. J Endotoxin Res. 2004;10:3–13. doi: 10.1179/096805104225003807. [DOI] [PubMed] [Google Scholar]
- 4.Yarovaya GA, Blokhina TB, Neshkova EA. Biochemistry (Moscow) 2002;67:13–24. doi: 10.1023/a:1013991828598. [DOI] [PubMed] [Google Scholar]
- 5.Gailani D, Broze GJJ. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 6.Naito K, Fujikawa K. J Biol Chem. 1991;266:7353–7358. [PubMed] [Google Scholar]
- 7.He R, Xiong S, He X, Liu F, Han J, Li J, He S. Thromb Haemost. 2001;85:1055–1059. [PubMed] [Google Scholar]
- 8.Keularts I. M. L. W., Zivelin A, Seligsohn U, Hemker HC, Beguin S. Thromb Haemost. 2001;85:1060–1065. [PubMed] [Google Scholar]
- 9.von dem Borne PAK, Meijers JCM, Bouma BN. Blood. 1995;86:3035–3042. [PubMed] [Google Scholar]
- 10.Bouma BN, Mosiner LO, Meijers JCM, Griffin JH. Thromb Haemost. 1999;82:1703–1708. [PubMed] [Google Scholar]
- 11.Von dem Borne PA, Bajzar L, Meijers JC, Nesheim ME, Bouma BN. J Clin Invest. 1997;99:2323–2327. doi: 10.1172/JCI119412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunnee T, La Porta C, Reddigari SR, Salerno VM, Kaplan AP, Silverberg M. Blood. 1993;81:580–586. [PubMed] [Google Scholar]
- 13.Scott CF, Colman RW. Proc Natl Acad Sci. 1992;89:11189–11193. doi: 10.1073/pnas.89.23.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baglia FA, Walsh PN. Biochemistry. 1998;37:2271–2281. doi: 10.1021/bi972113+. [DOI] [PubMed] [Google Scholar]
- 15.Baglia FA, Walsh PN. J Biol Chem. 2000;275:20514–20519. doi: 10.1074/jbc.M000464200. [DOI] [PubMed] [Google Scholar]
- 16.Oliver JA, Monroe DM, Roberts HR, Hoffman M. Arterioscler Thromb Vasc Biol. 1998;19:170–177. doi: 10.1161/01.atv.19.1.170. [DOI] [PubMed] [Google Scholar]
- 17.Greengard JS, Heeb MJ, Eva E, Walsh PN, Griffin JH. Biochemistry. 1986;25:3884–3890. doi: 10.1021/bi00361a022. [DOI] [PubMed] [Google Scholar]
- 18.Baglia FA, Jameson BA, Walsh PN. J Biol Chem. 1995;270:6734–6740. doi: 10.1074/jbc.270.12.6734. [DOI] [PubMed] [Google Scholar]
- 19.Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, Gailani D, Nieswandt B. J Exp Med. 2005;202:271–281. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.wuillemin WA, Minnema MC, Meijers JCM, Roem D, Eerenberg AJM, Nuijens JH, ten Cate H, Hack EC. Blood. 1995;85:1517–1526. [PubMed] [Google Scholar]
- 21.Smith RP, Higuchi DA, Broze GJJ. Science. 1990;248:1126–1128. doi: 10.1126/science.2111585. [DOI] [PubMed] [Google Scholar]
- 22.Van Nostrand WE, Wagner SL, Farrow JS, Cunningham DD. J Biol Chem. 1990;265:9591–9594. [PubMed] [Google Scholar]
- 23.Hojima Y, Pierce JV, Pisano JJ. Thromb Res. 1980;20:149–162. doi: 10.1016/0049-3848(80)90381-3. [DOI] [PubMed] [Google Scholar]
- 24.Rand MD, Lock JB, van't Veer C, Gaffney DP, Mann KG. Blood. 1996;88:3432–3445. [PubMed] [Google Scholar]
- 25.Pedicord DL, Seiffert D, Blat Y. Biochemistry. 2004;43:11883–11888. doi: 10.1021/bi048964g. [DOI] [PubMed] [Google Scholar]
- 26.Hemker HC, Al Dieri R, De Smedt E, Beguin S. Thromb Haemost. 2006;96:553–561. [PubMed] [Google Scholar]
- 27.Wielders SJ, Beguin S, Hemker HC, Lindhout T. Arterioscler Thromb Vasc Biol. 2004;24:1138–1142. doi: 10.1161/01.ATV.0000128125.80559.9c. Epub 2004 Apr 1138. [DOI] [PubMed] [Google Scholar]
- 28.Ramstrom S. Blood Coagul Fibrinolysis. 2005;16:447–452. doi: 10.1097/01.mbc.0000178827.52242.89. [DOI] [PubMed] [Google Scholar]
- 29.von dem Borne PAK, Koppelman SJ, Bouma BN, Meijers JCM. Thromb Haemost. 1994;72:397–402. [PubMed] [Google Scholar]
- 30.Minnema MC, Pajkrt D, wuillemin WA, Roem D, Bleeker WK, Levi M, van Deventer SJH, Hack EC, ten Cate H. Blood. 1998;92:3294–3301. [PubMed] [Google Scholar]
- 31.Minnema MC, Peters RJG, de Winter R, Lubbers YPT, B. B., Bauer KA, Rosenberg RD, Hack EC, ten Cate H. Arteriosclr Thromb Vasc Biol. 2000;20:2489–2493. doi: 10.1161/01.atv.20.11.2489. [DOI] [PubMed] [Google Scholar]
- 32.Sinha D, Seaman FS, Koshy A, Knight LC, Walsh PN. J Clin Invest. 1984;73:1550–1556. doi: 10.1172/JCI111361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi T, Iverson GM, Qi JC, Cockerill KA, Linnik MD, Konecny P, Krilis SA. Proc Natl Acad Sci USA. 2004;101:3939–3944. doi: 10.1073/pnas.0400281101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gailani D, Lasky NM, Broze GJ., Jr Blood Coagul Fibrinolysis. 1997;8:134–144. doi: 10.1097/00001721-199703000-00008. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Cheng Q, Xu L, Feuerstein GZ, Hsu MY, Smith PL, Seiffert DA, Schumacher WA, Ogletree ML, Gailani D. J Thromb Haemost. 2005;3:695–702. doi: 10.1111/j.1538-7836.2005.01236.x. [DOI] [PubMed] [Google Scholar]
- 36.Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, et al. Proc Natl Acad Sci USA. 2007;104:6388–6393. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colman RW. J Exp Med. 2006;203:493–495. doi: 10.1084/jem.20060217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seiffert D, Pedicord DL, Kieras CJ, He B, Stern AM, Billheimer JT. Thromb Res. 2002;108:181–189. doi: 10.1016/s0049-3848(02)00395-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}