

Abstract
Arginine methylation is a prevalent modification found in many RNA binding proteins, yet little is known about its functional consequences. Using a monoclonal antibody, 1E4, we have shown that the yeast NPL3 gene product Npl3p, an essential RNA binding protein with repeated RGG motifs, is arginine-methylated in vivo. The 1E4 epitope can be generated by incubating recombinant Npl3p with partially purified bovine arginine methyltransferase, and peptides that specifically inhibit arginine methyltransferases block this reaction. Npl3p methylation requires S-adenosyl-l-methionine and also occurs in yeast extracts. An Npl3p deletion mutant lacking the RGG domain is not a substrate for methylation, suggesting that the methylation sites lie within the RGG motifs. The discovery of arginine methylation in a genetically tractable organism provides a powerful entrée to understanding the function of this modification, particularly in view of the many roles postulated for Npl3p in RNA processing and transport. The recent discovery of phosphorylated serine residues within the RGG domain suggests a hypothesis in which a molecular switch governed by methylation and phosphorylation regulates the biochemical properties of the Npl3p RGG domain.
Protein methylation is a common enzymatically generated modification that can regulate the activity of the target protein or create new types of amino acids (1, 2). The catalysts for these modifications, protein methyltransferases, transfer a methyl group from S-adenosyl-l-methionine (SAM or AdoMet) to nucleophilic oxygen, nitrogen, or sulfur atoms on the substrate protein. The effects of protein methylation fall into two general categories. In the first, the relative levels of methyltransferases and methylesterases can control the extent of methylation at a particular carboxyl group, which in turn regulates the activity of the target protein. This reversible modification is thus analogous to protein phosphorylation, governed by kinases and phosphatases. This category includes the following enzymes: type I methyltransferases, which function in bacterial chemotaxis by modifying glutamate residues in the chemoreceptor; type II methyltransferases, which function in metabolizing damaged proteins; type III methyltransferases, which modify C-terminal isoprenylated cysteine residues, most notably those found in some fungal mating factors, many members of the G-protein family, some nuclear lamins and retinal cGMP phosphodiesterase; and type IV methyltransferases, which modify a C-terminal serine residue, most notably on protein phosphatase 2A.
The second general group of protein methylation reactions involves the apparent irreversible modification of sulfur or nitrogen atoms in the substrate protein (1, 2). These reactions generate new amino acids with altered biochemical properties that are proposed to alter the activity of the target protein. These amino acids include methyl amines of alanine, methionine, phenylalanine, proline, and lysine, methyl amides of glutamine and asparagine, S-methyl thioethers of cysteine and methionine and NG-mono- and dimethyl amines of arginine. In general, the biological function of these methylations is unclear but is being analyzed on a case-by-case basis in the individual proteins in which the modification is found.
Arginine methylation appears to play a number of roles depending upon its context. The modification is catalyzed by protein methyltransferase I, isolated in two immunologically distinct forms in partial purifications from calf brain cytosol (3, 4). The first form monomethylates and symmetrically (NG, N′G) dimethylates arginine 107 of myelin basic protein, whereas the second form monomethylates and asymmetrically (NG, NG) dimethylates arginine residues within glycine/arginine-rich domains found in many RNA binding proteins. The sequenced arginine methylation sites all contain an adjacent glycine residue, suggesting that this arrangement is required for methylation (5). Recently, a mammalian protein arginine methyltransferase (PRMT1) was identified in a yeast two-hybrid screen (6). Sequence data base searches revealed that PRMT1 is related to a protein arginine methyltransferase (RMT1) in yeast (7). This same yeast gene was recently independently isolated as an hnRNP (heterogeneous nuclear ribonucleoprotein particle) methyltransferase (HMT1) (8). The substrate specificity of recombinant Rmt1p/Hmt1p indicates that this enzyme is a protein methyltransferase I that methylates glycine/arginine-rich domains (7, 8).
The prevalence of methylated arginine residues in RNA binding proteins has led to the proposal that this modification may have profound effects on RNA metabolism (9). For example, most of the pre-mRNA-binding hnRNP proteins in HeLa cells are methylated, and approximately 65% of nuclear NG,NG-dimethylarginine resides in hnRNPs (10). One type of methylated RNA binding domain is the RGG box, 20- to 25-amino acids long and defined as closely repeated arginine-glycine-glycine (RGG) tripeptides interrupted by other amino acids, usually aromatics (11). The RGG box protein family is a large one, including members postulated to function in diverse areas of RNA metabolism, such as mRNA splicing, mRNA export, rRNA processing and RNA packaging. Most of these family members contain additional RNA binding domains, such as the RNA recognition motif (RRM or RNP-CS). This observation, together with RNA binding studies of nucleolin (12), suggests that RNA binding by the RGG box may be fairly nonspecific, mediated primarily by the positive charge on arginine. This model, however, does not address why arginine is used exclusively in this domain. An attractive hypothesis is that arginine methylation may regulate RGG box activity by blocking hydrogen bonding or introducing steric constraints that are predicted to hinder interactions between arginine and RNA (13). Alternatively, the observations that arginine can recognize particular RNA backbone structures (13) or bases (14, 15) suggest that some RGG boxes may direct specific RNA binding.
The study of arginine methylation would benefit greatly from the ability to molecularly dissect its function in a system amenable to genetic analysis. Recent work has identified an arginine methyltransferase and substrates in budding yeast (7, 8). One candidate substrate in yeast, encoded by the NPL3 gene, contains two RRM RNA binding domains and a C-terminal RGG box (Fig. 1) (16, 17). Interestingly, we noted that the RGG box overlaps with a domain containing eight arginine-serine (RS) or serine-arginine (SR) dipeptides, with the sequence SRGG repeated six times. Such RS-rich domains are found in many pre-mRNA splicing factors, including members of the SR protein family, characterized in metazoans as essential splicing factors that also affect splice site selection (18). The extensive serine phosphorylation observed in this domain has been proposed to regulate SR protein activity by affecting interactions between SR proteins and RNA or other splicing factor proteins (18). The presence of an RS domain in Npl3p, as well as sequence similarities between the RRMs in Npl3p and some SR proteins (19), has led to the proposal that Npl3p functions in splicing (C.W.S. and C.G., unpublished results).
Figure 1.
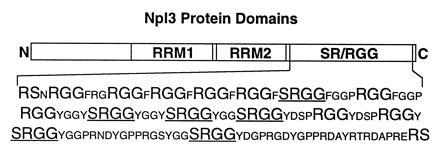
Schematic diagram of the Npl3 protein domains. The Npl3 protein, represented as a rectangle with the N terminus at left and the C terminus at right, has an N-terminal domain of unknown function, two central RRMs, and a C-terminal RGG box RNA binding domain that overlaps with an SR domain containing eight SR or RS dipeptides. The sequence of this SR/RGG domain is listed below, with the RS, SR, and RGG peptides in larger font and the SRGG tetrapeptides underlined. One or more serine residues within the SR or RS dipeptides appear to be phosphorylated and one or more arginine residues within the RGG tripeptides appear to be methylated (see text).
NPL3 also has been implicated in a variety of other reactions. Originally identified in a screen for mutations that affect nuclear protein import (16), NPL3 has also been independently identified: (i) as NOP3, in a low-stringency DNA hybridization screen for genes containing a glycine/arginine-rich domain; depletion of Nop3p in vivo appears to disrupt rRNA processing (17); (ii) as Nab1p, a protein that crosslinks in vivo to nuclear polyadenylylated [poly(A)+] RNA (20); (iii) in a screen for mutations that disrupt mating-type silencing (21); (iv) as a suppressor of a mutation that disrupts protein import into mitochondria (22); and (v) in a screen for mutations that lead to nuclear accumulation of polyadenylylated RNA, indicative of a possible mRNA export defect (23).
The pleiotropy of npl3 mutations, as well as the lack of in vitro assays, has made it difficult to assess whether NPL3 functions directly or indirectly in these different pathways. For example, it appears that NPL3 plays only an indirect role in protein import (16) and silencing (21). In contrast, recent work suggests that one function of NPL3 may be in mRNA export (24). Npl3p is a predominantly nuclear protein that shuttles between the nucleus and cytoplasm in a transcriptionally dependent fashion; mutations in the Npl3p RRM RNA binding domains prevent Npl3p from shuttling and lead to the accumulation of poly(A)+ RNA in the nucleus. These results have led to a model in which Npl3p functions as a carrier for mRNA being exported from the nucleus to the cytoplasm (24).
We sought to determine whether the RGG box of Npl3p is arginine-methylated, by exploiting a monoclonal antibody (mAb) 1E4 (20) that we discovered recognizes Npl3p expressed in yeast but not Npl3p [recombinant (r) Npl3p] expressed in bacteria, suggesting that it recognizes a modified epitope. Herein, we report that Npl3p is methylated on arginine in vivo, probably within the RGG box domain, and that this modification can be reconstituted in vitro. Thus, the widely used but poorly understood modification of arginine methylation can be studied in a genetically powerful system, and mAb 1E4 provides a valuable tool to simply assay methylation of an essential RNA binding protein. Moreover, taken together with our recent discovery that Npl3p is serine phosphorylated by a kinase specific for RS domains (C.W.S., L. Feng, X.-D. Fu, and C.G., unpublished results), these results suggest that serine and arginine residues within the Npl3p SR/RGG domain are targets for two different modifications. The potential juxtaposition of distinctly modified residues raises the intriguing possibility that one modification regulates the appearance of the other or that both modifications function together to bestow a dynamic range of new biochemical properties to the Npl3p SR/RGG domain.
MATERIALS AND METHODS
Yeast Extracts.
Yeast extracts were prepared as described (25). For Fig. 5, extracts were prepared for immunoblot analysis as described (26).
Figure 5.
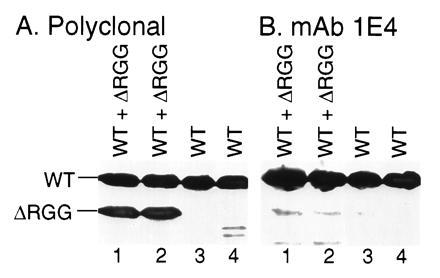
Npl3p lacking the C-terminal domain is expressed in vivo without the mAb 1E4 epitope. Duplicate extracts from cells carrying either a plasmid expressing an Npl3p deletion mutant lacking the C-terminal domain (ΔRGG) (lanes 1 and 2) or a control plasmid lacking an insert (lanes 3 and 4) were examined by immunoblot analysis using anti-Npl3p polyclonal antibodies (A) or mAb 1E4 (B). WT and ΔRGG on the left mark the positions of full-length Npl3p expressed from the wild-type chromosomal gene and the deletion mutant expressed from the plasmid, respectively.
Recombinant Npl3p.
The polymerase chain reaction (PCR) was used to amplify NPL3 or npl3-ΔRGG DNA and subclone the sequence from the initiation codon to the DraI site 58 base pairs downstream of the stop codon into the BamHI and PvuII sites of pRSETA (Invitrogen). Overlap extension PCR reactions (27) were used to create the npl3-ΔRGG mutant using the following primers: 5′A, CTGGATATTTAACAGACCCA; 3′A, CTCAACTATATAAATGGCTTATCTGATTGGTGGAGGATTGTCATCTC; 5′B, TAAGCCATTTATATAGTTGAG; 3′B, ATTAACCCTCACTAAAG. The template was pRS315-NPL3, made by subcloning the NPL3 HaeII–AflIII fragment (16) into the SmaI site of pRS315 (28). rNpl3p and rNpl3ΔRGGp were purified as described (29).
Immunoblot Analysis.
Immunoblot analysis using enhanced chemiluminescence was performed according to the manufacturer (Amersham) using 5% milk as the blocking agent. Anti-Npl3p polyclonal antibodies were prepared against purified rNpl3p (Berkeley Antibody, Richmond, CA). mAbs 1E4 and 2F1 were gifts from M. Swanson (University of Florida, Gainesville) and G. Dreyfuss (University of Pennsylvania, Philadelphia), respectively.
Methylation in Vitro.
For the immunoblot experiments, 25-μl reactions were as described (30) using 6.75 μg of partially purified arginine methyltransferase and 1 μg of rNpl3p. For the label transfer experiments, 10-μl reactions contained 14.5 μCi of [3H]SAM at 14 Ci/mmol (New England Nuclear; 1 Ci = 37 GBq), 2.7 μg of enzyme or 60 μg of yeast extract, and 400 ng of rNpl3p. The methyltransferase and the peptides were gifts from D. Aswad (University of California, Irvine).
RESULTS
Nuclear polyadenylylated RNA binding (Nab) proteins were previously purified from yeast by a method that uses UV light to crosslink proteins contacting poly(A)+ RNA in vivo (20, 31). The genes encoding Nab2p and Nab3p were cloned using mAbs raised against the purified Nab proteins to screen a bacterially expressed yeast protein library. mAb 1E4 against Nab1p, later shown to be identical to Npl3p (20), consistently failed to recognize any bacterially expressed protein in such screens. This observation hinted that mAb 1E4 might recognize an epitope that required an Npl3p modification made in yeast but not bacteria.
To determine whether mAb 1E4 recognizes a modified epitope on Npl3p, we used immunoblot analysis to compare the mAb 1E4 reactivity of Npl3p expressed in yeast (yNpl3p) with that of rNpl3p expressed in bacteria. As a control, anti-Npl3p mAb 2F1 (M. Matunis, P. O’Connor, and G. Dreyfuss, personal communication) (Fig. 2A) as well as anti-Npl3p polyclonal antibodies (data not shown) were used and shown to recognize both yNpl3p and rNpl3p. In contrast, mAb 1E4 efficiently recognized yNpl3p but failed to recognize even the highest amount of rNpl3p (Fig. 2B). These results suggest that mAb 1E4 recognizes an Npl3p epitope that includes a post-translational modification added to Npl3p in yeast but not in bacteria. The presence in Npl3p of an RS domain, known to be serine phosphorylated in metazoan SR splicing factors (18), suggested that the modification could be phosphorylation, whereas the presence of RGG tripeptides pointed to arginine methylation (Fig. 1).
Figure 2.
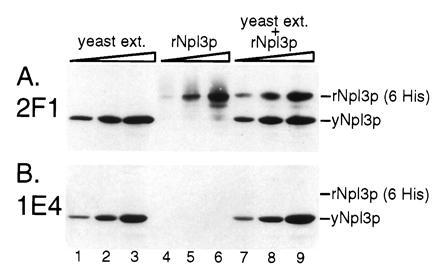
mAb 1E4 recognizes a modified epitope on Npl3p. Duplicate immunoblots were probed with anti-Npl3p mAbs 2F1 (A) or 1E4 (B). Lanes 1–3 and 7–9 contain 0.25, 0.5, or 1.0 μl of whole-cell yeast extract (approximately 10 mg of total protein per ml), respectively. Lanes 4–6 and 7–9 contain 12.5, 25, or 50 ng of recombinant Npl3p (rNpl3p) expressed in bacteria and purified exploiting a six-histidine tag that also increases the molecular mass of the protein relative to untagged Npl3p expressed in yeast (yNpl3p).
We tested whether mAb 1E4 recognition of Npl3p requires phosphorylation in two ways (data not shown). (i) Treatment of yNpl3p with calf intestinal phosphatase, which efficiently removes phosphates from SR splicing factors (32), had no effect on mAb 1E4 reactivity. (ii) The human SR protein kinase SRPK1 (33) failed to restore the mAb 1E4 epitope to rNpl3p. Therefore, the mAb 1E4 epitope appeared to require a modification other than or in addition to phosphorylation.
To test whether the mAb 1E4 epitope required arginine methylation, we first asked whether a partially purified bovine arginine methyltransferase would restore the mAb 1E4 epitope on rNpl3p. This enzyme has been shown to catalyze the methylation of arginine residues present in model RGG-containing peptides (30). After incubation with the methyl donor SAM and the methyltransferase, rNpl3p was efficiently recognized by mAb 1E4 on immunoblots (Fig. 3, lanes 7, 11, and 15). No mAb 1E4 signal was detected when SAM (lane 1), the methyltransferase (lane 2), or rNpl3p (lane 3) were left out of the reaction, strongly suggesting that restoration of the mAb 1E4 epitope involves the transfer of a methyl group from SAM to rNpl3p.
Figure 3.
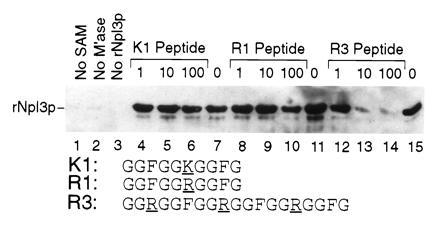
Arginine methyltransferase generates the mAb 1E4 epitope on rNpl3p. The mAb 1E4 reactivity of 1 μg of purified rNpl3p treated in vitro with a bovine arginine methyltransferase in the presence of the methyl donor SAM was examined by immunoblot analysis. Lanes: 7, 11, and 15, rNpl3p methylation reactions performed in parallel; 1–3, reactions lacking the methyl donor, enzyme, or substrate, respectively; 4–6, 8–10, and 12–14, reactions that included the indicated micromolar concentrations of the competitor peptides listed at the bottom.
To determine whether the epitope restoration reflected the activity of the arginine methyltransferase and not a contaminating activity present in the partially purified enzyme preparation, we asked whether specific peptide inhibitors of the arginine methyltransferase would block the reaction. The R3 peptide, containing three RGG boxes, is an efficient inhibitor of NG,NG-dimethylarginine formation on hypomethylated protein substrates. A 10-fold higher concentration of the R1 peptide, containing one RGG box, and a 100-fold higher concentration of the K1 peptide, identical to R1 except for a single R → K change, were required to achieve the same level of inhibition (30). Consistent with these previous results, 10 μM R3 peptide efficiently inhibited restoration of the mAb 1E4 epitope (lanes 12–14), 100 μM R1 peptide (lanes 8–10) slightly inhibited the reaction, and 100 μM K1 peptide (lanes 4–6) had little or no effect. Thus, these results further support the conclusion that the mAb 1E4 epitope requires methylation of Npl3p, most likely on arginine residues within the RGG boxes. Since the mAb 1E4 epitope is present on endogenous Npl3p expressed in yeast (Fig. 2), these results indicate that Npl3p is methylated in vivo.
As proof that Npl3p can be methylated in vitro, we found that the bovine arginine methyltransferase catalyzed the transfer of a 3H-labeled methyl group from SAM to rNpl3p (Fig. 4A, lane 3). Again, this reaction required the methyl donor SAM (lane 1). Methylation occurred on rNpl3p, as opposed to another protein substrate present in the methyltransferase fraction, because no labeled protein species of the correct gel mobility appeared in the absence of added rNpl3p (lane 2). The reaction also required the arginine methyltransferase because the R3 peptide (lanes 4–6), but not the control K1 peptide (lanes 7–9), inhibited the label transfer.
Figure 4.
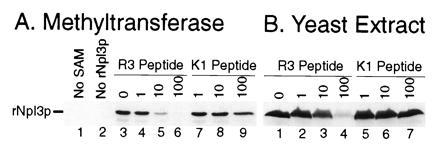
Arginine methyltransferase and yeast extract transfer a methyl group from SAM to rNpl3p. Methylation of purified rNpl3p was analyzed by fluorography after denaturing gel electrophoresis of rNpl3p that had been incubated with a bovine arginine methyltransferase (A) or yeast extract (B) in the presence of SAM carrying a tritiated methyl group. Labels above the lanes are as in Fig. 3.
Interestingly, yeast extracts contained a similar methyltransferase activity (Fig. 4B, lane 1). This activity likely methylated arginine residues because it also was inhibited by the R3 peptide (lanes 2–4) but not the K1 peptide (lanes 5–7). In addition to the rNpl3p that was added to these reactions, a number of endogenous proteins, including one that comigrated with yNpl3p, were also methylated (data not shown). These proteins likely represent hypomethylated substrates, with one or more methylation sites left unmodified in vivo, that can be further methylated in vitro.
To test whether the RGG boxes within the C-terminal region of Npl3p were the sites for arginine methylation (Fig. 1), an Npl3p deletion mutant (ΔRGG) lacking this domain was constructed. Npl3ΔRGGp expressed in yeast did not carry the mAb 1E4 epitope (Fig. 5). Although immunoblot experiments using anti-Npl3p polyclonal antibodies revealed that full-length Npl3p and Npl3ΔRGGp were expressed in yeast at similar levels (Fig. 5A), mAb 1E4 recognized only the full-length protein and failed to react with the ΔRGG deletion mutant (Fig. 5B). These results suggest that the methylation site(s) recognized by mAb 1E4 lie within the RGG domain.
One caveat of this interpretation, however, is that the ΔRGG deletion may cause Npl3p to be mislocalized in the cell. In fact, a larger Npl3p deletion removing the RGG domain as well as the second RRM RNA binding domain causes Npl3p to be mislocalized to the cytoplasm (34). Such mislocalization could prevent Npl3ΔRGGp methylation simply by physically separating Npl3ΔRGGp from the methyltransferase, which is predominantly nuclear (8). To address this possibility, we determined whether purified bacterially expressed Npl3ΔRGGp was a substrate for methylation in vitro using the labeled methyl group transfer assay (Fig. 6). Although equal amounts of rNpl3p and rNpl3ΔRGGp were tested (Fig. 6A, lanes 2–4), only full-length rNpl3p and not the ΔRGG mutant was a methylation substrate for the bovine arginine methyltransferase (Fig. 6B, lanes 2–4) or methyltransferase activity endogenous to yeast extracts (Fig. 6B, lanes 6–8). Again, the appearance of the methylated product depended on the addition of rNpl3p (Fig. 6B, lanes 1 and 5). Moreover, a methyltransferase inhibitor was not present in the rNpl3ΔRGGp preparation because addition of this preparation did not affect methylation of the wild-type protein (Fig. 6B, lanes 4 and 8). Together with the immunoblot results (Fig. 5), these results demonstrate that methylation requires the RGG domain and strongly suggest that methylation of Npl3p occurs within this domain.
Figure 6.
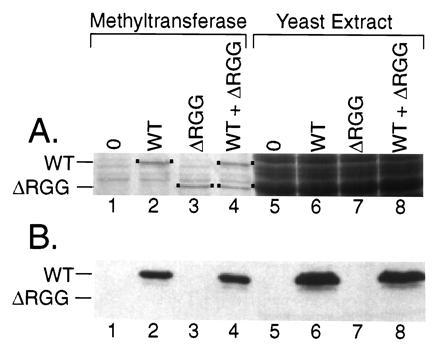
Npl3p lacking the C-terminal domain is not a substrate for methylation in vitro. Purified recombinant full-length Npl3p (WT) or truncated Npl3ΔRGGp (ΔRGG) were tested as substrates for methylation in vitro by the bovine arginine methyltransferase or yeast extract as in Fig. 4. Methylation was assayed by fluorography (B) of the gel stained for protein with Coomassie blue (A) (dots mark the locations of the WT and ΔRGG substrates). Assays were performed without substrate (lanes 1 and 5), with either substrate separately (lanes 2, 3, 6, and 7), and with the substrates together (lanes 4 and 8).
DISCUSSION
Our data indicate that the yeast RNA binding protein Npl3p is methylated in vivo, probably on one or more arginine residues within the Npl3p RGG RNA binding domain. We first discovered that Npl3p was modified in vivo by noting that mAb 1E4 recognized Npl3p expressed in yeast but not in bacteria. A bovine arginine methyltransferase restored the mAb 1E4 epitope by transferring a methyl group from SAM to bacterially expressed Npl3p, demonstrating that the modification was methylation. Importantly, yeast extracts displayed a similar methyltransferase activity. Npl3p lacking the RGG RNA binding domain was not methylated in vivo or in vitro, demonstrating that methylation requires this domain and strongly suggesting that the RGG tripeptides are the methylation sites.
The conclusion that mAb 1E4 recognizes an arginine methyl epitope rests primarily on two types of assays. (i) A characterized partially purified bovine arginine methyltransferase generated the mAb 1E4 epitope on recombinant Npl3p expressed in bacteria in a reaction dependent on SAM, an established methyl donor for arginine methyltransferases. (ii) This same enzyme transferred a labeled methyl group from SAM to rNpl3p. Importantly, both reactions were blocked by known peptide inhibitors of the arginine methyltransferase activity and not by control peptides, suggesting that the reactions are catalyzed by an arginine methyltransferase and not a contaminating methyltransferase present in the partially purified enzyme preparation.
Although we have not mapped the site(s) of methylation to specific amino acid residues, our results together with previous findings (8) strongly suggest that one or more of the arginine residues found within the RGG boxes in the C-terminal third of Npl3p is methylated (Fig. 1). Although the Npl3ΔRGGp deletion mutant, which lacks all of the RGG tripeptides, was expressed in vivo at the same level as wild-type Npl3p, Npl3ΔRGGp lacked the mAb 1E4 epitope, suggesting that the methylation site(s) map to the deleted domain. This result is consistent with a recent in vivo labeling experiment showing that a labeled methyl group maps to the C-terminal half of Npl3p within RRM2 or the RGG domain (8). Furthermore, our demonstration that purified rNpl3ΔRGGp failed as a methylation substrate in vitro for both the bovine arginine methyltransferase and yeast extracts argues strongly against the possibilities that the lack of Npl3ΔRGGp methylation simply reflects methylation at other sites not involving the mAb 1E4 epitope or an intracellular separation of Npl3ΔRGGp from the methyltransferase. We cannot eliminate two other possibilities, that the deleted domain merely functions as a site for enzyme recognition but the methylation sites lie elsewhere or that arginine residues located within the SR/RGG domain but outside of the RGG tripeptides are the relevant methylation sites. However, the precise mapping by peptide sequencing of a number of methylation sites in other proteins to arginine residues within RGG tripeptides and the observation that arginine methylation sites appear to require an adjacent glycine residue (5) argue against these explanations.
mAb 1E4 provides a valuable tool to study methylation using a simple assay. Using immunoblot analysis, we discovered that mAb 1E4 recognizes a methylated epitope on Npl3p. Although many yeast proteins possess methylated arginine residues (7) (Fig. 4), mAb 1E4 appears to recognize Npl3p specifically, both on immunoblots of whole-cell yeast extracts and in immunoprecipitation reactions (data not shown). The lack of cross-reactivity with other methylarginine-containing proteins likely reflects the fact that the mAb 1E4 epitope is composed of methylarginine as well as other amino acids in a sequence or context unique to Npl3p; in other words, methylarginine is necessary but not sufficient for mAb 1E4 reactivity. Alternatively, the apparent mAb 1E4 specificity may be due to a large number of potential methylation sites, namely, the 15 RGG tripeptides (Fig. 1), and the cellular abundance of Npl3p (e.g., see Fig. 2); indeed, mAb 1E4 may recognize methyl epitopes on other yeast proteins that escape detection because of a paucity of mAb 1E4 epitopes or low-expression levels. However, given that a number of yeast RGG box proteins are major components of the nucleolus and thus probably abundant (35), this explanation seems unlikely. In either case, mAb 1E4 greatly simplifies the study of arginine methylation of an essential RNA binding protein by providing a much quicker and simpler assay than the radiolabeling techniques previously employed.
We detected a methyltransferase activity in yeast whole-cell splicing extracts. Like the bovine enzyme, this activity was blocked by peptide inhibitors of arginine methyltransferases. Thus, an arginine methyltransferase activity that modifies RGG RNA binding domains appears to be widely conserved evolutionarily, having been found in budding yeast (Fig. 3) (7, 8), fission yeast, Drosophila, and various mammals (4, 9). A previous study failed to detect such an activity in Saccharomyces cerevisiae extracts (9), perhaps because of differences in extract preparation. Indeed, recent independent studies have identified a yeast arginine methyltransferase. RMT1 (protein arginine methyltransferase) was discovered as the yeast homolog of a human arginine methyltransferase, isolated in a protein interaction trap assay (7). Rmt1p appears to be the predominant NG,NG-dimethylarginine methyltransferase in yeast, since deletion of RMT1 causes an 85% reduction in the level of this modification. Furthermore, the RMT1 deletion decreases the methylation of a number of proteins, suggesting that Rmt1p methylates many substrates. At least some of these are likely to be RGG box proteins because purified recombinant Rmt1p can methylate recombinant hnRNP A1 in vitro; Rmt1p can also methylate histones, cytochrome c, and myoglobin but not myelin basic protein (7).
RMT1 has also been recently identified as HMT1 (hnRNP methyltransferase) (8). Importantly, HMT1 was identified in a screen for mutations that are lethal only in combination with the temperature-sensitive npl3-1 mutation, suggesting that Npl3p methylation is biologically relevant. Furthermore, HMT1 overexpression suppresses a number of npl3 temperature-sensitive mutations. Deletion of HMT1 disrupts Npl3p methylation assayed by labeling in vivo, and recombinant Hmt1p methylates Npl3p in vitro, suggesting that Hmt1p is an Npl3p methyltransferase (8). Thus, the Npl3p arginine methyltransferase that we detected in yeast extracts likely reflects, at least in part, Rmt1p/Hmt1p activity. Deletion of HMT1/RMT1, however, generates no obvious growth phenotype (7, 8). Perhaps HMT1/RMT1 is redundant with one or more arginine methyltransferases that account for the remaining 15% of NG,NG-dimethylarginine found in the HMT1/RMT1 deletion strain. This model assumes that the residual level of arginine methylation is sufficient to support cell growth and predicts that loss-of-function mutations in the redundant gene(s) will be lethal in combination with the HMT1/RMT1 deletion.
At this stage, we can only speculate on the function of HMT1/RMT1 in general and Npl3p methylation in particular. Previous studies point to a number of models that are not mutually exclusive. (i) Given the established role of arginine in RNA binding, one obvious possibility is that arginine methylation decreases the RNA binding affinity of the modified RGG domain. Methylation would not alter the positive charge on arginine but is predicted to hinder the arginine–RNA interaction by disrupting hydrogen bonds or introducing steric blocks (13). In experiments using hnRNP A1, however, RNA binding was only modestly affected after methylation (36). (ii) Previous results indicating that the hnRNP A1 glycine/arginine-rich domain mediates protein–protein interactions (37, 38) raise the possibility that methylation regulates this activity. (iii) One study using hnRNP A1 suggests that methylation protects the substrate from digestion with trypsin, albeit weakly, leading to speculation that methylation could affect protein turnover (36). (iv) Given that Npl3p (24) as well as a number of hnRNP proteins (39) shuttle between the nucleus and cytoplasm, methylation may function as part of a localization, retention, or transport signal. For example, the nuclear localization of both Hmt1p/Rmt1p (8) and the mAb 1E4 epitope (20) may suggest that methylation serves as part of a nuclear retention signal. Reversible arginine methylation would make the regulatory possibilities governed by this modification even more dynamic; however, we are unaware of any arginine methylesterases.
We have recently discovered that Npl3p carries a second post-translational modification, serine phosphorylation (C.W.S. and C.G., unpublished results). Given that Npl3p has an SR domain that can be phosphorylated in vitro by the human SR protein kinase (SRPK1) (C.W.S., L. Feng, X.-D. Fu, and C.G., unpublished results), some or all Npl3p phosphorylation probably reflects modification of the SR dipeptides within the SR/RGG domain. Therefore, nearby and possibly adjacent serine and arginine residues appear to be targets for distinct modifications. In the simplest case, each modification functions independently from the other. However, it is intriguing to speculate that juxtaposed modifications could also function in concert to bestow a dynamic and wide range of activities to the SR/RGG domain. For example, the local chemical environment of an SRGG repeat could vary between four distinct states: (i) unmodified, (ii) phosphorylated, (iii) methylated, and (iv) phosphorylated and methylated. The hypothesis that each state could display a different activity is particularly interesting given evidence suggesting that Npl3p may function in more than one RNA processing event, including splicing (C.W.S. and C.G., unpublished results) and mRNA export (24).
In addition, the juxtaposition of two modifications could create a variety of regulatory possibilities. (i) One modification could regulate the functional effects of the other. For example, arginine methylation could prevent protein–protein interactions or SR domain conformations facilitated by serine phosphorylation. In particular, the ionic interactions between phosphoserine and arginine residues that provide a structural paradigm akin to the polar zipper (40) to explain protein–protein interactions between SR domains (41, 42, 43) and possibly intramolecular interactions within an SR domain could be sterically blocked by arginine methylation. (ii) One modification could influence the appearance of the other, creating a molecular switch controlling activity of the SR/RGG domain. The observation that rNpl3p can be modified by purified recombinant Rmt1p/Hmt1p (8) or SRPK1 (C.W.S., L. Feng, X.-D. Fu, and C.G., unpublished results) argues against the hypothesis that one modification serves as a prerequisite for the other. Instead, phosphorylation (methylation) may inhibit methylation (phosphorylation). Numerous cases in which phosphorylation regulates protein recognition support the notion that phosphorylation could block recognition of an adjacent arginine residue by a methyltransferase.
Arginine methylation is an evolutionarily conserved modification found in many RNA binding proteins of diverse function. Thus, this modification likely affects a variety of RNA processing reactions in functionally important ways. The discovery that the essential yeast RNA binding protein Npl3p is arginine-methylated and that this modification can be simply assayed provides a powerful genetic and biochemical entrée, in combination with the discovery of an Npl3p methyltransferase, to dissect the biological functions of this modification at the molecular level. The observation that serine residues adjacent to the methylation sites appear to be targets for phosphorylation suggests that some of these functions may involve interactions between modifications to regulate the activity or localization of the target protein.
Acknowledgments
We thank K. Duncan, A. Frankel, S. Misra, P. Raghunathan, and J. Staley for critically evaluating the manuscript; M. Swanson for mAb 1E4 and stimulating discussions; G. Dreyfuss for mAb 2F1; D. Aswad for the bovine arginine methyltransferase and peptides; and L. Esperas, H. Roiha, and C. Pudlow for technical support. This work was supported by Grant GM21119 to C.G. C.W.S. was supported by a Helen Hay Whitney Postdoctoral Fellowship. C.G. is an American Cancer Society Research Professor of Molecular Genetics.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: hnRNP, heterogeneous nuclear ribonucleoprotein particle; r, recombinant; y, yeast; SAM, S-adenosyl-l-methionine; RRM, RNA recognition motif.
References
- 1.Clarke S. Curr Opin Cell Biol. 1993;5:977–983. doi: 10.1016/0955-0674(93)90080-a. [DOI] [PubMed] [Google Scholar]
- 2.Paik W K, Kim S. Protein Methylation. Boca Raton, FL: CRC; 1990. [Google Scholar]
- 3.Ghosh S K, Paik W K, Kim S. J Biol Chem. 1988;263:19024–19033. [PubMed] [Google Scholar]
- 4.Kim S, Chanderkar L P, Ghosh S K. In: Protein Methylation. Paik W K, Kim S, editors. Boca Raton, FL: CRC; 1990. pp. 77–95. [Google Scholar]
- 5.Lischwe M A. In: Protein Methylation. Paik W K, Kim S, editors. Boca Raton, FL: CRC; 1990. pp. 97–123. [Google Scholar]
- 6.Lin W J, Gary J D, Yang M C, Clarke S, Herschman H R. J Biol Chem. 1996;271:15034–15044. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- 7.Gary J D, Lin W-J, Yang M C, Herschman H R, Clarke S. J Biol Chem. 1996;271:12585–12594. doi: 10.1074/jbc.271.21.12585. [DOI] [PubMed] [Google Scholar]
- 8.Henry M F, Silver P A. Mol Cell Biol. 1996;16:3668–3678. doi: 10.1128/mcb.16.7.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Q, Dreyfuss G. Mol Cell Biol. 1995;15:2800–2808. doi: 10.1128/mcb.15.5.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boffa L C, Karn J, Vidali G, Allfrey V G. Biochem Biophys Res Commun. 1977;74:969–976. doi: 10.1016/0006-291x(77)91613-8. [DOI] [PubMed] [Google Scholar]
- 11.Burd C G, Dreyfuss G. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 12.Ghisolfi L, Kharrat A, Joseph G, Amalric F, Erard M. Eur J Biochem. 1992;209:541–548. doi: 10.1111/j.1432-1033.1992.tb17318.x. [DOI] [PubMed] [Google Scholar]
- 13.Calnan B J, Tidor B, Biancalana S, Hudson D, Frankel A D. Science. 1991;252:1167–1171. doi: 10.1126/science.252.5009.1167. [DOI] [PubMed] [Google Scholar]
- 14.Yarus M. Science. 1988;240:1751–1758. doi: 10.1126/science.3381099. [DOI] [PubMed] [Google Scholar]
- 15.Michel F, Hanna M, Green R, Bartel D P, Szostak J W. Nature (London) 1989;342:391–395. doi: 10.1038/342391a0. [DOI] [PubMed] [Google Scholar]
- 16.Bossie M A, DeHoratius C, Barcelo G, Silver P. Mol Biol Cell. 1992;3:75–93. doi: 10.1091/mbc.3.8.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russell I D, Tollervey D. J Cell Biol. 1992;119:737–747. doi: 10.1083/jcb.119.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu X D. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- 19.Birney E, Kumar S, Krainer A R. Nucleic Acids Res. 1993;21:5803–5816. doi: 10.1093/nar/21.25.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson S M, Datar K V, Paddy M R, Swedlow J R, Swanson M S. J Cell Biol. 1994;127:1173–1184. doi: 10.1083/jcb.127.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loo S, Laurenson P, Foss M, Dillin A, Rine J. Genetics. 1995;141:889–902. doi: 10.1093/genetics/141.3.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellis E M, Reid G A. Gene. 1993;132:175–183. doi: 10.1016/0378-1119(93)90193-7. [DOI] [PubMed] [Google Scholar]
- 23.Singleton D R, Chen S, Hitomi M, Kumagai C, Tartakoff A M. J Cell Sci. 1995;108:265–272. doi: 10.1242/jcs.108.1.265. [DOI] [PubMed] [Google Scholar]
- 24.Lee M S, Henry M, Silver P A. Genes Dev. 1996;10:1233–1246. doi: 10.1101/gad.10.10.1233. [DOI] [PubMed] [Google Scholar]
- 25.Umen J G, Guthrie C. Genes Dev. 1995;9:855–868. doi: 10.1101/gad.9.7.855. [DOI] [PubMed] [Google Scholar]
- 26.Peter M, Gartner A, Horecka J, Ammerer G, Herskowitz I. Cell. 1993;73:747–760. doi: 10.1016/0092-8674(93)90254-n. [DOI] [PubMed] [Google Scholar]
- 27.Tao B Y, Lee K C P. In: PCR Technology. Griffin H G, Griffin A M, editors. Boca Raton, FL: CRC; 1994. pp. 69–83. [Google Scholar]
- 28.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siebel C W, Kanaar R, Rio D C. Genes Dev. 1994;8:1713–1725. doi: 10.1101/gad.8.14.1713. [DOI] [PubMed] [Google Scholar]
- 30.Najbauer J, Johnson B A, Young A L, Aswad D W. J Biol Chem. 1993;268:10501–10509. [PubMed] [Google Scholar]
- 31.Anderson J T, Wilson S M, Datar K V, Swanson M S. Mol Cell Biol. 1993;13:2730–2741. doi: 10.1128/mcb.13.5.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roth M B, Murphy C, Gall J G. J Cell Biol. 1990;111:2217–2223. doi: 10.1083/jcb.111.6.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gui J F, Lane W S, Fu X D. Nature (London) 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- 34.Flach J, Bossie M, Vogel J, Corbett A, Jinks T, Willins D A, Silver P A. Mol Cell Biol. 1994;14:8399–8407. doi: 10.1128/mcb.14.12.8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Girard J P, Lehtonen H, Caizergues-Ferrer M, Amalric F, Tollervey D, Lapeyre B. EMBO J. 1992;11:673–682. doi: 10.1002/j.1460-2075.1992.tb05099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajpurohit R, Paik W K, Kim S. Biochem J. 1994;304:903–909. doi: 10.1042/bj3040903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nadler S G, Merrill B M, Roberts W J, Keating K M, Lisbin M J, Barnett S F, Wilson S H, Williams K R. Biochemistry. 1991;30:2968–2976. doi: 10.1021/bi00225a034. [DOI] [PubMed] [Google Scholar]
- 38.Cobianchi F, Karpel R L, Williams K R, Notario V, Wilson S H. J Biol Chem. 1988;263:1063–1071. [PubMed] [Google Scholar]
- 39.Pinol-Roma S, Dreyfuss G. Nature (London) 1992;355:730–732. doi: 10.1038/355730a0. [DOI] [PubMed] [Google Scholar]
- 40.De Baere I, Liu L, Moens L, Van Beeumen J, Gielens C, Richelle J, Trotman C, Finch J, Gerstein M, Perutz M. Proc Natl Acad Sci USA. 1992;89:4638–4642. doi: 10.1073/pnas.89.10.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu J Y, Maniatis T. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 42.Kohtz J D, Jamison S F, Will C L, Zuo P, Lührmann R, Garcia-Blanco M A, Manley J L. Nature (London) 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 43.Amrein H, Hedley M L, Maniatis T. Cell. 1994;76:735–746. doi: 10.1016/0092-8674(94)90512-6. [DOI] [PubMed] [Google Scholar]