

Abstract
The N-terminal domain of HIV-1 integrase contains a pair of His and Cys residues (the HHCC motif) that are conserved among retroviral integrases. Although His and Cys residues are often involved in binding zinc, the HHCC motif does not correspond to any recognized class of zinc binding domain. We have investigated the binding of zinc to HIV-1 integrase protein and find that it binds zinc with a stoichiometry of one zinc per integrase monomer. Analysis of zinc binding to deletion derivatives of integrase locates the binding site to the N-terminal domain. Integrase with a mutation in the HHCC motif does not bind zinc, consistent with coordination of zinc by these residues. The isolated N-terminal domain is disordered in the absence of zinc but, in the presence of zinc, it adopts a secondary structure with a high alpha helical content. Integrase bound by zinc tetramerizes more readily than the apoenzyme and is also more active than the apoenzyme in in vitro integration assays. We conclude that binding of zinc to the HHCC motif stabilizes the folded state of the N-terminal domain of integrase and bound zinc is required for optimal enzymatic activity.
Zinc plays a key structural role in a large number of proteins (reviewed in ref. 1), and more than 10 classes of zinc binding domains have now been identified. These domains are diverse in their structure and function; many are involved in binding DNA, while others mediate protein–protein interactions. In each case, zinc is coordinated by His and Cys residues. The integrase protein of HIV-1 and other retroviruses contains a pair of highly conserved His and Cys residues (the HHCC motif) in the N-terminal domain. This motif does not correspond to any of the recognized zinc binding domains, although a zinc finger structure has been proposed based on the similarity of the HHCC motif to the CCHH motif found in transcription factor TFIIIA and related proteins (2).
Integration of a DNA copy of the HIV genome into host DNA is an essential step in the viral replication cycle (3, 4, 5). The key cutting and joining reactions that integrate the viral DNA into the host genome are carried by the virus-encoded integrase protein. Integrase cleaves two nucleotides from the 3′ ends of the viral DNA (3′-processing) and subsequently inserts these 3′-recessed ends into host DNA (DNA strand transfer). The 5′ ends of the viral DNA and the 3′ ends of host DNA at the site of insertion remain unjoined in the resulting integration intermediate (6, 7). Completion of integration requires removal of the unpaired nucleotides at the 5′ ends of the viral DNA and repair of the single strand gaps between viral and host DNA, steps that are probably carried out by cellular enzymes.
Integrase is a 32-kDa protein that is comprised of three structurally and functionally distinct domains: the N-terminal domain that contains the HHCC motif, the central core domain, and the C-terminal domain. The core domain contains a triad of acidic amino acid residues, the D,D-35-E motif, which is conserved among all retroviral and retrotransposon integrase proteins and even among some prokaryotic transposases (8, 9, 10, 11). Site-directed mutagenesis studies implicated these residues to be intimately involved in catalysis (8, 12, 13); mutation of any of these residues abolishes or severely diminishes all catalytic activity. They are located in close proximity in the crystal structures of the core domains of both HIV-1 integrase (14) and RSV integrase (15), consistent with a direct role in chemical catalysis. The C-terminal domain binds DNA nonspecifically (16, 17, 18), and the solution structure of the minimal fragment required for DNA binding has been determined by NMR (19, 20).
Although it is clear that the core domain contains the active site for chemical catalysis, the roles of the N-terminal domain and the C-terminal domain are less well understood. All three domains are required for catalysis of 3′-processing and DNA strand transfer (16, 21, 22). However, a pair of inactive proteins, one lacking the N-terminal domain and the other lacking the C-terminal domain, complement to restore activity (23, 24). This trans-complementation implies that the active form of integrase is a multimer. Integrase has been shown to form both dimers (25, 26, 27, 28, 29) and tetramers (27, 29) in solution, but the identity of the active multimer is unknown.
Here, we focus on the N-terminal domain of HIV-1 integrase and its interaction with zinc. The notion that the N-terminal domain of HIV integrase may normally bind zinc is supported by the observation that a peptide comprising the N-terminal 55 amino acids, which includes the HHCC motif, can bind zinc (2) and by the binding of zinc to full-length integrase in a “zinc blot” assay (30) and by atomic absorption spectroscopy (31). Binding of zinc to full-length HIV integrase, and the effects of bound zinc on the structure and function of the enzyme have been difficult to investigate because of the tendency of the protein to aggregate. In this report, we have exploited a soluble mutant of HIV-1 integrase (29) that retains full catalytic activity to examine the binding of zinc to integrase and its effect on the multimeric state and catalytic activity of the enzyme.
MATERIALS AND METHODS
Preparation of Proteins.
IN1–212/F185K was expressed and purified as described for IN50–212/F185K (32). IN1–270/F185K and IN1–288/F185K/C280S/H12N,H16N were expressed and purified as described for IN50–288/F185K/C280S and IN1–288/F185K/C280S (29). The superscripted numbers correspond to residue numbers from the N terminus of HIV-1 integrase; amino acid substitutions relative to wild-type integrase of HIV strain NL4-3 (33) integrase are indicated after the slashes. Protein concentration were determined by absorption at 280 nm using extinction coefficients calculated from the amino acid composition (34).
Determination of Multimeric State of Integrase in the Presence or Absence of Zinc.
Gel filtration was carried out at 4°C with a calibrated Superdex 200 column (HR 3.2/30; Pharmacia) on a Pharmacia Smart System at a flow rate of 50 μl/min. Proteins were first dialyzed overnight, under a helium blanket, at 4°C against buffer A (20 mM Hepes, pH 7.5/1 M NaCl/10% glycerol/1 mM DTT) containing 50 μM ZnCl2 or 1 mM EDTA. Protein samples were diluted to 10 μM with the same buffer and left at 4°C overnight before loading. Aliquots of 50 μl were then applied to the column equilibrated with buffer A.
CD Spectroscopy.
Measurements were obtained using a Jasco J-720 spectrometer (Jasco, Tokyo) at room temperature. Spectra in the far UV region (200–260 nm) were measured at protein concentrations of 10 μM in a quartz cuvette with a 0.1-mm path length. Spectra in the near UV region (250–320 nm) were measured at protein concentrations of 90–126 μM in a quartz cuvette of 0.5-mm path length.
Measurement of Zinc Concentration by Atomic Absorption Spectroscopy.
Zinc was measured using an atomic absorption spectrometer (model 3110; Perkin–Elmer), and zinc concentrations were determined using a zinc standard curve prepared from a standard zinc solution (J.T. Baker). In equilibrium dialysis experiments, samples were measured after a 500-fold dilution with 0.1% HCl (Optima grade; Fisher Scientific) in extensively deionized water. The zinc content was determined using the spectrometer in conjunction with a Perkin–Elmer HGA 600 graphite furnace. Parameters of the program steps were as follows (T, temperature; R, ramp; and H, hold). Step 1: T = 120°C, R = 5 sec, and H = 50 sec. Step 2: T = 250°C, R = 5 sec, and H = 30 sec. Step 3: T = 20°C, R = 1 sec, and H = 10 sec. Step 4 (atomization): T = 1800°C, R = 0 sec, and H = 3 sec. Step 5: T = 2600°C, R = 1 sec, and H = 5 sec. A gas flow of 300 ml/min was maintained throughout all steps except atomization, for which it was reduced to 200 ml/min. Maintaining gas flow during atomization decreases the sensitivity (which is otherwise inconveniently high) and increases the dynamic range over which measurements can be made. The zinc content of integrase–zinc complex devoid of free zinc was measured using the spectrometer in flame mode, which has a lower sensitivity for zinc (1 μM range); in this case, the sample was analyzed directly without dilution.
Equilibrium Dialysis Measurement of Zinc Binding to Full-Length Integrase (IN1–288/F185K/C280S).
IN1–288/F185K/C280S is relatively resistant to oxidation compared with integrase with a cysteine at position 280 (29). This enabled zinc binding experiments to be carried out in the absence of DTT, which would otherwise complicate the analysis by competing for zinc. However, dialysis buffers were sparged with helium and maintained under a helium blanket to minimize oxidation. Aliquots of 60 μl of integrase (166–233 μM) were placed in a dialysis button (Cambridge Repetition Engineers, Cambridge, U.K.) wrapped with dialysis membrane (cutoff molecular weight, 8000) and dialyzed at 4°C for 36 hr against 1 liter of buffer A containing ZnCl2, ranging from 1 to 10 μM. After dialysis, the concentrations of protein and zinc were measured, and the dissociation constant for zinc binding to integrase IN1–288/F185K/C280S was calculated.
Preparation of Zinc-Bound Integrase.
Zinc-bound integrase was formed by dialyzing the protein (150–190 μM) against buffer A containing 50 μM ZnCl2. To remove free zinc, zinc-bound integrase (400–500 μl) was then applied to a Superdex 75 column (HR10/30; Pharmacia) equilibrated with buffer A without ZnCl2. The column was run at a flow rate of 0.5 ml/min, and fractions were collected for protein and zinc measurements. IN1–270/F185K, due to its lower solubility, was first dialyzed at 11 μM against buffer A containing 50 μM ZnCl2 and then against buffer A to remove free zinc.
Integrase Activity Assays.
Double-stranded oligonucleotides that mimic the U5 end of HIV-1 DNA were used as the DNA substrate. For 3′-processing, oligonucleotide AE 118 (5′-GTGTGGAAAATCTCTAGCAGT), and for DNA strand transfer, AE 119 (5′-GTGTGGAAAATCTCTAGCA), were labeled at their 5′ ends with [γ-32P]ATP (3000 Ci/mmol, 1 Ci = 37 GBq; NEN) using T4 polynucleotide kinase (Pharmacia). Each labeled oligonucleotide was then annealed with AE 117 (5′-ACTGCTAGAGATTTTCCACAC) and separated from unincorporated label as described (29). 3′-Processing and strand transfer reaction mixtures (35) contained 25 mM 3-morpholinopropanesulfonic acid (Mops; pH 7.2), 10 mM DTT, 5% (wt/vol) polyethylene glycol-8000 (PEG-8000; Fluka), 5% (vol/vol) dimethyl sulfoxide (DMSO; Aldrich), 0.05% Nonidet P-40 (Sigma), 30 mM NaCl, and 5 mM MgCl2, with 20–150 nM DNA substrate and 0.94 μM integrase in a final 16-μl volume. Reaction products were analyzed by electrophoresis in 15% polyacrylamide/urea gels as previously described (32) and quantitated using a PhosphorImager (Molecular Dynamics).
Protein Secondary Structure Prediction.
The secondary structure of the N-terminal domain of HIV-1 integrase was predicted by the PHD method (36) using the PHD mail server at EMBL, Heidelberg, Germany (e-mail address: Predict-Help@embl-heidelberg.de).
RESULTS
HIV-1 Integrase Binds Zinc with a Stoichiometry of 1:1.
Preliminary experiments (data not shown) established that the protein purified from Escherichia coli contained undetectable levels of zinc. We note that EDTA was present throughout the purification, and, if zinc was originally present, it may have been extracted from the protein. The affinity of the apoprotein for zinc was measured by equilibrium dialysis as described in Materials and Methods, and an apparent Kd of ≈6 μM was determined at pH 7.5 in the presence of 1 M NaCl.
Since zinc remained associated with the full-length integrase protein during size exclusion chromatography in a buffer that does not contain zinc (Fig. 1A), gel filtration provided a convenient assay for measuring the stoichiometry of binding. Zinc was introduced into integrase by dialysis as described in Materials and Methods, and the protein was then subjected to gel filtration on a Superdex 200 column (Fig. 1A). The peak fraction protein concentrations were measured by A280, and the zinc concentration in the same fractions was measured by atomic absorption spectroscopy. The stoichiometry of bound zinc to integrase protomers was 1:1 in each fraction.
Figure 1.
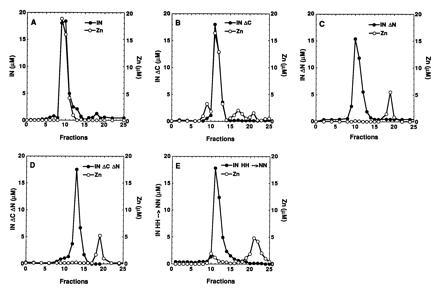
Coelution of zinc with HIV-1 integrase in gel filtration. Integrase was dialyzed against buffer A containing zinc chloride as described and applied to a Superdex 75 column equilibrated with buffer A not containing zinc. The protein concentration in each of the collected fractions was determined by measuring the optical density at 280 nm, and the zinc concentration in the same fractions was determined by atomic absorption spectroscopy. (A) Full-length integrase (IN1–288/F185K/C280S). (B) Integrase with a C-terminal deletion (IN1–212/F185K). (C) Integrase with an N-terminal deletion (IN50–288/F185K/C280S). (D) The central domain of integrase (IN50–212/F185K). (E) Integrase with the His residues of the HHCC motif changed to Asn (IN1–288/H12N/H16N/F185K/C280S).
Zinc Binding Requires the Presence of the HHCC Motif.
We next investigated the binding of zinc to deletion derivatives of integrase to determine which part of the protein is involved in binding. The same 1:1 stoichiometry of bound zinc was observed with integrase lacking the carboxyl terminal domain (Fig. 1B). However, zinc did not coelute with integrase that lacks the N-terminal domain (Fig. 1C) or with the core domain alone (Fig. 1D). These results locate the domain responsible for binding zinc to the N-terminal domain of integrase. Finally, mutant integrase in which the histidine residues of the HHCC motif were changed to asparagine failed to bind zinc (Fig. 1E), directly demonstrating the role of this motif in zinc binding. The zinc-binding stoichiometries of the wild-type and mutant integrase proteins are summarized in Table 1.
Table 1.
Zinc binds to HIV-1 integrase with a 1:1 molar ratio
| Protein | Molar ratio of bound zinc |
|---|---|
| IN | 0.99 |
| IN HH → NN | 0.04 |
| IN ΔC | 0.90 |
| IN ΔN | <0.01 |
Zinc Is Required for Folding of the Isolated N-Terminal Domain of HIV-1 Integrase.
CD was used to probe the effect of zinc on the structure of the N-terminal domain (residues 1–55). In the absence of zinc, the far UV CD spectrum of this domain indicated that the peptide was disordered (Fig. 2, solid line). In contrast, the zinc-bound peptide exhibited a CD spectrum characteristic of alpha helices (Fig. 2, dotted line). Neither Mg2+ nor Mn2+ could substitute for zinc in ordering the isolated N-terminal domain (data not shown). We conclude that zinc promotes the folding of this isolated domain into secondary structural elements that are mostly helical.
Figure 2.
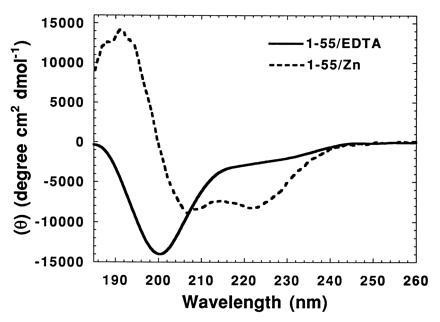
Folding of the N-terminal domain of HIV-1 integrase in the presence of zinc. Far UV CD spectra of the N-terminal domain of integrase (IN1–55) in the absence (solid line) and presence (dotted line) of bound zinc.
In contrast to the isolated N-terminal domain, the far UV CD spectrum of full-length HIV-1 integrase is indistinguishable in the presence or absence of zinc (data not shown). Since the N-terminal domain comprises ≈19% of the full-length protein, we would have expected to observe a perceptible change in the CD spectrum if the folding of this domain, in the context of the full-length protein, is dependent upon the binding of zinc. We infer that zinc is required for the folding of the isolated N-terminal domain, but contacts involving other parts of integrase may stabilize at least some of the secondary structure of this domain in the context of the full-length protein.
Although the far UV CD spectra of full-length integrase in the presence or absence of zinc were not detectably different, significant differences were observed in the near UV region (Fig. 3A). The origin of these differences is speculative, but they are likely due to differences in tertiary structure affecting the local environment of aromatic residues (37). Mutant integrase in which the histidine residues of the HHCC motif were changed to asparagine exhibited very similar near UV spectra in the presence or absence of zinc (Fig. 3B).
Figure 3.
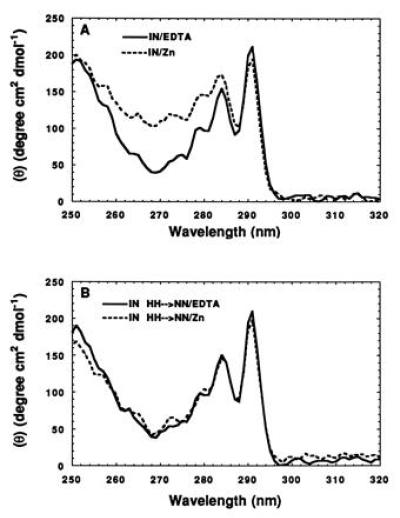
Zinc-binding alters the CD spectrum of HIV-1 integrase in the near UV region. (A) Near UV CD spectra of integrase (IN1–288/F185K/C280S) in the absence (solid line) and presence (dotted line) of bound zinc. (B) Near UV CD spectra of IN1–288/H12N/H16N/F185K/C280S) in the absence (solid line) and presence (dotted line) of bound zinc. The substitution of Asn for His in the HHCC motif abolishes the effect of zinc on the near UV CD spectrum.
Zinc Promotes Tetramerization of HIV-1 Integrase.
IN1–288/F185K/C280S exists in an equilibrium between dimer and tetramer species in the absence of zinc (29). The effect of zinc on multimerization was monitored by size exclusion chromatography on a Superdex 200 column after dialysis of the protein against a buffer containing 50 μM zinc chloride or 1 mM EDTA. When loaded at a concentration of 10 μM, the protein that had been dialyzed against EDTA eluted at a position corresponding to dimers (Fig. 4A, solid line), relative to globular protein standards, with a slight trail toward the tetramer position. In contrast, the protein dialyzed against zinc chloride eluted at the position expected for tetramers (Fig. 4A, dotted line). Preliminary equilibrium ultracentrifugation experiments supported the conclusion that zinc promotes multimerization of integrase (data not shown).
Figure 4.
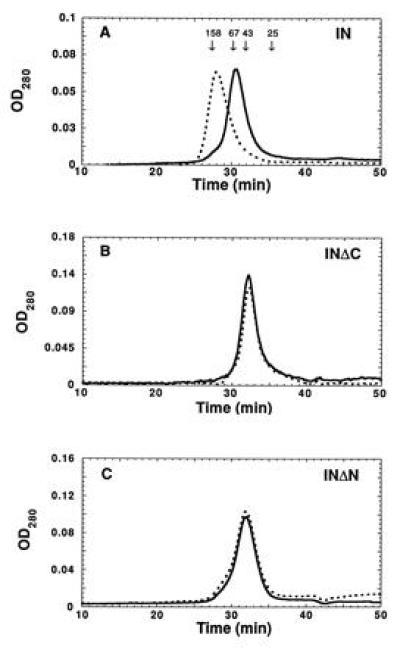
Zinc binding promotes tetramerization of HIV-1 integrase. (A) Gel filtration of integrase (IN1–288/F185K/C280S) on Superdex 200 after dialysis against buffer A containing EDTA (solid line) or zinc (dotted line). The zinc-dialyzed protein eluted at the expected position for tetramers (relative to globular protein standards). The EDTA-dialyzed protein eluted at the expected position for dimers. The peak elution times of chymotrypsinogen A (25 kDa), ovalbumin (43 kDa), bovine serum albumin (67 kDa), and aldolase (158 kDa) standards are indicated. (B) Integrase lacking the C-terminal domain (IN1–212/F185K) eluted exclusively as dimers after dialysis against EDTA or zinc. (C) Integrase lacking the N-terminal domain (IN50–288/F185K/C280S) eluted predominantly as dimers, with a trailing edge toward the tetramer position, after dialysis against EDTA or zinc.
To further investigate the effect of zinc on multimerization, size exclusion chromatography experiments were repeated with two deletion derivatives of integrase. We first examined IN1–212/F185K, which forms dimers, but not tetramers, in the absence of zinc (29). Dialysis of this protein against zinc chloride did not affect its multimerization properties when the protein was loaded at 10 μM (Fig. 4B) or at any of a variety of concentrations (data not shown). Secondly, zinc did not induce IN50–288/F185K/C280S, which is fully competent for tetramerization at higher protein concentrations (29), to form tetramers at lower protein concentrations (Fig. 4C).
Zinc-Bound Integrase Is More Active in in Vitro Assays.
The catalytic activities of the HIV-1 integrase (IN1–288/F185K/C280S) containing zinc were compared with the catalytic properties of the apoenzyme. Both 3′-processing and DNA strand transfer activities were found to be markedly stimulated by zinc (Fig. 5). Quantitation of the results revealed that zinc increased the rate of both 3′-processing and DNA strand transfer, and no significant lag period was observed with or without zinc (Fig. 5). This stimulation was observed regardless of whether free zinc was removed from the protein solution before reaction (data not shown). A 1:1 stoichiometry of bound zinc to integrase monomers is therefore sufficient for this stimulation. In contrast to the effect on 3′-processing and DNA strand transfer, disintegration activity (38) was not stimulated by bound zinc (data not shown). The absence of a stimulatory effect on disintegration was as expected, since the N-terminal domain is dispensable for this reaction.
Figure 5.
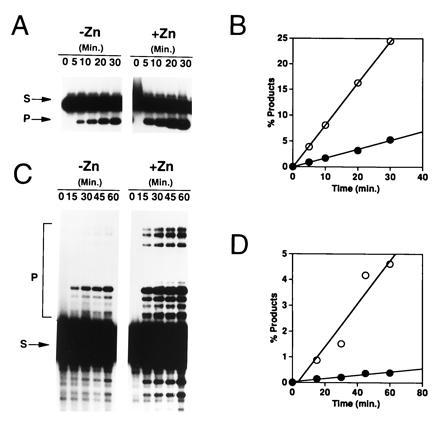
Zinc binding stimulates both the 3′-processing and DNA strand transfer activities of HIV integrase. (A) Autoradiogram of 3′-processing products of reactions catalyzed by integrase dialyzed against buffer A containing EDTA or zinc. Reaction mixtures containing 20 nM DNA substrate and 0.94 μM integrase were incubated at 37°C for 1 hr. The migration positions of the unreacted DNA substrate (S) and 3′-processing products (P) are indicated. (B) Quantitation of the 3′-processing time course shown in A. ○, Integrase dialyzed against zinc; •, integrase dialyzed against EDTA. (C) Autoradiogram of DNA strand transfer products of reactions catalyzed by integrase dialyzed against buffer A containing EDTA or zinc. Reaction conditions and labeling are the same as indicated for A, except that the concentration of DNA substrate was 150 μM. (D) Quantitation of the DNA strand transfer time course shown in C. ○, Integrase dialyzed against zinc; •, integrase dialyzed against EDTA.
DISCUSSION
The N-Terminal Domain of HIV-1 Integrase Is a Zinc Binding Domain.
Our results indicate that zinc is a normal structural component of the N-terminal domain of HIV-1 integrase. Although this domain is frequently referred to a “zinc finger” domain, there is no evidence that it is in fact structurally related to zinc fingers. On the contrary, circumstantial evidence suggests that it is not a zinc finger domain. First, whereas two antiparallel β strands are intrinsic features of zinc fingers, the secondary structure of the N-terminal domain of HIV-1 integrase is predicted by the PHD method (36) to be mostly alpha helices (data not shown); this prediction is in good agreement with the CD spectrum. Second, the counterparts of the residues that form the hydrophobic core of canonical zinc fingers mostly have charged side chains in the case in the of HIV-1 integrase. Third, most known zinc fingers are DNA binding domains, whereas the N-terminal domain of HIV-1 integrase does not appear to play a primary role in DNA binding. The N-terminal domain of HIV-1 integrase complexed with zinc is a good candidate for structural studies. Comparison of the structure of this domain with that of the numerous known zinc-binding domain structures may provide important clues regarding the functional role of this domain.
Although zinc is essential for folding the isolated N-terminal domain of HIV-1 integrase, the far UV CD spectra of full-length integrase is not detectably different in the presence or absence of bound zinc. The N-terminal domain may therefore have significant secondary structure in the full-length apoprotein, even in the absence of zinc. In contrast, the near UV CD spectra of the full-length integrase are quite different in the presence and absence of zinc, suggesting significant differences in tertiary structure. However, these differences are unlikely to simply reflect differences in multimeric state because the CD spectra were recorded at a protein concentration where both the apoprotein and zinc-associated protein are predominantly tetrameric.
Zinc-Induced Tetramerization.
Tetramerization of HIV-1 integrase requires only the core and C-terminal domains (29). In the absence of zinc, the Kd for tetramerization appears to be similar in the presence or absence of the N-terminal domain. However, when zinc is bound, the full-length protein forms tetramers at a lower protein concentration than in the absence of zinc. The N-terminal domain must either contribute additional intersubunit contacts within the tetramer or effect an allosteric change in the monomers that favors tetramerization.
Although Mn2+ and Mg2+ also promote multimerization of HIV-1 integrase (refs. 39, 40, 41; data not shown), the mechanism is different from that reported here for zinc. Zinc clearly binds to the N-terminal domain of HIV-1 integrase, whereas the sites of Mn2+ and Mg2+ binding appear to map to the core domain (data not shown).
We note that the integrase proteins used for these studies contained the F185K and C280S mutations. The F185K mutation lowers the Kd for dimerization (32), so the mutant protein is predominantly dimeric at a concentration where integrase without this substitution would be mostly monomeric. We confirmed that zinc- and Mn2+-induced tetramerization also occurs with the wild-type integrase (data not shown). The major difference is that the self-associating species for wild-type are monomers, dimers, and tetramers, rather than predominantly dimers and tetramers, as with the mutant protein.
Effect of Zinc on Catalytic Activity.
Zinc-bound integrase is more active than the zinc-free enzyme for both 3′-processing and DNA strand transfer. The reaction rate enhancements were between 5- and 15-fold with zinc-bound integrase; the degree of stimulation depended on the concentration of integrase and DNA substrate included in the reaction mixture (data not shown).
The integrase protein we have routinely used for in vitro 3′-processing and DNA strand transfer reactions does not contain bound zinc, yet the presence of the N-terminal domain is essential for catalytic activity. We cannot exclude the possibility that the Mn2+ or Mg2+ included in the reaction mixture introduces a trace amount of zinc. However, we speculate that the N-terminal domain of integrase does not acquire zinc from the other reaction components. The domain may be sufficiently well-folded in the context of the full-length protein to contribute partial function in the absence of bound zinc. However, the enhanced catalytic activity seen with the zinc-bound enzyme suggests that the presence of zinc is required for optimal catalytic activity. In fact, we have observed near complete conversion of DNA substrate to processed product with the zinc-bound enzyme (data not shown). Stimulation of integrase 3′-processing activity by zinc has also been recently reported by Lee and Han (42).
Although the mechanism of stimulation by zinc remains to be determined, the effects on the self-association properties of integrase suggest that one role of zinc may be to facilitate assembly of the active multimeric form of the enzyme.
Acknowledgments
We thank Marc Lewis for equilibrium ultracentrifugation studies and Martin Gellert, Kiyoshi Mizuuchi, and Alison Hickman for comments on the manuscript. This work was supported in part by the National Institutes of Health Intramural AIDS Targeted Antiviral Program.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Berg J M, Shi Y. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 2.Burke C J, Sanyal G, Bruner M W, Ryan J A, LaFemina R L, Robbins H L, Zeft A S, Middaugh C R, Cordingley M G. J Biol Chem. 1992;267:9639–9644. [PubMed] [Google Scholar]
- 3.Whitcomb J M, Hughes S H. Annu Rev Cell Biol. 1992;8:275–306. doi: 10.1146/annurev.cb.08.110192.001423. [DOI] [PubMed] [Google Scholar]
- 4.Goff S P. Annu Rev Genet. 1992;26:527–544. doi: 10.1146/annurev.ge.26.120192.002523. [DOI] [PubMed] [Google Scholar]
- 5.Vink C, Plasterk R H A. Trends Genet. 1993;9:433–438. doi: 10.1016/0168-9525(93)90107-s. [DOI] [PubMed] [Google Scholar]
- 6.Fujiwara T, Mizuuchi K. Cell. 1988;54:497–504. doi: 10.1016/0092-8674(88)90071-2. [DOI] [PubMed] [Google Scholar]
- 7.Brown P O, Bowerman B, Varmus H E, Bishop J M. Proc Natl Acad Sci USA. 1989;86:2525–2529. doi: 10.1073/pnas.86.8.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelman A, Craigie R. J Virol. 1992;66:6361–6369. doi: 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rowland S J, Dyke K G. Mol Microbiol. 1990;4:961–975. doi: 10.1111/j.1365-2958.1990.tb00669.x. [DOI] [PubMed] [Google Scholar]
- 10.Kulkosky J, Jones K S, Katz R A, Mack J P, Skalka A M. Mol Cell Biol. 1992;12:2331–2338. doi: 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fayet O, Ramond P, Polard P, Prere M F, Chandler M. Mol Microbiol. 1990;4:1771–1777. doi: 10.1111/j.1365-2958.1990.tb00555.x. [DOI] [PubMed] [Google Scholar]
- 12.van Gent D C, Groeneger A A M, Plasterk R H A. Proc Natl Acad Sci USA. 1992;89:9598–9602. doi: 10.1073/pnas.89.20.9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leavitt A D, Shiue L, Varmus H E. J Biol Chem. 1993;268:2113–2119. [PubMed] [Google Scholar]
- 14.Dyda F, Hickman A B, Jenkins T M, Engelman A, Craigie R, Davies D R. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 15.Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz R A, Skalka A M. J Mol Biol. 1995;253:333–346. doi: 10.1006/jmbi.1995.0556. [DOI] [PubMed] [Google Scholar]
- 16.Vink C, Oude Groeneger A A M, Plasterk R H A. Nucleic Acids Res. 1993;21:1419–1425. doi: 10.1093/nar/21.6.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engelman A, Hickman A B, Craigie R. J Virol. 1994;68:5911–5917. doi: 10.1128/jvi.68.9.5911-5917.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woerner A M, Marcus-Sekura C J. Nucleic Acids Res. 1993;21:3507–3511. doi: 10.1093/nar/21.15.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lodi P J, Ernst J A, Kuszewski J, Hickman A B, Engelman A, Craigie R, Clore G M, Gronenborn A M. Biochemistry. 1995;34:9826–9833. doi: 10.1021/bi00031a002. [DOI] [PubMed] [Google Scholar]
- 20.Eijkelenboom A P, Lutzke R A, Boelens R, Plasterk R H A, Kaptein R, Hard K. Nat Struct Biol. 1995;2:807–810. doi: 10.1038/nsb0995-807. [DOI] [PubMed] [Google Scholar]
- 21.Drelich M, Wilhelm R, Mous J. Virology. 1992;188:459–468. doi: 10.1016/0042-6822(92)90499-f. [DOI] [PubMed] [Google Scholar]
- 22.Schauer M, Billich A. Biochem Biophys Res Commun. 1992;185:874–880. doi: 10.1016/0006-291x(92)91708-x. [DOI] [PubMed] [Google Scholar]
- 23.Engelman A, Bushman F D, Craigie R. EMBO J. 1993;12:3269–3275. doi: 10.1002/j.1460-2075.1993.tb05996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Gent D C, Vink C, Groeneger A A M, Plasterk R H A. EMBO J. 1993;12:3261–3267. doi: 10.1002/j.1460-2075.1993.tb05995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherman P A, Fyfe J A. Proc Natl Acad Sci USA. 1990;87:5119–5123. doi: 10.1073/pnas.87.13.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vincent K A, Ellison V, Chow S A, Brown P O. J Virol. 1993;67:425–437. doi: 10.1128/jvi.67.1.425-437.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones K S, Coleman J, Merkel G W, Laue T M, Skalka A M. J Biol Chem. 1992;267:16037–16040. [PubMed] [Google Scholar]
- 28.Grandgenett D P, Vora A C, Schiff R D. Virology. 1978;89:119–132. doi: 10.1016/0042-6822(78)90046-6. [DOI] [PubMed] [Google Scholar]
- 29.Jenkins T, Engelman A, Ghirlando R, Craigie R. J Biol Chem. 1996;271:7712–7718. doi: 10.1074/jbc.271.13.7712. [DOI] [PubMed] [Google Scholar]
- 30.Bushman F D, Engelman A, Palmer I, Wingfield P, Craigie R. Proc Natl Acad Sci USA. 1993;90:3428–3432. doi: 10.1073/pnas.90.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haugan I R, Nilsen B M, Worland S, Olsen L, Helland D E. Biochem Biophys Res Commun. 1995;217:802–810. doi: 10.1006/bbrc.1995.2843. [DOI] [PubMed] [Google Scholar]
- 32.Jenkins T M, Hickman A B, Dyda F, Ghirlando R, Davies D R, Craigie R. Proc Natl Acad Sci USA. 1995;92:6057–6061. doi: 10.1073/pnas.92.13.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adachi A, Gendelman H E, Koenig S, Folks T, Willey R, Rabson A, Martin M A. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wetlaufer D E. Adv Protein Chem. 1962;17:303–390. [Google Scholar]
- 35.Engelman A, Craigie R. J Virol. 1995;69:5908–5911. doi: 10.1128/jvi.69.9.5908-5911.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 37.Strickland E H. CRC Crit Rev Biochem. 1974;2:113–175. doi: 10.3109/10409237409105445. [DOI] [PubMed] [Google Scholar]
- 38.Chow S A, Vincent K A, Ellison V, Brown P O. Science. 1992;255:723–726. doi: 10.1126/science.1738845. [DOI] [PubMed] [Google Scholar]
- 39.Ellison V, Gerton J, Vincent K A, Brown P O. J Biol Chem. 1995;270:3320–3326. doi: 10.1074/jbc.270.7.3320. [DOI] [PubMed] [Google Scholar]
- 40.Pemberton I K, Buckle M, Buc H. J Biol Chem. 1996;271:1498–1506. doi: 10.1074/jbc.271.3.1498. [DOI] [PubMed] [Google Scholar]
- 41.Wolfe A L, Felock P J, Hastings J C, Blau C U, Hazuda D J. J Virol. 1996;70:1424–1432. doi: 10.1128/jvi.70.3.1424-1432.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee S P, Han M K. Biochemistry. 1996;35:3837–3844. doi: 10.1021/bi952056p. [DOI] [PubMed] [Google Scholar]