

Abstract
Low-barrier hydrogen bonds have recently been proposed as a major factor in enzyme catalysis. Here we evaluate the feasibility of transition state (TS) stabilization by low-barrier hydrogen bonds in enzymes. Our analysis focuses on the facts that (i) a low-barrier hydrogen bond is less stable than a regular hydrogen bond in water, (ii) TSs are more stable in the enzyme active sites than in water, and (iii) a nonpolar active site would destabilize the TS relative to its energy in water. Combining these points and other experimental and theoretical facts in a physically consistent framework shows that a low-barrier hydrogen bond cannot stabilize the TS more than an ordinary hydrogen bond. The reason for the large catalytic effect of active site hydrogen bonds is that their formation entails a lower reorganization energy than their solution counterparts, due to the preorganized enzyme environment.
Keywords: enzymes, transition state, subtilisin, electrostatics, evolution
The origin of the enormous catalytic power of enzymes is a problem of great fundamental and practical importance. It is becoming increasingly clear that this catalytic power is mainly due to transition state (TS) stabilization, but there is yet to be a consensus on how the stabilization is provided. The uncatalyzed versions of reactions that are catalyzed by enzymes often proceed extremely slowly in aqueous solution. Associating the polarity of water with this slowness, a nonpolar environment has often been thought to be necessary for accelerating those reactions (1, 2). This has lead to proposals of enzymatic reaction mechanisms that are viable only in nonpolar media, whereas enzyme active sites are usually polar. One such proposal suggests that the enzyme forms a partial covalent bond with the ionic TS through a low-barrier hydrogen bond (LBHB) that is stabilized by quantum resonance interactions (3, 4, 5). Hydrogen bonds (HBs) do contribute substantially to enzyme catalysis. This fact has been gaining increasingly wider recognition, due, in part, to mutation experiments (6, 7) that confirmed earlier theoretical estimates of the catalytic HBs and the prediction that “preorganized local dipoles” (e.g., HBs and carbonyls) are very important in TS stabilization (8, 9, 10). However, the physical reasons for the importance of catalytic HBs are apparently still subject to debate (3, 4, 5, 11, 12, 13, 14, 15).
Using a valence bond (VB) description of hydrogen bonding and analyzing energetic requirements, we conclude that LBHBs do not offer extra TS stabilization over regular HBs. This conclusion is independent of specific computational or experimental methodologies, although they are used to provide examples, hopefully making the arguments clearer.
Hydrogen Bonding
Below we define ordinary HB (OHB) and LBHB in a way that will hopefully make the analysis amenable to a clear discussion. Note that terms such as “short” or “low-barrier” by themselves do not add any new physics to the regular HB description—i.e., they cannot define a new kind of HB. As we describe below, the only relevant and novel concept introduced by LBHBs is a more covalent character, accompanied by a more disperse charge distribution. Only to the extent that this distribution is significantly different than an OHB and has different energetics, especially in response to its environment, that we can make sure that we are considering a distinct class of HB.
When a hydrogen atom is between two electronegative atoms, an attractive interaction develops. This attraction is stronger than a van der Waals interaction, and is called a “hydrogen bond” (16). Hydrogen bonding can simplistically be explained as the electrostatic attraction between the partial charges on the atoms involved. For example, the dipole formed by X−δ − H+δ would have a fairly strong interaction with the charge on Y−Δ. Since a restricted electrostatic description would use the unperturbed charge distribution of the isolated fragments (i.e., X−δ − H+δ and Y−Δ), it would be unable to account for the distortions in those distributions when the HB donor and acceptor approach each other. However, that process can be partly represented by polarizability and handled by the methods of classical electrostatics (17). The remaining factors are strictly quantum mechanical effects, such as charge transfer between proton donor and acceptor. The exact nature of such “partitioning” of the hydrogen bonding and the size of the individual contributions depend on personal perspective and the quantum mechanical methodology employed (18, 19). Our choice for describing hydrogen bonding is based on a VB formalism and is motivated by the ease with which the environmental effects and the degree of covalent interaction can be incorporated into the empirical VB (EVB) formulation (17).
The starting point of this approach is a VB model of a HB in vacuum. The corresponding Hamiltonian is based on the three-orbital four-electron model of Coulson and Danielsson (20) which involves three VB states as follows (17):
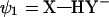 |
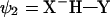 |
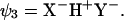 |
1 |
The effective VB Hamiltonian involves diagonal (diabatic) energies of the three states (Fig. 1) and off-diagonal terms that represent the resonance interaction between those states. The ground state-energy (Eg) of the system is obtained from the effective VB Hamiltonian (17). The free energy corresponding to Eg is denoted by ḡ. An effective Hamiltonian can be obtained by fitting a three-state model to the results of gas phase ab initio (AI) calculations as is done in the EVB approach (21, 22). The three VB states can usefully be projected onto an effective two-state model (Fig. 1) that simplifies the analysis and discussion of HB systems. Thus the “quantum” or “resonance” contribution is condensed into a mixing term between the two “unpure” VB structures. The deformation in the charge distribution of the interacting species as they approach each other is captured by the mixing term H12 and the polarizability (which is a property of the individual fragments and can be treated classically once its value is known) of each species. Most importantly for our discussion, the VB model provides a simple way for examining the effect of the environment. The ground-state free energy in a given environment can be expressed as (17, 22, 23):
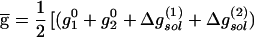 |
2 |
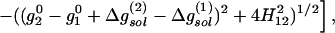 |
Figure 1.
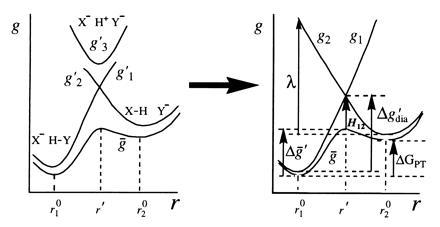
A three-state VB system with E1, E2, and E3 and the corresponding diabatic free energies g1, g2, and g3 can be projected onto a two-state VB representation. The resulting surfaces E′1 and E′2 and free energies g′1 and g′2 no longer represent pure resonance structures, but their mixing results in the same ground-state potential surface Eg (with a corresponding free energy surface ḡ). When the donor and the acceptor are held at a distance R, the stabilization resulting from the mixing of the two unpure states is given by H12(R,r), and its value at the barrier (r = r′) is denoted simply by H12. The coordinate r corresponds to proton movement at fixed R.
where the superscript 0 designates gas-phase properties and Δgsol(i) is the free energy of stabilization of the ith state of the “solute” (e.g., the HB system) by its environment, which is referred to here as the “solvent.” Eq. 2 allows one to assess the effect of the environment [Δgsol(i)] and the mixing term H12 on the HB free energy surface ḡ. This EVB equation is the basis of our analysis.
The difference between an OHB and a more covalent LBHB can be quantified by considering the free energy surface ḡ in terms of the modified Marcus relationship (21). In this approach, one uses the EVB two-state model of Fig. 1 and expresses the free energy at r′ as:
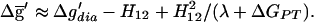 |
3 |
The term quadratic in H12 constitutes a small correction in the context of this article. ΔGPT is the free energy of proton transfer (PT) from the donor to the acceptor and is zero when the pKa values match. Δg′dia is the difference between the intersection of the diabatic free energy curves (g1, g2) and g1(r10). This intersection is approximated by the Marcus expression:
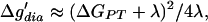 |
4 |
where λ is the so-called “reorganization energy,” which is defined in Fig. 1. The last two terms in Eq. 3 are due to the “covalent” mixing of two VB states and are missing in the Marcus formula, which was developed for electron transfer reactions where the mixing is small. Now we can classify HBs according to whether Δḡ′ is larger or smaller than zero. For this purpose, it is convenient to define a parameter θ by:
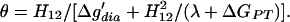 |
5 |
Using the equality Δḡ′ = H12(1 − θ)/θ, a straightforward definition of LBHB corresponds to θ ≥ 1 (i.e., Δḡ′ ≤ 0). Similarly, we can define an OHB as one with θ < 1. Thus the existence of an LBHB is defined in terms of the competition between Δḡ′dia and the resonance mixing term H12. This is also related to the competition between solvation and H12, since solvation effects increase Δgdia.
The existence of LBHB can also be formulated in terms of a charge
distribution. For example, a conventional HB with a negatively charged
acceptor can be represented as X −
H···Y− (or, more precisely,
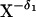 −
−
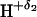 ···
···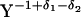 ),
where the charge is concentrated around one atom. This basically
corresponds to the resonance structure ψ1 (in the
effective two-state model). In contrast, the charge is spread out in an
LBHB
−½X···H···Y−½
(or, more precisely,
−½−γX···H···Y−½+γ),
because of a “charge transfer” effect. In other words, when
H12 at the minimum of ḡ is
larger than the corresponding difference between
g2 and g1, the resonance
structures [X− H—Y] and [X—H Y−]
contribute equally. Since the resonance mixing in the LBHB case is
accompanied by significant charge rearrangement, its extent is
solvation-dependent. A polar environment will favor either of the pure
resonance structures because a concentrated charge is solvated better,
whereas a nonpolar environment would compensate for reduced solvation
by increasing covalent mixing. The balance between these two effects
determines whether or not we have an LBHB in a given environment. (See
Fig. 2 and the discussion below).
),
where the charge is concentrated around one atom. This basically
corresponds to the resonance structure ψ1 (in the
effective two-state model). In contrast, the charge is spread out in an
LBHB
−½X···H···Y−½
(or, more precisely,
−½−γX···H···Y−½+γ),
because of a “charge transfer” effect. In other words, when
H12 at the minimum of ḡ is
larger than the corresponding difference between
g2 and g1, the resonance
structures [X− H—Y] and [X—H Y−]
contribute equally. Since the resonance mixing in the LBHB case is
accompanied by significant charge rearrangement, its extent is
solvation-dependent. A polar environment will favor either of the pure
resonance structures because a concentrated charge is solvated better,
whereas a nonpolar environment would compensate for reduced solvation
by increasing covalent mixing. The balance between these two effects
determines whether or not we have an LBHB in a given environment. (See
Fig. 2 and the discussion below).
Figure 2.
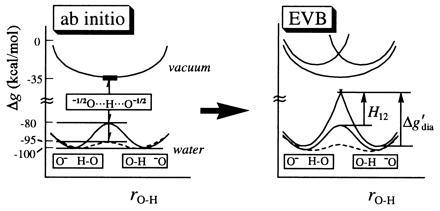
Demonstrating the effect of the environment on the nature of hydrogen bonding and the independence of our conclusions on the model used. The figure considers the HO− HOH system in vacuum and in water, representing the results of ref. 22 in a schematic way. Gas-phase calculations are presented for a single distance R = 2.4 Å between the oxygen atoms, while the calculations in solution are presented for both R = 2.8 Å (——) and for a least energy path where R is allowed to change upon displacement of the hydrogen (---). The distance R is short in vacuum, and the corresponding potential for proton motion is flat, reflecting the fact that H12 is similar in magnitude to Δg′dia. On the other hand, in polar medium, a barrier is induced because the concentrated charge of the O−1 H—O configuration is solvated more than the delocalized −½O···H···O−½ charge distribution. Now Δg′dia has a large solvent contribution and becomes larger than H12. Both the AI and the EVB calculations confirm that the short bond in vacuum is indeed strong and that in water a longer OHB is the most stabilized form. Furthermore, it is clear that the ordinary bond in water is more stable than the “strong” bond in vacuum.
Free Energy Surfaces of Ionic HBs in Different Environments
To clarify the effect of the environment on an ionic HB, we study HO− HOH system in vacuum and in water. The energetics of this system were evaluated by the EVB approach and a recently developed hybrid AI/molecular mechanics approach (22). The results of the calculations are presented qualitatively in Fig. 2. The system has a LBHB character in the gas phase, but it loses most of it in polar solvent. The solvent provides a larger stabilization to the “product” and “reactant” configurations (O− H—O and O—H O−) than to the delocalized charge distribution of the proposed TS −½O···H···O−½. This result agrees with experiments (ref. 25; note that the barrier is reduced significantly upon compression of the O···O distance).
In the gas phase, where Δḡ′ ≈ 0 and ΔGPT = 0, we have from Eqs. 3 and 4:
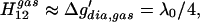 |
6 |
while in solution, we have from Eqs. 4 and 6:
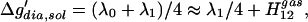 |
7 |
where Δg′dia,sol is the diabatic barrier height in solution, λ0 is the reorganization energy in the gas phase, and λ1 is the solvent contribution to the total reorganization energy λ. Thus we establish that in polar solution:
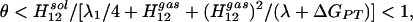 |
8 |
because H12sol < H12gas (donor–acceptor distance is larger in solution, resulting in smaller mixing of VB states) and since λ1 > 0. λ1/4 is the change in solvation energy of the [O− H—O] configuration when the solvent moves from a geometry that stabilizes the reactant state to one that stabilizes the TS. λ1 can be estimated from different models (17, 21, 26) and is ≈20 kcal/mol for the case presented in Fig. 2, where the O–O distance is allowed to relax during the proton transfer process (note that Δḡ′sol − Δḡ′gas ≈ λ1/4). λ has similar values for various polar solvents, including water. As Fig. 2 indicates, our conclusions do not depend on a specific model, in that both AI and EVB approaches give similar results. In general, when a gas-phase LBHB is placed in a polar environment, it becomes an ordinary double-well HB due to solvation effects. In other words, as indicated by the absence of symmetric HBs in recent experiments (25) on a variety of systems, an OHB in a polar solvent has a lower free energy than an LBHB in a polar or nonpolar medium.
The analysis presented above focuses on energetics and charge distributions, yet one might wonder how this is related to the common association of a low barrier with a small separation between electronegative atoms. One might also wonder about the relationship of our analysis and the fact that any HB can become an LBHB if sufficiently compressed (15). Neither the strength of a HB nor its degree of stabilization can be correlated with the barrier height. There are two obvious reasons for that. First, in many cases (e.g., HBs in water) the short distance required for a low barrier is obtained only when the system is compressed at the expense of HB strength. More importantly, bond strengths in different environments cannot be used for deducing relative stabilities of the systems—i.e. the stabilization provided by a bond in a given environment (e.g., vacuum) is not portable to a different environment (e.g., enzyme) because the stability of the dissociated fragments are different (see the discussion on Fig. 3, below).
Figure 3.
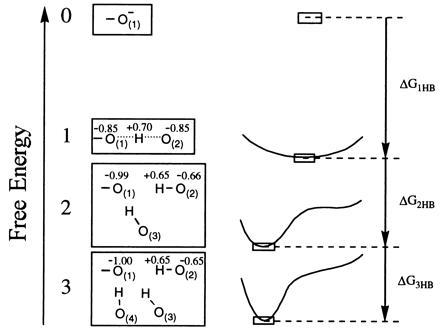
The effect of multiple hydrogen bonding on the energy of a negatively charged proton acceptor. While a single HB to O− will form an LBHB, successive HB donors pull it away from that configuration because of the larger stabilization provided by several OHBs, and the potential curve for proton movement takes on a double-well character. The numbers 0, 1, 2, and 3 correspond to the number of HB donors. The figure presents schematically the results of AI calculations with OH− and 0–3 water molecules. Note that the partial charges of hydrogens that are not explicitly shown are added to those of the shown oxygen atoms to which they are bonded.
Finally, before we discuss the energetics of LBHBs in enzymes, we must define what is meant by an “LBHB-assisted” reaction. Since the LBHB formation is supposed to stabilize an otherwise separate reacting system, the coordinate of the LBHB proton is by definition “orthogonal” to the reaction coordinate that needs acceleration. The TS is at a saddle point corresponding to a free-energy maximum along the reaction coordinate of the desired reaction and at the minimum of a “stabilizing” coordinate. The reaction profile would be a slice of the free-energy surface through the lowest point on the stabilizing HB (that is claimed to be an LBHB) free-energy curve. The TS is stabilized by lowering the minimum of this curve. The proton involved in forming the LBHB cannot itself be an essential, formal part of the reaction coordinate, although it can be regarded as a part of the solvent or environment coordinate of the reaction.
A General Analysis of Catalytic LBHB
In the LBHB-driven catalysis hypothesis, a covalent interaction between an HB donor from the enzyme and the TS is the main source of catalysis. This covalent interaction is said to be enhanced when the pKa values of the HB donor and acceptor match. Thus the stabilization of R—X− TS by a HB donor H—Y is attributed to a covalent interaction between resonance structures [R—X− H—Y] and [R—X—H Y−]. Similarly, the stabilization of an H—B+ part of a TS by a HB acceptor A is attributed to the resonance interaction of the form [A− H—B+] ↔ [A—H B]. This requires the LBHB-stabilized TS to be more stable than the corresponding TS in water or in a non-LBHB enzyme active site, a condition that is nontrivial to satisfy.
To realize the problems with catalytic LBHBs it is useful to make a simple observation: Even when the pKa values of the donor and acceptor match, LBHB is not formed in water. Actually, convincing cases of LBHBs in liquids are rare (25), with the possible exception of donor–acceptor systems with very favorable electron delocalization (27). This means, as discussed in the previous section, that a solvated OHB is more stable than an LBHB, and any extra covalent stabilization in the LBHB is not large enough to win over the largely electrostatic solvation effects; otherwise, the covalent part would dominate in spite of the polar environment. The actual situation in solution corresponds, in the notation of Eq. 5, to Δg′dia > H12. As indicated by Eq. 7, this requirement is satisfied in any solvent as long as 4H12gas is not much larger than λ0. The so-called “resonance-assisted HBs” (27) in systems with extensive π-electron conjugation might have large enough H12 and form LBHBs despite environmental effects. If the substrate and active site groups are appropriate, such conjugation might lead to a LBHB in an enzymic TS. Such systems, if they occur, are better characterized as special cases.
In view of the above discussion, we conclude that an LBHB can only exist in a nonpolar or weakly polar environment where the solvation is reduced so that the covalent stabilization becomes more important. Unfortunately, the increased covalent interactions do not fully compensate for the lost solvation, and the idea of extra-stable LBHBs to the TS is not fruitful‡ because (i) a charged TS with a stabilizing LBHB in a hypothetical nonpolar enzyme active site will be less stable than the same TS with an OHB in water, and (ii) in polar active sites, an LBHB to the TS will be less stable than a regular HB.
The discussion so far is sufficient for concluding that an LBHB-stabilized enzymic TS cannot be more stable than the same TS stabilized by OHBs. Nevertheless, in the following sections we provide perhaps a more tangible and quantitative analysis of specific systems to further clarify the arguments presented above.
Specific Analysis of Catalytic LBHBs: TS in Subtilisin Cannot Be Stabilized by an LBHB
After establishing by a general analysis that LBHBs cannot be effective in enzymes, we turn to specific cases that might help in clarifying our discussion. We start by considering the hypothesis that LBHBs provide major stabilization to negatively charged TSs.
We first address the question of what the optimal environment for a negatively charged TS should be. Fig. 3 shows the energetics of —O− as we go from a nonpolar environment to a polar one by adding HB donors near the negative charge. The TS in vacuum is clearly very unstable. Then we consider the TS with a single HB in vacuum, a configuration that promotes LBHB formation. A single HB will supply a large amount of energy (≈30 kcal/mol) and form a LBHB with the TS (gas-phase surface of Fig. 2). However, a single HB in vacuum or in a nonpolar environment still provides much less stabilization than that provided by water (the “in water” case of Fig. 2). Thus an enzyme with a single LBHB in a nonpolar environment will not be a catalyst, since the TS will be extremely unstable relative to its energy in water. Now we may try to stabilize the TS by surrounding it with several HB donors, as is done in the lower part of the left side of Fig. 3. If we form, for example, an active site with three HBs, it will provide more stabilization than an active site with a single HB. However, now we will not have any LBHB, since it is much more beneficial to have an asymmetric HB to each donor [e.g., O(2)] than to delocalize the charge and thus lose the stabilization of the fully charged —O(1)− by the other HBs. This is in fact the reason why we will not have an LBHB in an arrangement that involves the TS and three or more water molecules in the gas or condensed phase.
In the case of an enzyme active site, the tendency to form an OHB (rather than an LBHB) is even greater than in the case of a water cluster. This point is demonstrated more directly in Fig. 4, which considers the “oxyanion hole” (ref. 17, pp. 170–188) in subtilisin, a serine protease, while including one of the HBs in the explicit quantum system. Along with His and Ser, an Asp residue is invariably present in the active sites of serine proteases, forming what is often called a “catalytic triad.” The reaction involves the formation of a TS that includes the oxyanion “tetrahedral intermediate” —O—C—O− (designated as t−) in an environment (particularly two HBs from Ser-221 and Asn-155) that is designed to give it an optimal stabilization. As shown in Fig. 4, since an LBHB in water is already less stable than a regular HB, forming an LBHB destabilizes the oxyanion and therefore cannot help in catalysis. The enzyme can, however, stabilize the TS more than water by solvating the t− configuration better. The delocalized charge of the LBHB configuration, associated with more covalent stabilization but less solvation, does not lead to a HB that is more catalytic than an OHB. This point can be made clearer if one considers the general point of Fig. 3 and the specific case of Fig. 4. An OHB is clearly preferred, since the free energy of proton transfer from Asn-155 is positive (i.e., no pKa matching in the enzyme). This reflects the fact that the main chain Ser-221 HB has to spend a large amount of free energy to move and stabilize a negative charge on Asn-155.
Figure 4.
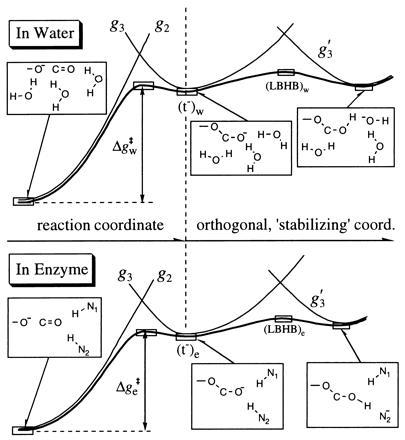
Schematic description of the energetics of the nucleophilic step in the reaction of serine protease in a reference solvent cage and in the active site of subtilisin. The reaction is usually described in terms of the states (His Ser C=O), (HisH+ Ser− C=O), and (HisH+ t−) with the free-energy functions g1, g2, and g3,respectively. Here we consider for simplicity only g2 and g3 as well as the energy g′3 of the additional LBHB state. The route to the tetrahedral intermediate (t−) in the serine proteases involves a higher barrier if it goes through an LBHB-like configuration. For pictorial simplicity, we show a LBHB to t− rather than to the TS obtained at the intersection of g2 and g3 (this is justified since the TS and the t− are energetically and structurally similar). The figure corresponds to the results of EVB calculations (R. P. Muller and A.W., unpublished results). The subscripts w and e denote water and enzyme environments, respectively. The label LBHB denotes a configuration that would form an LBHB in vacuum rather than a real LBHB, since it is unstable in polar media. The figure indicates that the enzyme stabilizes t− more than water does. This is done by taking what is already an OHB and moving it to an environment that is preoriented to stabilize the localized C—O− charge. Now the LBHB-like configuration becomes even less stable than in water (relative to [t−)e], since the main chain Ser-221 HB (designated as N1—H) has to pay too much free energy for moving toward the ionized Asn-155 (designated by N2−.)
We now turn to the [X−···H—Y+] ↔ [X—H Y] type systems. Here, again, we can take the serine protease as our test system. This time we look at the problem of stabilizing a TS defined to include the catalytic Asp and His groups together with the tetrahedral intermediate. That is, the above mentioned catalytic reaction of the serine protease actually involves the formation of the Asp− His+ t− state. The ion pair Asp− His+ has been originally implicated in the so called charge-relay mechanism (28, 29) and more recently introduced as a major example of a catalytic LBHB where the histidine proton partially transfers to the Asp residue (5). Previous EVB studies have demonstrated, on the basis of energy considerations, that the charge-relay mechanism does not have a catalytic advantage (30); the ion pair is already ≈7 kcal/mol more stable than the neutral form in water, and the gap widens even further in the enzyme. Exactly the same considerations show that an LBHB could not augment the catalytic power of a serine protease (Fig. 5). As explained in the caption of Fig. 5, ΔGPT and the pKa difference between the donor and acceptor increases in the protein relative to the corresponding situation in water, contrary to the presumed “pKa equivalence” (for LBHB formation.) This fact is established by the experimental observation of reduced pKa of Asp in the protein (31) and by computational studies whose robustness is proven by reproducing experimentally known pKa values (30, 32). Thus, the consideration of Fig. 5 (see also the detailed discussion in ref. 30) indicate clearly that only the Asp− HisH+ configuration can help in catalysis, and, again, the LBHB formation at the TS has an anticatalytic effect.
Figure 5.
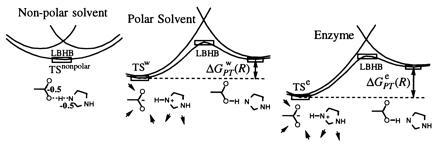
A schematic representation of the energetics of the Asp− HisH+ system (as a part of the TS in serine proteases) in different environments. In a nonpolar environment, the LBHB configuration [Asp− HisH+] ↔ [AspH His] is more stable than the Asp− HisH+ (OHB) configuration. However, in a polar solvent, the OHB is more stable than the LBHB configuration in any environment. The enzyme can interact strongly only with the polar [Asp− HisH+] configuration, and, since it has to stabilize the TS more than water does, it can only do this by pushing the energy of this state down. This increases ΔGPT and makes the LBHB configuration even less likely than in water. Note that the TS of the actual reaction is in the lowest point of the corresponding surface in our figure and not at its maximum. Also note that the TS is most stable in the enzyme and least stable in the nonpolar solvent.
Is There Any Experimental Evidence for LBHB Involvement in Enzyme Catalysis?
An LBHB-stabilized TS has never been directly observed. The LBHB hypothesis refers to a HB formed between the enzyme and the TS, which is by definition an unstable species, and, as such, it cannot be observed in x-ray structures. It can be argued that a so-called TS analogue could be used instead of the reactant substrate. Reasonable as it may seem, that proposal has serious flaws. A true TS–enzyme complex is an unstable configuration even with TS stabilization, and in any case is located at a free-energy “saddle point” instead of a minimum in all coordinates. Therefore, although a “TS analogue” superficially can have the same shape as the true TS, it must have a different electronic structure and interact with the active site differently. Therefore, the relevance of x-ray LBHBs is highly questionable. This applies to any experimental method (such as NMR) that does not provide information about the TS itself.
One may also ask whether any of the experimental evidence points to a catalytically important ground state LBHB. The attempts so far to invoke LBHBs as a major catalytic factor involve somewhat arbitrary interpretations of experiments (such as NMR or x-ray); e.g., NMR shifts that are proposed as indications of LBHB formation can readily be interpreted in terms of perturbations on OHBs. Recent AI (MP2/6-31+G** level) calculations (J. Florian and A.W., unpublished results) produced very large 1H chemical shifts in asymmetric HBs that were previously interpreted as indicators of LBHB formation. As discussed by Guthrie (33), interpreting large chemical shifts as unambiguous LBHB indicators is fundamentally flawed. Although it has been implied (12) that any HB-induced changes in molecular spectra must be explained by unusually covalent hydrogen bonding, such an assumption is simply incorrect. Identifying every change in wavefunctions as covalent effects because everything ultimately is of quantum mechanical nature is as futile as trivializing electrostatic interactions by interpreting the entire molecular science as “electrostatic,” since quantum mechanics contains only electrostatic forces (12). As far as evaluating the LBHB hypothesis is concerned, the relevant question is not about the exact energy decomposition of ordinary double-well HBs, but about whether unusually covalent HBs are necessary to explain the observed spectra§. Moreover, the strongest experimental “evidence” (5) cited for ground state LBHB in serine proteases is in conflict with coupling constant measurements (34) and other recent experiments that clearly locate the proton on His rather than between Asp and His (W. W. Bachovchin, personal communication).
Model compounds are sometimes used for supporting the LBHB hypothesis. For example, the large NMR shifts in model compounds were interpreted (13) to indicate stabilization by LBHB formation, while it appears that the shifts are due to the destabilization of their unprotonated forms (33) and do not require one “to invent a new physicochemical phenomenon” (13).
What we have tried to convey in this section is that extra caution should be exercised before declaring an experimental result as evidence for the LBHB hypothesis. Of course, this also applies to claims of experimental evidence against the LBHB hypothesis (33, 35). For example, experimental results on model compounds in solution, although interesting, cannot by themselves be used to exclude catalytic LBHBs in enzymes. Only combining such results with an energy analysis of enzymic catalysis can exclude catalytic LBHBs. Furthermore, some experimental analyses are not necessarily justified. For example, ref. 35 argued that the electrostatic contribution to HB should depend linearly on the ΔpKa between the donor and the acceptor and implied that formation of an LBHB should be signaled by a different dependence (causing deviation from the line corresponding to the OHB). Then the observed linearity of the relationship between HB strength and ΔpKa was used to exclude LBHB formation in solutions. Although we may agree with its conclusion, the basis of such an analysis is unclear. Using an EVB description of HBs (Eq. 2) and the considerations that lead to the modified Marcus equation, we obtain for an ordinary double-well HB:
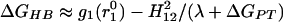 |
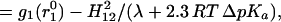 |
9 |
and for an LBHB:
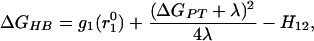 |
10 |
where g1(r10) is the free energy at the minimum of the VB surface corresponding to the proton formally bonded to the donor, without the covalent mixing (H12) contribution. In this formulation, H12 is a function of donor–acceptor distance only. For a double-well HB (Eq. 9), as long as λ ≫ 2.3RTΔpKa, which is the case in solution, the HB strength depends on pKa only through the dependence of g1; same as in the case of a LBHB (Eq. 10). Therefore, LBHB formation would not lead to a detectable deviation from the ΔpKa dependence of OHBs, especially for small ΔpKa (and at large ΔpKa values, we have ordinary, asymmetric HB).
What Is Special About the HBs in Enzymes?
Although a LBHB-specific contribution to enzyme catalysis can be ruled out, one still might wonder what is so special about the HBs in enzymes that make them more effective than those in water. A plausible answer to this issue has been provided quite early (8, 9) by observing that the difference between HBs in water and in proteins is that catalytic HBs in proteins are preoriented toward the atoms that would be charged at the TS so that the enzyme does not have to “pay” in reorienting these HBs (36). This represents a very significant catalytic advantage because of the free-energy cost of polarizing a “random” environment. Avoiding having to pay for a significant part of this energy (instead paid for by the folding energy of the enzyme during its synthesis) increases the solvation energy and can be readily used to stabilize intermediates or the TS (17, 26). Therefore, the nature of the individual HBs in the enzyme active site matters little and need not be much different than those formed in water. Even a TS that has exactly the same interactions with the enzyme as with water would benefit from the reduction in reorganization energy. Interestingly, even if the nature of enzymic HBs were more covalent (but of energy comparable with HBs in water), the source of catalysis would still be in the preorganization of the enzyme; for electrostatic HBs, the prepaid reorganization cost is mostly in the form of unfavorable dipole–dipole interactions within the protein, whereas with covalent HBs, it would be in the form of the entropic cost of placing the HB donor in the right configuration¶.
Conclusions
The importance of HBs in enzyme catalysis has been determined quite early, and the only new element in the proposal of LBHB-assisted catalysis is the idea that covalent, rather than electrostatic, effects are responsible for the catalytic effect of HBs. The primary problem with the LBHB idea is associated with the effect of the environment on HB systems. The “unusual” covalent stabilization of a gas-phase LBHB falls short of the electrostatic stabilization provided to conventional HBs by a polar environment, as evidenced by the absence of covalently stabilized LBHBs in water. A simple accounting of the energy costs involved dictates that the formation of an LBHB to a TS does not lead to reaction rates faster than with an OHB.
Enzymes seem to work by providing polar interactions similar to those in water, including conventional hydrogen bonding, but without all of the accompanying reorganization free-energy “penalty.” By avoiding a significant fraction of this energy cost via having preoriented bonds, and solvating polar TSs better, very “difficult” reactions can indeed be catalyzed without resorting to esoteric schemes.
Acknowledgments
We would like to thank Dr. Jan Florian and Dr. Richard P. Muller for fruitful discussions and for providing the results of their calculations relevant to this paper. This work was supported by the Grant GM24492 of the National Institutes of Health.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: TS, transition state; HB, hydrogen bond; LBHB, low-barrier HB; OHB, ordinary HB; VB, valence bond; EVB, empirical VB; AI, ab initio.
It can be argued that the heterogeneous microenvironment of the active site could have evolved to stabilize a spread-out charge distribution, as in an LBHB, better than a concentrated charge. Although theoretically possible, that argument has several serious problems. (i) This surprising solvation behavior must be proven for individual enzymes, case by case, as it is not an a priori expected solvation behavior. A general explanation of enzyme activity cannot be based on such a premise. (ii) The partial charge magnitudes and their topology in the delocalized LBHB are still very similar to those in an OHB, making it extremely difficult to have an environment that can stabilize the delocalized distribution more than the localized, ordinary one in the same environment and in water. (iii) It goes strictly against the spirit of the LBHB stabilization concept, which suggests that the covalent interaction within the LBHB is the main catalytic factor. If electrostatic stabilization by an exotic active site configuration is necessary for the LBHB to exist, it would be incorrect to assign the catalytic driving force to the covalentness of the LBHB.
Spectral shifts can often be reasonably captured by an electrostatic model that includes polarization effects. For instance, if we consider the Asp− His+ pair in serine proteases and allow His+ and Asp− to be mutually polarized by each others electric fields (by assigning the the proper polarizabilities), we can obtain the changes in electronic properties (including chemical shifts) without considering the covalent part of the bonding between Asp and His. Moreover, even if the covalent part of HBs were necessary to explain spectral changes, that would not necessitate an LBHB-based explanation. Thus, the LBHB hypothesis which requires the existence of an approximately even mixture of [Asp− HisH+] and [AspH His] is, in general, not essential at all for explaining the observed NMR shifts or other spectral changes.
As pointed out by Perrin (25), even if an LBHB-like configuration is of similar or slightly lower energy (or enthalpy) than others, because of its geometry constraints, it will be entropically unfavorable (hence will have a higher free energy) and will not be observed in a random environment. The preorganized environment of an enzyme could, in principle, provide the required geometry for such an LBHB to exist, but then the covalent nature of the HB would be largely a consequence of the enzyme structure rather than the cause of catalysis. To assign catalytic significance to the covalentness, the covalent HB has to be of significantly lower energy as well as free energy than an OHB in water or in the enzyme.
References
- 1.Wolfenden R. Science. 1983;222:1087–1093. doi: 10.1126/science.6359416. [DOI] [PubMed] [Google Scholar]
- 2.Dewar M J S, Dieter K M. Biochemistry. 1988;27:3302–3308. doi: 10.1021/bi00409a027. [DOI] [PubMed] [Google Scholar]
- 3.Cleland W W. Biochemistry. 1992;31:317–319. doi: 10.1021/bi00117a001. [DOI] [PubMed] [Google Scholar]
- 4.Cleland W W, Kreevoy M M. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 5.Frey P A, Whitt S A, Tobin J B. Science. 1994;264:1927–1930. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson A J, Fersht A R, Blow D M, Winter G. Biochemistry. 1983;22:3581–3586. doi: 10.1021/bi00284a007. [DOI] [PubMed] [Google Scholar]
- 7.Krebs J F, Fierke C A. J Biol Chem. 1993;268:948–954. [PubMed] [Google Scholar]
- 8.Warshel A. Proc Natl Acad Sci USA. 1978;86:5820–5824. doi: 10.1073/pnas.86.15.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warshel A, Russell S, Weiss R M. In: Studies in Organic Chemistry. Green B S, Ashani Y, Chipman D, editors. Amsterdam: Elsevier; 1981. pp. 267–273. [Google Scholar]
- 10.Rao S N, Singh U C, Bash P A, Kollman P A. Nature (London) 1987;328:551–554. doi: 10.1038/328551a0. [DOI] [PubMed] [Google Scholar]
- 11.Warshel A, Papazyan A, Kollman P A. Science. 1995;269:102–104. doi: 10.1126/science.7661987. [DOI] [PubMed] [Google Scholar]
- 12.Cleland W W, Kreevoy M M. Science. 1995;269:104. doi: 10.1126/science.269.5220.104. [DOI] [PubMed] [Google Scholar]
- 13.Frey P A. Science. 1995;269:104–106. doi: 10.1126/science.269.5220.104-a. [DOI] [PubMed] [Google Scholar]
- 14.Guthrie J P, Kluger R. J Am Chem Soc. 1993;115:11569–11572. [Google Scholar]
- 15.Scheiner S, Kar T. J Am Chem Soc. 1995;117:6970–6975. [Google Scholar]
- 16.Kollman P A. In: Methods of Electronic Structure Theory. Schaefer H F, editor. New York: Plenum; 1977. pp. 109–151. [Google Scholar]
- 17.Warshel A. Computer Modeling of Chemical Reactions in Enzymes and Solutions. New York: Wiley; 1991. [Google Scholar]
- 18.Glendening E D, Streitwieser A. J Chem Phys. 1994;100:2900–2909. [Google Scholar]
- 19.Martinov M, Cioslowski J. Mol Phys. 1995;85:121–129. [Google Scholar]
- 20.Coulson C A, Danielsson Ark Fys. 1954;8:239–245. [Google Scholar]
- 21.Hwang J-K, King G, Creighton S, Warshel A. J Am Chem Soc. 1988;110:5297–5311. [Google Scholar]
- 22.Muller R P, Warshel A. J Phys Chem. 1995;99:17516–17524. [Google Scholar]
- 23.Warshel A, Russell S T. Q Rev Biophys. 1984;17:283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- 24.Timoneda J J, Hynes J T. J Phys Chem. 1991;95:10431–10442. [Google Scholar]
- 25.Perrin C L. Science. 1994;266:1665–1668. doi: 10.1126/science.266.5191.1665. [DOI] [PubMed] [Google Scholar]
- 26.Hwang J-K, Chu Z T, Yadav A, Warshel A. J Phys Chem. 1991;95:8445–8448. [Google Scholar]
- 27.Gilli P, Bertolasi V, Ferretti V, Gilli G. J Am Chem Soc. 1994;116:909–915. [Google Scholar]
- 28.Blow D M, Birktoft J J, Hartley S S. Nature (London) 1969;221:337–340. doi: 10.1038/221337a0. [DOI] [PubMed] [Google Scholar]
- 29.Kraut J. Annu Rev Biochem. 1977;46:331–358. doi: 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- 30.Warshel A, Naray-Szabo G, Sussman F, Hwang J-K. Biochemistry. 1989;28:3629–3637. doi: 10.1021/bi00435a001. [DOI] [PubMed] [Google Scholar]
- 31.Brayer G D, Delbaere J T J, James M N G. J Mol Biol. 1978;124:261–283. doi: 10.1016/0022-2836(78)90159-6. [DOI] [PubMed] [Google Scholar]
- 32.Warshel A, Russell S. J Am Chem Soc. 1986;108:6569–6579. [Google Scholar]
- 33.Guthrie J P. Chem Biol. 1996;3:164–170. doi: 10.1016/s1074-5521(96)90258-6. [DOI] [PubMed] [Google Scholar]
- 34.Bachovchin W W. Proc Natl Acad Sci USA. 1985;82:7948–7951. doi: 10.1073/pnas.82.23.7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shan S O, Loh S, Herschlag D. Science. 1996;272:97–101. doi: 10.1126/science.272.5258.97. [DOI] [PubMed] [Google Scholar]
- 36.Shoichet B K, Baase W A, Kuroki R, Matthews B W. Proc Natl Acad Sci USA. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]