

Abstract
The heterologous transcription factors NFAT and AP-1 coordinately regulate cytokine gene expression through cooperative binding to precisely juxtaposed DNA recognition elements. The molecular origins of cooperativity in the binding of NFAT and AP-1 to DNA are poorly understood. Herein we have used yeast one-hybrid screening and alanine-scanning mutagenesis to identify residues in AP-1 that affect cooperative interactions with NFAT on DNA. Mutation of a single conserved Arg residue to Ala in the cJun spacer region (R285A) led to a virtually complete abolition of cooperative interactions with NFAT. The DNA-binding activity of AP-1 alone was unaffected by the cJun R285A mutation, thus indicating that this residue influences cooperative binding only. Ala-scanning mutations elsewhere in AP-1, including the cFos subunit, revealed no other strongly interacting single positions. We thus conclude that NFAT contacts AP-1 in the spacer region of the cJun subunit, making an especially important contact to R285, and that these interactions drive formation of the cooperative NFAT/AP-1/DNA complex. These results provide a general strategy for selectively ablating cooperativity between transcription factors without affecting their ability to act alone and yield insights into the structural basis for coordinate regulation of gene expression.
Antigen recognition by the T-cell receptor results in transmission of a signal across the plasma membrane and activation of mitogenic signaling pathways, leading to the induced expression of interleukin 2 (IL-2) (1). The sequence elements that control antigen-dependent induction of the IL-2 gene, located in an enhancer region within 300 bp upstream of the transcription start point (2), have been extensively studied (1). A sequence in the IL-2 enhancer termed the upstream antigen receptor response element, or ARRE2, serves as a composite recognition site for the nuclear factor of activated T cells (NFAT) and AP-1 (1, 3, 4, 5, 6). Whereas AP-1 transcription is induced upon activation of the Ras/Map kinase pathway (1), NFAT is sequestered in the cytoplasm of resting T cells and translocates to the nucleus in response to Ca2+ mobilization (7). Translocation of NFAT is blocked by the clinically important immunosuppressive drugs cyclosporin A (CsA) and FK506 (7), which inhibit the Ca2+-activated Ser/Thr phosphatase calcineurin (8). In the course of T-cell activation, calcineurin is believed to dephosphorylate NFAT directly (9), thereby releasing by an unknown mechanism the blockade to nuclear translocation.
Originally isolated from cytoplasmic extracts of a T-cell hybridoma (9), NFAT is now known to define a family of transcription factors containing at least four members (10, 11, 12, 13, 14). NFAT family members differ in their tissue distribution—none are localized exclusively in T cells—and appear not to be functionally redundant, as judged by the distinctive phenotype of NFAT1-knockout mice (15, 16). These proteins share a ≈300-amino acid domain that may be distantly related to the Rel homology region (RHR), a DNA-binding and dimerization module present in the NF-κB transcription factors (17, 18). The putative RHR of NFAT proteins is both necessary and sufficient for their binding to DNA and cooperative interactions with AP-1 (13, 18). NFAT family members, unlike the Rel proteins, bind DNA as monomers (13, 17). Contained within ARRE2 is a consensus recognition site for NFAT family members, 5′-GGAAAA-3′, closely juxtaposed with a nonconsensus AP-1 site, 5′-TGTTTCA-3′ (compare Fig. 1 A and B).
Figure 1.
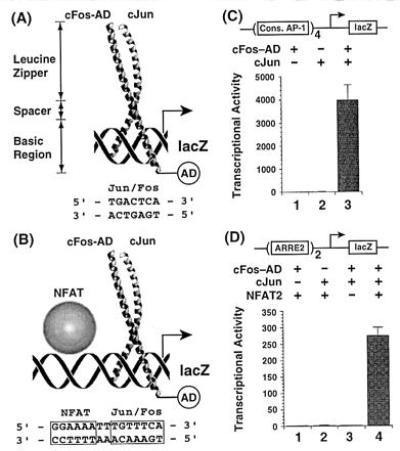
Activation of lacZ reporter gene transcription by AP-1 acting alone and in cooperation with NFAT. (A) Structure of the AP-1 bZIP domain (22), with functional domains denoted, bound to a consensus AP-1 site. (B) NFAT, bound to a polypurine tract in ARRE2, cooperatively recruits AP-1 to the adjacent nonconsensus AP-1 site, thereby activating reporter gene expression. AD denotes the B42 activation domain, which is fused to the basic region of Fos. (C) AP-1-driven reporter assay detects the binding of AP-1 alone to a consensus site in yeast. (D) ARRE2-linked reporter assay detects the reconstitution of the cooperative AP-1/NFAT/ARRE2 complex in yeast. Transcriptional activity refers to units of β-galactosidase activity.
Whereas NFAT binds ARRE2 with modest affinity, and the binding of AP-1 to ARRE2 is very weak (see below), the two proteins together form a highly stable complex on ARRE2 (19). In the cooperative complex, AP-1 is bound to the nonconsensus element in ARRE2 (21). Most if not all heterodimeric combinations of the two AP-1 subunits of Fos (cFos, FosB, Fra-1, and Fra-2) and Jun (cJun, JunB, and JunD) appear capable of cooperating with NFAT family members identified thus far (5). The cooperative interaction between NFAT and AP-1 on ARRE2 is essential for IL-2 gene induction (12, 20).
X-ray crystallographic studies (22) have revealed that the basic region–leucine zipper (bZIP) domains of cFos and cJun pair to form continuous helices that splay to present a Y-shaped crevice into which the DNA binds (Fig. 1A). Each of the helices contains three functionally distinct segments: the leucine zipper dimerization domain, the basic region DNA-binding domain, and the intervening spacer region, which controls DNA half-site separation (23). Whereas AP-1 alone binds DNA in two orientations related by interchange of cFos and cJun (21, 22), only one of these orientations is observed in the cooperative AP-1/NFAT/DNA complex (21) (Fig. 1B). Although the mechanism of this orientational locking is not well-understood, chemical crosslinking experiments suggest that NFAT and AP-1 contact each other directly in the cooperative NFAT/AP-1/DNA complex (21). AP-1 fragments containing only the bZIP motif cooperate with NFAT as effectively as full-length AP-1, thus indicating that the structural determinants required for cooperative DNA binding with NFAT are contained entirely within this ≈60-amino acid stretch (9).
Herein we report the reconstitution of the core NFAT/AP-1/ARRE2 complex in a yeast genetic system (24, 25) and the use of alanine-scanning mutagenesis (26) to identify positions in AP-1 that make essential contacts to NFAT. We find that mutation of a single arginine residue in the cJun spacer (R285) to alanine virtually abolishes cooperative binding to NFAT/ARRE2 but leaves unaffected the affinity of AP-1 alone for DNA. These and other data that we present suggest that cooperative binding results from contacts between the spacer region of cJun and the putative RHR of NFAT.
MATERIALS AND METHODS
Yeast Strains.
Saccharomyces cerevisiae FY250 (MATα, ura3-52, his3Δ200, leu2Δ1, trp1Δ63) was the generous gift of M. Ptashne (Harvard University). The gcn4− derivative of FY250 was generated by insertion of TRP1 into the GCN4 locus (27).
β-Galactosidase Reporter Gene Assays.
Strains FY250 and FY250 gcn4− were used to assay ARRE2- and AP-1-driven reporter gene expression, respectively. Yeast transformants were grown at 30°C to midlogarithmic phase in appropriate selection media containing 2% raffinose as the sole carbon source. Protein expression was induced by the addition of galactose to a final concentration of 20 g/liter. After 6 h, β-galactosidase activity was quantified as described (28) with 2.5 mM chlorophenol red-β-d-galactopyranoside. Units were calculated as described (29); those shown in Figs. 1 C and D and 2 represent the mean of at least three experiments. Assays performed on different days were normalized to the average wild-type activity obtained.
Oligonucleotides.
A blunt-ended 80-bp duplex probe containing wild-type ARRE2 (XY = TT) or an ARRE2 variant containing a consensus AP-1 site (consARRE2, XY = AC) was used in electrophoretic mobility-shift assay (EMSA) experiments: 5′-ATTGTTGAATTCCCGGGATCCCCAAAGAGGAAAATTTGXYTCATACAGGATCCTCGAGCTCGGTTCTAGAGTCGACTGAT-3′. The ARRE2 reporter was constructed by insertion of a synthetic oligonucleotide duplex containing tandemly repeated ARRE2 sites: 5′-tcgaC(AGAGGAAAATTTGXYTCATAC)nAG-3′ (XY = TT; n = 2; lowercase type denotes a single-stranded DNA overhang). A related oligonucleotide containing four consensus ARRE2 sites was used to construct the AP-1 reporter (XY = AC; n = 4).
Plasmid Construction.
LacZ reporter plasmids were derived from pLR1Δ1 (30). Synthetic oligonucleotides were inserted into the unique XhoI site of pLR1Δ1. A fusion protein encoding an hemagglutinin (HA) epitope tag (YPYDVPDYA), nuclear localization sequence (NLS) of the simian virus 40 large tumor antigen and the putative RHR of human NFAT2 (residues 415–710) was expressed under the control of the GAL1 promoter and ADH1 transcription terminator on the LEU2-selectable plasmid pBC103 (B. Cohen and R. Brent, personal communication). The bZIP domain of cFos was expressed as an N-terminal fusion to the B42 activation domain on the yeast plasmid pJG4-5 (25). cJun was expressed as an N-terminal NLS/C-terminal HA-tag fusion protein on the yeast plasmid pBP1. pBP1, which was constructed by replacement of the pUC19-derived parts of pJP190 (J. Pearlberg and M. Ptashne, personal communication) with the corresponding parts of pUK21, provides kanamycin-resistance and HIS3 selection markers, a constitutive ACTIN promoter, and GAL11 transcription terminator. In the cFos and cJun constructs, a single oxidation-sensitive Cys residue was mutated to Ser (C154S and C278S, respectively); the DNA-binding activity of the mutant proteins is identical to wild type (31). For the AP-1 assay, LEU2-selectable constructs expressing cFos and cFos Ala mutants were generated by subcloning the B42-activation-domain-tagged cFos sequences from pJG4-5 into pBC103. All mutants of cFos and cJun were prepared by PCR megaprimer mutagenesis using Pfu polymerase (Stratagene) (32). All new constructs and mutations were confirmed by dideoxynucleotide DNA sequencing.
Recombinant Proteins.
The recombinant NFAT1 RHR (residues 1–297) was expressed and purified to homogeneity as described (10). Recombinant cJun, cJun R285A-(247–340), and cFos-(118–211) bZIP fragments were expressed as (His)6 fusion proteins, solubilized in guanidinium hydrochloride, purified by chromatography on Ni2+-NTA-agarose (Qiagen), and quantified by BCA assay (Pierce).
Gel EMSAs.
Protein–DNA complexes studied by EMSA were formed by incubation at room temperature for 1 h with <100 pM end-labeled DNA probe in 20 μl of binding buffer [16 mM Hepes·KOH, pH 7.5/60 mM KCl/10% glycerol/1 mM DTT/BSA (10 μg/ml)/poly(dI·dC) (5 μg/ml)].
RESULTS
Reconstitution of the Core NFAT/AP-1/ARRE2 Complex in Yeast.
As a rapid and efficient means of screening AP-1 mutants for those that affect cooperative DNA binding with NFAT, and for distinguishing these mutants from those that affect DNA-binding by AP-1, we developed parallel modified yeast one-hybrid assays (24) (Fig. 1 A and B) based on the interaction trap system (25). For the assays that examined cooperativity between NFAT and AP-1, we chose the well-characterized NFAT2 isoform (also known as NF-ATc) (12). Truncated forms of NFAT2, cJun, and cFos containing the core domains required for DNA-binding and cooperative complexation on ARRE2 (putative RHR and bZIP domains, respectively) simultaneously coexpressed in yeast were used to drive the expression of a lacZ reporter linked to tandem ARRE2 sites (Fig. 1B). Even though NFAT binds ARRE2 with moderate strength in the absence of AP-1, the core NFAT2 RHR alone is incapable of activating reporter gene expression (data not shown). Because cFos is unable to homodimerize yet forms a stable heterodimer with cJun, we chose to fuse the B42 activation domain onto the cFos bZIP (Fos-AD), so as to detect exclusively DNA binding by cFos-AD/cJun.
To validate the cooperative binding assay in yeast, we expressed various combinations of NFAT2, cFos-AD, and cJun in yeast and measured reporter gene expression. NFAT2 in combination with only cFos-AD (Fig. 1D, lane 1) or cJun (lane 2) failed to activate the lacZ reporter. Furthermore, cFos-AD and cJun gave no activation in the absence of NFAT2 (lane 3), consistent with the low affinity of AP-1 for ARRE2. However, when NFAT2, cFos-AD, and cJun were all present in the yeast cells, the lacZ reporter was activated roughly 100-fold above basal levels (lane 4). Since reporter gene activation is dependent upon all three polypeptides that make up the cooperative complex, we conclude that the assay detects the recruitment of AP-1 to ARRE2 by NFAT.
The lack of reporter gene activation by NFAT2 alone (data not shown) or by NFAT in the presence of only one AP-1 subunit (Fig. 1D, lanes 1 and 2) indicates that the endogenous yeast AP-1 homolog GCN4 (27) does not interfere in the in vivo cooperativity assay through functional replacement of AP-1; this is consistent with the observation that the GCN4 bZIP cooperates poorly if at all with NFAT/ARRE2 in vitro (L. Chen and G.L.V., unpublished results). Reporter constructs incorporating multiple copies of consensus AP-1 sites, however, were evaluated with a gcn4− yeast strain (27). Under these conditions, neither cFos-AD nor cJun alone activated lacZ reporter gene expression (Fig. 1C, lanes 1 and 2, respectively), but cFos-AD and cJun together stimulated reporter gene expression roughly 4000-fold over basal levels (lane 3). This consensus AP-1 reporter system thus provided an in vivo assay for the DNA-binding activity of mutant AP-1 proteins.
Alanine-Scanning Mutagenesis Identifies Regions in AP-1 That Are Critical for Cooperation with NFAT.
To identify regions of the AP-1 bZIP that play an important role in formation of the cooperative NFAT/AP-1/DNA complex, we initially converted blocks of residues to alanines and determined the effect of these block mutations on cooperativity in the yeast ARRE2-driven reporter assay. Alanine substitution was chosen to eliminate interactions beyond the β-carbon while favoring the formation of α-helical structure in the bZIP protein (26). Because all but a few of the residues in the basic region contact DNA directly (22), and point mutations in this region of bZIP proteins disrupt DNA binding (33, 34), we avoided alanine-scanning this portion of cFos or cJun. Furthermore, we did not mutate any of the functionally important leucine residues that define the heptad repeats of the leucine zipper dimerization domain. Of the 14 block alanine mutants tested, each containing more than three mutated positions, only four resulted in a greater than 10-fold loss of transcriptional activity when assayed for cooperativity with NFAT2 (Fig. 2): cJun284–288 and cJun290–295 have blocks of alanines in the cJun spacer and first heptad repeat, respectively; cFos132–137 and cFos138–143 have block Ala substitutions in the region immediately N-terminal to the cFos DNA-binding domain. Immunoblot analysis demonstrated that the mutant proteins were expressed at roughly the same levels as wild type (data not shown).
Figure 2.

Effects of alanine-scanning mutations on AP-1 activity in vivo. Sequence of the bZIP region of the AP-1 subunits cJun (A) and cFos (B), with positions mutated to alanine denoted below. To the right of the sequences are shown the transcriptional activity of wild-type and mutant AP-1 proteins in the ARRE2 reporter assay, and (for selected examples) in the AP-1 reporter assay. ND, not determined. The standard errors in the measurements of transcriptional activity assays are estimated at ±25%.
To localize more precisely the positions within the four deleterious blocks that had the greatest effect on cooperative binding, we analyzed mutant proteins containing fewer Ala substitutions within these same regions (Fig. 2). In two regions of the cFos N terminus, (Ala)3 block mutations (cFos135–137 and cFos141–143) led to an ≈3-fold reduction in the ability to activate the ARRE2-driven reporter. These same constructs had a ≈3-fold diminished ability to activate the AP-1 reporter, suggesting that the defect in their ability to cooperate with NFAT2 results from impaired DNA-binding activity.
In cJun, although a block of Ala residues from positions 290 to 295 had a greater than 20-fold effect on ARRE2 reporter gene expression, mutant proteins having fewer Ala substitutions in this region were only moderately affected. These results rule out a strong interaction between NFAT2 and any single amino acid in the first heptad repeat of cJun but leave open the possibility that this region may contribute one or more weak contacts that facilitate cooperative binding. cJun mutants containing single Ala substitutions at each of the four spacer positions altered in cJun284–288 were also assayed. Three of these Ala mutations only slightly affected cooperative reporter gene activation. The fourth, containing an Arg → Ala substitution at position 285 (R285A), virtually abolished ARRE2-driven reporter gene expression but had no effect in the AP-1 reporter system (Fig. 2, cJun285). We thus conclude that of the positions studied herein, R285 is the single most critical one for cooperative interactions with NFAT2.
AP-1 Containing the R285A Mutant cJun Subunit Is Significantly Impaired in Cooperative DNA Binding with NFAT1 in Vitro.
To characterize further the effect of the R285A mutation in cJun on AP-1 activity, we carried out electrophoretic mobility assays (EMSA) on bacterially expressed bZIP fragments of cFos, cJun, and cJun R285A and the recombinant NFAT1 RHR domain. The RHR domains of NFAT1 and NFAT2 are virtually identical in their DNA binding affinity and ability to cooperate with AP-1 (13). Both cJun and R285A exhibited essentially indistinguishable Kd values of ≈80 nM for a consensus site and ≈300 nM for the nonconsensus site in ARRE2 (Fig. 3 A and B, respectively). Thus, the presence of the two nonconsensus positions in ARRE2 weakens the AP-1 affinity relative to a consensus site by ≈4-fold (0.8 kcal/mol; 1 cal = 4.184 J) under the conditions of these assays.
Figure 3.
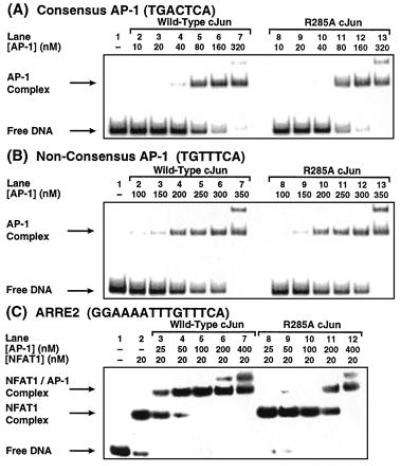
EMSA analysis of DNA-binding activity in vitro. (A and B) Wild-type AP-1 and R285A cJun mutant AP-1 bind with essentially identical affinities to a consensus AP-1 site (Kd = ≈80 nM) (A) and to the nonconsensus site in ARRE2 (Kd = ≈300 nM) (B). (C) The R285A cJun mutant AP-1 shows a substantially diminished ability to cooperate with NFAT1. From the data in C, the Kd of wild-type AP-1 for the NFAT1/ARRE2 complex is estimated at ≈30 nM.
To quantify cooperative binding by the wild-type and R285A cJun mutant bZIP domains of AP-1, we measured the effective Kd for binding of AP-1 to the NFAT1/ARRE2 complex (Fig. 3C). In these experiments, the NFAT1 concentration was held fixed at 20 nM to afford greater than 70% binary NFAT1/DNA complex in the absence of AP-1 (lane 2). Whereas only 30 nM wild-type AP-1 results in ≈50% formation of the ternary AP-1/NFAT1/ARRE2 complex (lanes 3 and 4), roughly 225 nM of the R285A cJun mutant is required to produce the same amount of ternary complex (lane 11). Thus, mutation of Arg-285 to Ala in cJun reduces the ability to cooperate with NFAT1 by ≈8-fold. By comparing the apparent affinity of AP-1 for ARRE2 in the presence and absence of NFAT1 (compare Fig. 3 C, lane 3, with B, lane 6), we deduce that the affinity of AP-1 is stabilized by ≈10-fold in the presence of NFAT1. These data are consistent with kinetics measurements on the NFAT1/ARRE2 complex that have shown (19) that the dissociation half-life is increased approximately 10-fold by AP-1.
DISCUSSION
S. cerevisiae as a Powerful Genetic System for the Study of Cooperative Interactions Among Transcription Factors.
Eukaryotic promoter/enhancers and the proteins that bind them rely extensively on cooperativity to achieve complex patterns of gene expression. The importance of cooperativity in DNA binding by dimeric transcription factors containing identical or homologous subunits has long been recognized, but only more recently has it become apparent that even heterologous proteins often bind adjacent DNA sites cooperatively. Whereas the structural basis for protein–protein interactions in transcription factor dimers has been extensively studied, relatively little detailed mechanistic information is available about cooperative interactions involving heterologous DNA binding proteins (35, 36). Information is especially lacking about composite regulatory elements, genetic loci that redefine the functional identity of transcription factors through cooperative juxtaposition with other regulatory proteins. A prototypical example of a composite regulatory element is ARRE2, which converts the ubiquitous mitogen-inducible transcription factor AP-1 into a T-cell receptor-specific responder by templating its cooperative complexation with NFAT. The present studies were undertaken to gain insight into the nature of the mutually stabilizing interactions between AP-1 and NFAT on ARRE2.
Analysis of a transcription complex assembled from three polypeptide chains and two DNA strands represents a formidable technical challenge. As a simplifying strategy, we used a modified version of the yeast one-hybrid system to assemble the core NFAT/AP-1/ARRE2 complex in the yeast cell nucleus, where its presence could be detected via the expression of a linked reporter element. The use of the one-hybrid system and related yeast expression assays to map functional domains on transcription factors (27, 29), to clone DNA binding proteins from a fusion cDNA library (24), and to identify the preferred DNA-binding site for a known protein (37) is widely documented. The present report expands the scope of the yeast one-hybrid assay to include the mutational analysis of cooperative interactions between proteins on a composite regulatory element.
Key to our design strategy was the need to ensure that the system selectively detects cooperative interactions between NFAT and AP-1. To accomplish this objective, we coexpressed all three DNA-binding domains of the cooperative complex in yeast but fused the B42 activation domain onto the one subunit (cFos) that is incapable of binding DNA on its own. Fortuitously, cJun/cFos-AD is unable to substantially activate expression from the nonconsensus AP-1 site in ARRE2, even with a reporter containing two tandemly repeated sites. In addition, we found that the putative RHR domain of NFAT2 alone, though bound to ARRE2 under the conditions of our in vivo assay, could not activate reporter gene expression. Thus, we demonstrated that the activation of lacZ transcription in our system is critically dependent upon the recruitment of cJun/cFos-AD to ARRE2 through cooperative interactions with NFAT2. A second design aspect involved the use of tandem assays, an ARRE2 screen and a consensus AP-1 screen, to distinguish between mutations affecting protein–protein versus protein–DNA interactions.
An Acidic cFos Segment Stabilizes the AP-1/DNA Complex.
Deletion of an acidic sequence several residues N-terminal to the cFos basic region (refer to Figs. 1A and 2) has been reported to affect assembly of the cooperative NFAT1/AP-1/ARRE2 complex, without influencing the strength of AP-1/DNA interactions (38). This interpretation is inconsistent with earlier demonstrations that point mutations affecting the acidic cFos N-terminal region strongly weaken the affinity of AP-1 for DNA (33). We have revisited this issue using alanine-scanning mutagenesis in the yeast reporter system, in which we find that blocks of alanine mutations in this region substantially decrease lacZ expression in the cooperative ARRE2 reporter system. For example, block replacement of cFos residues Glu-135, Glu-136, and Glu-137 with Ala reduced ARRE2-driven β-galactosidase activity greater than 2.5-fold (Fig. 2B, cFos135–137). However, mutations in the cFos N-terminal region that significantly decrease the ARRE2 signal also affect the AP-1-driven reporter assay to a similar extent. The present data, and in vitro results (33), indicate that mutations in the region N-terminal to the cFos bZIP domain diminish cooperative NFAT/AP-1/ARRE2 complex formation primarily if not exclusively by weakening AP-1/DNA interactions. The influence of this acidic segment seems likely to originate from electrostatic stabilization of α-helical structure in the basic region that indirectly influences the strength of DNA binding.
Contacts Between NFAT and the Spacer Region of the AP-1 bZIP Play an Essential Role in Cooperative Binding to ARRE2.
The observation that NFAT and AP-1 can be chemically cross-linked when bound to ARRE2 has suggested that the two proteins contact each other in the ternary NFAT/AP-1/ARRE2 complex (21). Nonetheless, there remained a small but real possibility that the cross-linking reagent had spanned a gap between the two (noninteracting) proteins in the ternary complex. Herein we have shown that the removal of side-chain functionality in AP-1 results in a loss of cooperative binding affinity of AP-1 for the NFAT/ARRE2 complex, without affecting the strength of AP-1/DNA interactions. These data thus provide biochemical evidence in favor of a model wherein AP-1 is recruited to ARRE2 through direct cooperative contacts with NFAT.
Although high-resolution structural information on the NFAT/AP-1/ARRE2 complex has not yet been forthcoming, its structure has been modeled on the basis of biochemical data and the x-ray structures of AP-1 and the RHR of p50 NF-κB (17). This model revealed that the so-called insert region of the p50 RHR, a ≈60-amino acid stretch that in p50 forms a compact helical subdomain (39, 40), lies closest to the AP-1 bZIP. We have recently obtained NMR and other data that indicate that the corresponding insert region of the NFAT RHR is completely different in structure from that of p50, being a ≈20 amino acid loop that may contact the 3′ end of the recognition site in the minor groove (S. A. Wolfe, P. Zhou, L. Chen, and G.L.V., unpublished results). Incorporation of these recent observations yields the model shown in Fig. 4A. In this model, the insert region of NFAT lies closest to the spacer region of cFos/cJun, suggesting these regions represent the locus of mutual contact between NFAT and AP-1. Indeed, we observed that alanine-scanning mutations within the spacer region and first heptad repeat of cJun (residues 284–295) strongly affect cooperativity without perturbing AP-1/DNA interactions; such cooperativity-specific mutational effects were not observed at any other region within AP-1 (Fig. 2). The guanidinium-bearing side chain of cJun R285, truncation of which practically abrogates cooperative binding with NFAT, projects out from the cJun helix directly toward the NFAT insert (Fig. 4A). Although the nature of the selection system precludes mutational analysis of the cFos and cJun basic regions, it is very unlikely that these are involved directly in contacts to NFAT, because the few residues of the basic region that are not already tied up in contacts to DNA appear too distant from NFAT and project in the wrong direction to make favorable contacts (Fig. 4A). The same distance problem applies to the C-terminal portion of the leucine zipper of AP-1, which also appears too distant from NFAT to represent a likely site of interactions.
Figure 4.
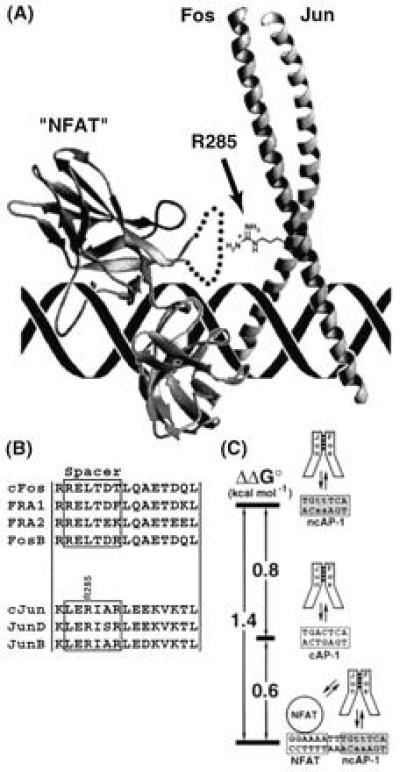
Cooperative contacts between NFAT and AP-1. (A) Notional model of the NFAT/AP-1/DNA complex, constructed by docking the x-ray structure of AP-1 (22) in the correct orientation (21) with that of an NF-κB p50 RHR monomer (“NFAT”) (39). Because the insert region of p50 is not homologous to the corresponding sequence in NFAT, it was replaced in the model with a schematic loop (dotted line), part of which is believed to contact the DNA minor groove (L. Chen and G.L.V., unpublished results). The side chain of R285 in cJun, shown explicitly, projects toward NFAT. (B) Sequence alignments of mammalian Jun and Fos family members in the spacer region and first heptad repeat, which in Jun contacts NFAT. (C) Free-energy diagram illustrating the influence of sequence composition and NFAT cooperativity on the energetics of AP-1/DNA interactions. Stability of the complexes increases from top to bottom.
We have previously reported that NFAT1 orients AP-1 on DNA in the cooperative complex, such that cFos is localized over the NFAT-distal half-site and cJun is localized over the NFAT-proximal half-site (as depicted in Fig. 4A). NFAT2 is known to display the same orientation preference as NFAT1, as might now be expected, because the insert regions of all NFAT family members are highly conserved (10, 11, 12, 13, 14). The present observation that cJun R285 makes an important contact to NFAT provides some insight into the structural basis of this orientational preference. Whereas the residue corresponding to cJun R285 is conserved in all known Jun family members, all Fos family members contain a conserved Leu residue at this position (Fig. 4B). In addition to the gain of favorable contacts made with cJun R285 and perhaps other nearby AP-1 residues in the preferred orientation, it is also possible that orientation selectivity results to some extent from the avoidance of unfavorable contacts in the dispreferred orientation. Although the precise mechanism for achieving orientational selectivity is unknown, the high degree of sequence similarity among Fos and Jun family members in the region that contacts NFAT suggests that most if not all AP-1 heterodimeric forms should be capable of cooperating with NFAT to form an oriented complex (Fig. 4B).
The Energetics of Cooperativity in NFAT/AP-1/DNA Interactions.
Although it has been widely recognized that NFAT and AP-1 cooperate on DNA, the magnitude of the cooperative effect was unknown. Herein we report that the presence of NFAT on ARRE2 stabilizes the binding of AP-1 by roughly 10-fold, corresponding to 1.4 kcal/mol (Fig. 4C). This mutual stabilization results from multiple contributions: the favorable energy of protein–protein contacts in the NFAT/AP-1 interface, balanced against potentially unfavorable costs of induced distortions in DNA structure and induced folding in the NFAT insert, which may be necessary to bring about contacts between the two proteins. This being said, it is remarkable that the side chain of a single residue, cJun R285, can account for greater than 85% of the net cooperative binding energy (≈1.2 kcal/mol). Interestingly, the presence of NFAT converts ARRE2 into a “superconsensus site,” one that binds AP-1 even more tightly (by 0.6 kcal/mol) than an isolated ideal AP-1 site. This enhanced affinity concentrates AP-1 on the IL-2 enhancer under conditions of T-cell activation.
Acknowledgments
We are grateful to S. Harrison, G. Crabtree, A. Rao, R. Brent, and M. Ptashne for clones and reagents; and to X. Zhang, L. Gaudreau, B. Cohen, and S. Saha for experimental advice. This work was supported by the Hoffmann–La Roche Institute of Chemistry and Medicine. B.R.P. was supported by the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation Fellowship (DRG-1337).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: IL-2, interleukin 2; ARRE2, antigen receptor response element; RHR, Rel homology region; EMSA, electrophoretic mobility-shift assay; bZIP, basic region–leucine zipper.
References
- 1.Crabtree G R, Clipstone N A. Annu Rev Biochem. 1994;63:1045–1083. doi: 10.1146/annurev.bi.63.070194.005145. [DOI] [PubMed] [Google Scholar]
- 2.Durand D B, Bush M R, Morgan J G, Weiss A, Crabtree G R. J Exp Med. 1987;165:395–407. doi: 10.1084/jem.165.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaw J-P, Utz P J, Durand D B, Toole J J, Emmel E A, Crabtree G R. Science. 1988;241:202–205. [PubMed] [Google Scholar]
- 4.Durand D B, Shaw J-P, Bush M R, Replogle R E, Belagaje R, Crabtree G R. Mol Cell Biol. 1988;8:1715–1724. doi: 10.1128/mcb.8.4.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rao A. Immunol Today. 1994;15:274–281. doi: 10.1016/0167-5699(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 6.Jain J, McCaffrey P G, Valge-Archer V E, Rao A. Nature (London) 1992;356:801–804. doi: 10.1038/356801a0. [DOI] [PubMed] [Google Scholar]
- 7.Flanagan W M, Corthésy B, Bram R J, Crabtree G R. Nature (London) 1991;352:803–807. doi: 10.1038/352803a0. [DOI] [PubMed] [Google Scholar]
- 8.Liu J, Farmer J D, Jr, Lane W S, Friedman J, Weissman I, Schreiber S L. Cell. 1991;66:1–9. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 9.Jain J, McCaffrey P G, Miner Z, Kerppola T K, Lambert J N, Verdine G L, Curran T, Rao A. Nature (London) 1993;365:352–353. doi: 10.1038/365352a0. [DOI] [PubMed] [Google Scholar]
- 10.McCaffrey P G, Luo C, Kerppola T K, Jain J, Badalian T M, Ho A M, Burgeon E, Lane W S, Lambert J N, Curran T, Verdine G L, Rao A, Hogan P G. Science. 1993;262:750–754. doi: 10.1126/science.8235597. [DOI] [PubMed] [Google Scholar]
- 11.Ho S N, Thomas D J, Timmerman L A, Li X, Francke U, Crabtree G R. J Biol Chem. 1995;270:19898–19907. doi: 10.1074/jbc.270.34.19898. [DOI] [PubMed] [Google Scholar]
- 12.Northrop J P, Ho S N, Chen L, Thomas D J, Timmerman L A, Nolan G P, Admon A, Crabtree G R. Nature (London) 1994;369:497–502. doi: 10.1038/369497a0. [DOI] [PubMed] [Google Scholar]
- 13.Hoey T, Sun Y-L, Williamson K, Xu X. Immunity. 1995;2:461–472. doi: 10.1016/1074-7613(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 14.Masuda E S, Naito Y, Tokumitsu H, Campbell D, Saito F, Hannum C, Arai K-I, Arai N. Mol Cell Biol. 1995;15:2697–2706. doi: 10.1128/mcb.15.5.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodge M R, Ranger A M, de La Brousse F C, Hoey T, Grusby M J, Glimcher L H. Immunity. 1996;4:397–405. doi: 10.1016/s1074-7613(00)80253-8. [DOI] [PubMed] [Google Scholar]
- 16.Xanthoudakis S, Viola J P B, Shaw K T Y, Luo C, Wallace J D, Bozza P T, Curran T, Rao A. Science. 1996;272:892–895. doi: 10.1126/science.272.5263.892. [DOI] [PubMed] [Google Scholar]
- 17.Chytil M, Verdine G L. Curr Opin Struct Biol. 1996;6:91–100. doi: 10.1016/s0959-440x(96)80100-x. [DOI] [PubMed] [Google Scholar]
- 18.Jain J, Burgeon E, Badalian T M, Hogan P G, Rao A. J Biol Chem. 1995;270:4138–4145. [PubMed] [Google Scholar]
- 19.Jain J, Miner Z, Rao A. J Immunol. 1993;151:837–848. [PubMed] [Google Scholar]
- 20.Boise L H, Petryniak B, Mao X, June C H, Wang C, Lindsten T, Bravo R, Kovary K, Leiden J M, Thompson C B. Mol Cell Biol. 1993;13:1911–1919. doi: 10.1128/mcb.13.3.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Oakley M G, Glover J N M, Jain J, Dervan P B, Hogan P G, Rao A, Verdine G L. Curr Biol. 1995;5:882–889. doi: 10.1016/s0960-9822(95)00178-3. [DOI] [PubMed] [Google Scholar]
- 22.Glover J N M, Harrison S C. Nature (London) 1994;373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- 23.Metallo S J, Schepartz A. Chem Biol. 1994;1:143–151. doi: 10.1016/1074-5521(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 24.Wang M M, Reed R R. Nature (London) 1993;364:121–126. doi: 10.1038/364121a0. [DOI] [PubMed] [Google Scholar]
- 25.Gyuris J, Golemis E, Chertkov H, Brent R. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- 26.Cunningham B C, Wells J A. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 27.Hope I A, Struhl K. Cell. 1986;46:885–894. doi: 10.1016/0092-8674(86)90070-x. [DOI] [PubMed] [Google Scholar]
- 28.L-Ausubel F M, Brent R, Kingston R, Moore D, Seidman J, Smith J A, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1994. [Google Scholar]
- 29.Iwabuchi K, Li B, Bartel P, Fields S. Oncogene. 1993;8:1693–1696. [PubMed] [Google Scholar]
- 30.West R W, Jr, Yocum R R, Ptashne M. Mol Cell Biol. 1984;4:2467–2478. doi: 10.1128/mcb.4.11.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abate C, Patel L, Rauscher F J, III, Curran T. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 32.Sarkar G, Sommer S S. BioTechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 33.Neuberg M, Schuermann M, Müller R. Oncogene. 1991;6:1325–1333. [PubMed] [Google Scholar]
- 34.Suckow M, Von Wilcken-Bergmann B, Müller-Hill B. Nucleic Acids Res. 1993;21:2081–2086. doi: 10.1093/nar/21.9.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nikolov D B, Chen H, Halay E D, Usheva A A, Hisatake K, Lee D K, Roeder R G, Burley S K. Nature (London) 1995;377:119–128. doi: 10.1038/377119a0. [DOI] [PubMed] [Google Scholar]
- 36.Geiger J H, Hahn S, Lee S, Sigler P B. Science. 1996;272:830–836. doi: 10.1126/science.272.5263.830. [DOI] [PubMed] [Google Scholar]
- 37.Li J J, Herskowitz I. Science. 1993;262:1870–1874. doi: 10.1126/science.8266075. [DOI] [PubMed] [Google Scholar]
- 38.Yaseen N R, Park J, Kerppola T, Curran T, Sharma S. Mol Cell Biol. 1994;14:6886–6895. doi: 10.1128/mcb.14.10.6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Müller C W, Rey F A, Sodeoka M, Verdine G L, Harrison S C. Nature (London) 1995;373:311–317. doi: 10.1038/373311a0. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh G, Van Duyne G D, Ghosh S, Sigler P B. Nature (London) 1995;373:303–310. doi: 10.1038/373303a0. [DOI] [PubMed] [Google Scholar]