

Abstract
Fidelity of DNA and protein synthesis is regulated by a proofreading mechanism but function of a similar mechanism during RNA synthesis has not been demonstrated. Analysis of transcriptional fidelity and its control has been hampered by the necessity to employ complex DNA templates requiring either a promoter and initiation factors or 3′-extended templates. To circumvent this difficulty, we have created an RNA–DNA dumbbell template that can be recognized as a template-primer and extended by RNA polymerase II. By employing this system, we demonstrate that RNA polymerase II can misincorporate a nucleotide and carry out template-dependent elongation at the mispaired end. The transcripts containing misincorporated residues can be cleaved by the very slow 3′ → 5′ ribonuclease activity of the RNA polymerase II, but enhancement of this activity by the elongation factor TFIIS generates RNA with a high degree of fidelity. This enhanced preferential cleavage of misincorporated transcripts suggests an important role for TFIIS in maintaining transcriptional fidelity.
Keywords: RNA–DNA dumbbell, transcriptional proofreading
Maintenance of fidelity in macromolecular information transfer is important for precise replication and differentiation of an organism. DNA is replicated with astounding precision (10−9 to 10−10 errors per base replicated); the high fidelity is achieved by a DNA polymerase capable of both base-pair discrimination and proofreading (1). The analogous question of how transcriptional fidelity might be maintained remains largely unanswered. Maintenance of fidelity during elongation is likely to be an important target for gene regulation in eukaryotes. As has been shown for Escherichia coli, the elongation/cleavage factor GreA controls the fidelity of RNA polymerase transcription (2, 3, 4). Several eukaryotic elongation factors, TFIIF (5), TFIIS (6, 7, 8, 9, 10), P-TEFb (11), and elongin SIII (12), have been shown to enhance the ability of RNA polymerase II (pol II) to produce full-length mRNA transcripts, but their role in transcriptional fidelity has not yet been explored. The recent discovery that pol II harbors an intrinsic 3′ → 5′ ribonuclease activity that can be potentiated by TFIIS (9, 10, 13, 14) led us to investigate the role of this factor in transcriptional fidelity.
Assessment of transcriptional fidelity has been impeded by the difficulties in generating functional elongation complexes halted at specific sites and in precise analysis of the RNA products. While promoter-dependent transcription by E. coli RNA polymerase of a DNA template containing λPR promoter allowed study of the fidelity of transcription and its control by elongation factor GreA, these studies are complex and require sophisticated kinetic analysis to resolve the data (2). Other templates not requiring any accessory factors such as 3′-extended DNAs and the synthetic RNA–DNA bubble duplexes can be accurately initiated and elongated by pol II but they also pose difficulties in simple biochemical analysis of the RNA products to identify misincorporation (7, 15).
In the present studies, we have designed a simplified mimic of the nucleic acid framework of an elongation complex that has a novel RNA–DNA dumbbell architecture. The dumbbell can be accurately initiated and elongated by purified pol II, without any accessory factors, by recognizing it as a template–primer complex. By single nucleotide addition, we demonstrate misincorporation of a cytidine opposite deoxyadenosine and uridine opposite deoxyguanosine. Elongation of the mispaired end by the addition of the next correct residue occurs at a considerably reduced level thereby stalling elongation. Such impediment imposed by reduced fidelity in RNA chain extension can be relived by TFIIS, which potentiates pol II cleavage of transcripts containing misincorporated residue and thus restores the transcriptional fidelity.
MATERIALS AND METHODS
RNA–DNA Dumbbells.
The RNA–DNA dumbbell oligonucleotides were chemically synthesized and deprotected on a Biosearch RNA–DNA synthesizer according to the manufacturer’s protocol. The deprotected oligonucleotides were purified by 8% polyacrylamide/8 M urea gel electrophoresis. Sequence of the RNA was confirmed by nucleotide sequence analysis.
Proteins.
Calf thymus pol II was purified as described (16) and was determined to be free from TFIIS by immunoblot analysis using rabbit anti-human TFIIS antibodies. Human recombinant His6 TFIIS was purified as described (8). T4 polynucleotide kinase was purchased from United States Biochemical and used under the conditions of the manufacturer.
Transcription.
The 5′ terminus of the RNA–DNA dumbbell oligonucleotide was labeled with [γ-32P]ATP (3000 Ci/mmol; 1 Ci = 37 GBq) and T4 polynucleotide kinase and purified by 8% polyacrylamide/8 M urea gel electrophoresis. Transcription reactions were performed as described (14) except that the dC-tailed template was replaced by dumbbell oligonucleotide (4 nM in 12.5 μl of buffer). Pol II (4–10 nM) was added and the mixture was either preincubated for 5 min at 30°C or used directly. The reaction was initiated by addition of indicated 200 μM NTP or an indicated combination of NTPs. His6 TFIIS, which lacks ribonuclease activity (17), was added in saturating amounts equivalent to 40–60 nM. The reaction mixture was incubated for the indicated time at 37°C. The reaction products were analyzed by 10% polyacrylamide/8 M urea gel electrophoresis.
RNA Cleavage.
A typical RNA cleavage reaction (12.5 μl) contained 25 mM Hepes (potassium salt) at pH 7.9, 0.5 mM EDTA, 0.5 mM DTT, 5 mM spermidine, 100 mM potassium glutamate, 10% glycerol, 4 nM 5′-labeled dumbbell oligonucleotide, and 4–10 nM pol II. The mixture was incubated for 5 min at 30°C; 6.25 mM MgCl2 (final) was added with and without His6 TFIIS (40–60 nM) and incubated at 37°C for the indicated times. The reaction products were analyzed by 10% polyacrylamide/8 M urea gel electrophoresis.
RESULTS AND DISCUSSION
The detailed rationale for the design of an RNA–DNA dumbbell as a mimic of the nucleic acid framework in an elongation complex will be presented elsewhere. Briefly, in a DNA dumbbell oligonucleotide, a stretch of 2–4 deoxynucleotides were replaced by ribonucleotides and linked to the 3′ end of the DNA (Fig. 1A). The 3′ end of the RNA segment and the unpaired deoxynucleotide at the middle of the dumbbell serve as the primer site and NTP-binding site, respectively, thus mimicking the RNA–DNA bubble duplex associated with the pol II elongation complex (15, 18). A 41-nt sequence was derived from a group of extensively studied DNA dumbbells (19). The stretch of four thymidines at the middle of the dumbbell were substituted by uridines to generate an RNA–DNA oligonucleotide of dumbbell architecture (Fig. 1A). The chimeric oligonucleotides were chemically synthesized and shown to fold into the predicted dumbbell structure by chemical and enzymatic methods (data not shown).
Figure 1.
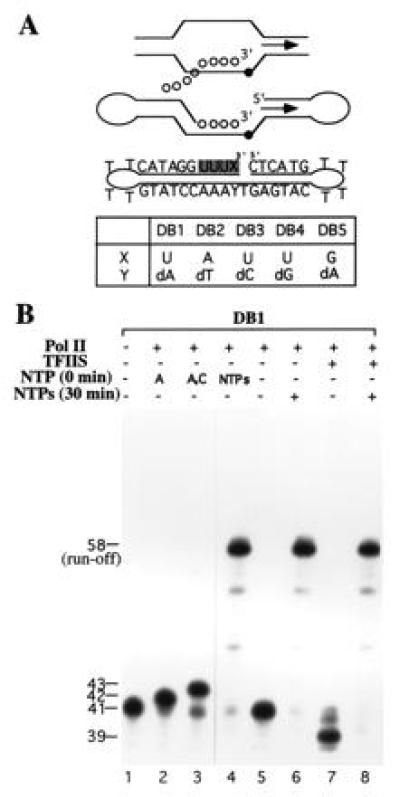
Architecture and transcription of RNA–DNA dumbbell by RNA pol II with and without TFIIS. (A) Schematics of the nucleic acid framework in the elongation bubble and its minimalistic mimic dumbbell. Arrows indicate transcription direction, and solid and open circles represent deoxy- and ribonucleotides, respectively. In the sequence of the dumbbell presented, the ribonucleotides are shaded. Several dumbbells in which the nature of the 3′-terminal ribonucleotide X and deoxynucleotide Y were varied are shown. (B) Elongation and cleavage of DB1 by pol II with and without TFIIS. RNA synthesis was carried out by pol II without preincubation (0 min) or after preincubation (30 min) with DB1 followed by addition of indicated NTP. The template-dependent elongation of the 41-nt dumbbell DB1, in the presence of four NTPs and pol II generates a run-off 58-nt RNA–DNA duplex. The ribonuclease activity of pol II cleaves DB1 into 39-nt dumbbell. In the RNA cleavage reaction, NTPs were omitted.
The RNA–DNA dumbbell template is efficiently utilized by pol II in transcript elongation and in TFIIS-potentiated RNA hydrolytic activity (Fig. 1B). The addition of ATP, ATP and CTP, or all four NTPs resulted in 42-, 43-, and 58-nt RNA–DNA hybrids, respectively, demonstrating the elongation competence of the DB1 dumbbell (Fig. 1B, lanes 2–4). The highly efficient elongation of DB1 (>90%) in the presence of all NTPs relative to the 3′-extended DNAs (10–15%, ref. 7) and the synthetic RNA–DNA bubble duplexes (<50%, figures 3 and 4 in ref. 15) demonstrates that dumbbells are superior templates for investigation of elongation. In the absence of elongation, excision of the terminal dinucleotide by a very slow pol II 3′ → 5′ ribonuclease activity (see Fig. 3, DB1) and its enhancement by TFIIS (Fig. 1B, lane 7) is observed, as has been seen previously using conventional stalled elongation complexes (10). The efficient RNA cleavage (>90%, Fig. 1B, lane 7) relative to previously employed templates (20–30%, refs. 9, 10, and 14) demonstrates that dumbbells are also effective templates for the study of 3′ → 5′ ribonuclease activity. Since dumbbells can be readily synthesized and manipulated, they offer significant advantages over commonly used templates in the investigation of elongation.
Figure 3.

Hydrolysis of mispaired RNA–DNA 3′ ends by RNA pol II with and without TFIIS. The nucleotide sequences of the paired and mispaired dumbbells is given in Fig. 1A and their abbreviated identification is presented on the left. The 5′-labeled dumbbells were incubated with pol II, with and without TFIIS for the indicated times. All cleavage reactions were carried out under identical conditions. The DB2 reaction contained 5% of 40-nt product that could not be removed by two gel electrophoresis purifications. Production of a band corresponding to the 38-nt product demonstrates the presence of the 40-nt product. For quantitation, the radioactivity in each gel band was measured by phosphorimaging (Fuji). The percent of cleavage by pol II in 60 min and pol II with TFIIS in 30 sec and 3 min is presented as a histogram.
The dumbbell can serve as an effective template in the investigation of transcriptional fidelity by employing the single-nucleotide addition strategy. To determine the misincorporation of a given nucleotide and extension of the misincorporated product, the elongation region of the dumbbell was specifically designed. A 39-nt dumbbell containing a terminal diribonucleotide and a gap of 3 nt across from the dGdAdG sequence was used as a template to assess misincorporation (Fig. 2). The diribonucleotide primer site itself is resistant to ribonuclease activity but is rendered hydrolyzable upon elongation; thus this feature of the dumbbell offers a sensitive assay for monitoring misincorporation. The failure to elongate the dumbbell and to generate the TFIIS-stimulated hydrolyzed product when adenosine or guanosine was added indicated that purine–purine mismatches did not allow elongation by pol II (Fig. 2A, lanes 2–5). However, pyrimidine–purine mismatches are produced, leading to a relatively low level of further elongation. Elongation by pol II with UTP produced 40- and 41-nt products (Fig. 2B, lane 3); an incorporation pattern similar to the E. coli RNA polymerase (lane 2). The misincorporation of uridine opposite deoxyguanosine was demonstrated by nucleotide sequence analysis of the 41-nt products from each of the polymerases (Fig. 2C Center and Right) whereas incorporation of the correct nucleotide cytidine at this position was shown by similar analysis of the 45-nt product prepared by using UTP, CTP, and GTP (Fig. 2C Left). Unexpectedly, a product, marked with a star in Fig. 2B, was also produced by both E. coli RNA polymerase and pol II (lanes 2 and 3). Nucleotide sequence analysis of this product was consistent with the incorporation of uridine. The distinct electrophoretic mobility of the star product suggests incorporation of a modified uridine residue, however further characterization of this minor product will be needed to verify this.
Figure 2.

Fidelity of RNA pol II transcription with and without TFIIS. Shown at the top is the nucleotide sequence of the dumbbell in which the 3′-terminal ribonucleotides are shaded. The mispaired nucleotides are shown by open letters. (A) RNA pol II misincorporation by a single nucleotide addition. The 5′-labeled dumbbell, pol II, and each of the indicated NTP (0.2 mM) was incubated as described in Fig. 1 with and without saturation amounts of His6 TFIIS. (B) Analysis of uridine incorporation by pol II and E. coli RNA polymerase. Transcription reactions were carried out as in A. The product with unusual electrophoretic mobility is marked by an asterisk. E.C. refers to E. coli RNA polymerase. (C) Characterization of the 41-nt products from B by RNA sequence analysis. Each of the 41-nt bands from lanes 2 and 3 was isolated and subjected to enzymatic sequencing. As a control, the sequence of the dumbbell elongated to 45 nt by pol II and 0.2 mM UTP, 0.2 mM ATP, and 0.2 mM CTP (Fig. 4B, lane 5) was determined and is shown on the left. A nucleotide ladder produced by partial hydrolysis of 5′-labeled 45-nt product by NaOH is shown on the left. In each case, the nature of the 40th nucleotide is indicated by an arrow.
Similar analysis on the addition of cytidine produced 40-, 41-, and 42-nt products (Fig. 2A, lane 6). The nucleotide sequence analysis showed that 40- and 41-nt products were produced by the expected incorporation of cytidine opposite deoxyguanosine and the generation of 41-nt product was due to misincorporation of cytidine opposite deoxyadenosine (data not shown). Furthermore, it is unlikely that CTP that may have been contaminated by UTP allowed incorporation of uridine opposite deoxyadenosine because nucleotide sequence identified cytidine at this position and such an addition would have been expected to produce a major correctly paired 42-nt product instead of the observed relatively minor product (lane 6). Thus, pol II can misincorporate uridine opposite deoxyguanosine and cytidine opposite deoxyadenosine in elongation reactions involving single-nucleotide additions. The increased level of misincorporation of cytidine opposite deoxyadenosine relative to uridine opposite deoxyguanosine may reflect the mechanism by which pol II discriminates base pairs. Similar misincorporation of uridine opposite deoxyguanosine by E. coli RNA polymerase, as shown previously (2) and by this study, suggests a mechanistic similarity in the elongation activities of the two polymerases.
The nucleotide misincorporation by pol II impedes further elongation and thus generates stalled complexes. Could TFIIS-potentiated pol II ribonuclease activity recognize misincorporated transcripts in the stalled complex and selectively excise misincorporated residue to relieve the elongation block? The misincorporated uridine from the 40-, 41-, and 42-nt products was removed in dinucleotide increments by TFIIS to generate an original 39-nt and a new 38-nt product (Fig. 2B, lane 3 versus 4). Similarly, misincorporated cytidine from the 40-, 41-, and 42-nt products was removed as a dinucleotide to generate 38-, 39-, and 40-nt dumbbells. Of these hydrolytic products, the 39-nt DB1 template is efficiently elongated by addition of the correct residue cytidine to a 40-nt product, so the 39 oligonucleotide does not accumulate whereas the 38 nt produced from 42 and 40 nt is not elongated because of the absence of ATP (Fig. 2A, lane 7 versus 6). Because of the competing ribonuclease and elongation activities produced identical length oligonucleotides, the significance of TFIIS-stimulated hydrolytic activity can be arguably demonstrated by production of 38-oligonucleotide from 42- and 40-nt products (lane 7 versus 6). Thus, TFIIS stimulates pol II diribonuclease activity, which preferentially removes misincorporated nucleotides. This finding is reminiscent of GreA stimulating E. coli RNA polymerase ribonuclease activity to prevent misincorporation (2).
Since both TFIIS-stimulated pol II ribonuclease activity and the 3′ → 5′ exonuclease activity of DNA polymerase (1) can recognize and excise correctly and incorrectly paired nucleotides, the mechanism of ribonuclease function may also involve discrimination between relative degree of pairing between the terminal nucleotides (i.e., preference for weakly paired over strongly paired residues). This idea was tested by investigation of several dumbbells representing a variety of paired and mispaired ends for preferential trend in nucleotide excision (Fig. 1A). The dinucleotide removal by pol II was 3- to 6-fold faster for the mispaired ends, U·dG, G·dA, and U·dC, relative to the paired A·dT end (Fig. 3, compare DB3, DB4, and DB5 with DB2). Within the paired ends, the U·dA pair was hydrolyzed slightly faster (2-fold) than the A·dT, whereas the C·dG pair was not hydrolyzed. This slow ribonuclease activity of pol II was enhanced 104-fold by TFIIS, as estimated from the kinetic analysis of the A·dT pair (DB2). Similar analysis showed that the U·dA pair was hydrolyzed 2-fold faster than the A·dT, the mispaired ends were hydrolyzed 4- to 7-fold faster than the A·dT, and the C·dG pair was not hydrolyzed (data not shown). These preferential effects among paired and mispaired ends, with the exception of C·dG pair, while small, are consistent and highly reproducible (A. Sitikov, personal communication). The cleavage of relatively weakly paired U·dA over A·dT pair (20) and no cleavage of stably paired C·dG suggests that within the paired ends, the weakly paired are hydrolyzed preferentially over the strongly paired ends. While all possible combinations of mispaired ends have not been described herein, a trend in cleavage preference of dinucleotides emerges within this group: U·dC > U·dG > G·dA > U·dA > A·dT > C·dG. Similar cleavage preference based upon relative pairing of bases is exhibited by the 3′ → 5′ exonuclease of DNA polymerase; dA·dT pairs are cleaved preferentially over dC·dG pairs (1). Thus in a stalled elongation complex, the relative weakness in 3′ ribonucleotide pairing in the RNA–DNA hybrid may be a determinant of stimulation of pol II ribonuclease activity that is further potentiated by TFIIS. This leads to preferential excision of mispaired and weakly paired nucleotides.
While the single nucleotide addition strategy revealed limited pol II fidelity and TFIIS-stimulated cleavage of the misincorporated nucleotide, a similar analysis with all four NTPs may determine what happens in vivo. The elongation analysis with four NTPs may not be technically feasible because the misincorporation step is exceedingly slower than the extension from a correctly incorporated nucleotide residue and thus poses difficulty in identification of a misincorporated residue and its fate in the presence of TFIIS. However, this difficulty can be circumvented by employing templates designed with built-in site-specific mispaired ends, such as the templates DB3, DB4, and DB5 that were useful in pol II ribonuclease activity analysis. Elongation by pol II of the terminal G·dA mispaired DB5 dumbbell was compared with the correctly paired U·dA DB1 (Fig. 1A). Both DB5 and DB1 dumbbells produced a major full-length 58-nt run-off product but the efficiency of elongation of DB5 was half as that of DB1 (40% for DB5 versus 98% for DB1, Fig. 4A, lane 2 versus Fig. 1B, lane 4). The preincubation of DB5 with pol II increases the amount of 58-nt run-off product (40% versus 52%, Fig. 4A, lane 2 versus 4). A straightforward interpretation of this result is that mispaired ends are poorly extended relative to the correctly paired ends but the level of mispaired ends decreases on preincubation with pol II in the absence of nucleotides, which allows the excision of mispaired residue and the newly generated paired ends are then efficiently elongated. This idea is supported by the observation that with TFIIS, pol II elongates DB5 to 58-nt run-off product as efficiently as DB1 (Fig. 4A, lane 6 versus Fig. 1B, lane 4).
Figure 4.
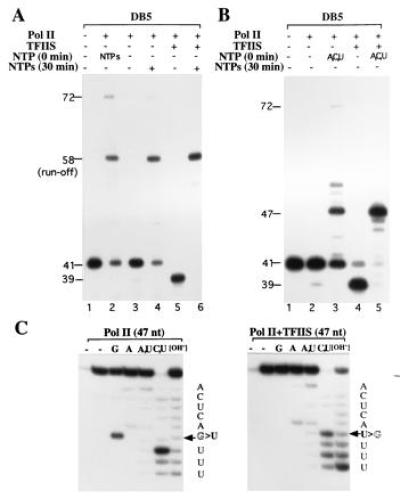
RNA pol II elongation at a mispaired 3′ end with and without TFIIS. (A) Elongation of the G·dA mispaired dumbbell DB5. The 5′-labeled DB5 was elongated by pol II in the absence of preincubation as indicated. Each of the four NTPs at 0.2 mM was used. For cleavage reactions, NTPs were omitted. His6 TFIIS was included as indicated. The unexpected 72-nt product is marked. (B) Same as A except that the mixture of ATP, CTP, and UTP was used instead of all four NTPs. (C) Characterization of the 47-nt bands from B by RNA sequence analysis. The 47-nt bands from lanes 3 and 5 were isolated and their RNA sequence was determined by enzymatic methods. The nucleotide sequence is presented on the right and the relative band intensity of the paired and mispaired nucleotides is indicated.
To determine whether the 58-nt run-off products contained mispaired G and/or correctly paired uridine, they were analyzed by sequencing. Numerous attempts to sequence this product were unsuccessful, perhaps due to difficulty in denaturation of long RNA–DNA hybrids. Shorter RNA–DNA hybrids of 47 nt were prepared by limited elongation of DB5 by pol II with and without TFIIS in the presence of ATP, CTP, and UTP, as expected for elongation up to the first template dC residue (Fig. 4B, lane 3). The 47-nt products generated by pol II (Fig. 4B, lane 3) and by pol II with TFIIS (lane 5) were analyzed by sequencing. The data show that pol II can elongate from G·dA mismatch because G was still present in the elongated transcript (Fig. 4C Left) whereas, pol II with TFIIS, elongation of only correctly paired ends was inferred, based on the presence of uridine at this position (Fig. 4C Right).
Unexpectedly, however, a minor 72-nt product (4%) was also produced from DB5 but not from DB1 (Fig. 4A, lane 2 versus Fig. 1B, lane 4). That the synthesis of 72-nt product appears to be specific for mispaired ends is supported by the observation that TFIIS stimulation of pol II ribonuclease activity prevents synthesis of this product (Fig. 4A, lanes 5 and 6). Further studies would be required to understand the mechanism of synthesis of a discrete-sized 72-nt product.
Our data on RNA–DNA dumbbell templates may impact on the long-standing debate on the architecture of the nucleic acid framework in the catalytic zone of the elongating RNA polymerase. Currently, two models are being debated: One model proposes a 12-nt RNA–DNA hybrid at the 3′ terminus (model 1, ref. 21) and the other proposes a 2- to 3-nt RNA–DNA hybrid in the same region (model 2, ref. 22). Experiments showing effective elongation of a dumbbell with only 2 ribonucleotides at the 3′ terminus (Fig. 2), cleavage of this dumbbell when elongated to 3 ribonucleotides (Fig. 2, lane 7) and cleavage of the 4 ribonucleotide dumbbell (Fig. 3) tend to support model 2 since as few as 2 or 3 ribonucleotides are sufficient for transcript elongation and cleavage by polymerase. The other observation of this work is that the proposed second RNA binding site located 10 ribonucleotides from the 3′ terminus (model 2) does not have to be composed of ribonucleotides; deoxyribonucleotides can be effective substitutes.
The potential for reduced transcription fidelity and its restoration by TFIIS may have important implications on our understanding of the mechanism of elongation and termination. The poor elongation rate of mismatched end implies that reduced level of transcription fidelity could impede template-dependent elongation by forming ternary and binary complexes. The relatively weaker binary complexes may lead to transcription termination (23). It is tempting to speculate that TFIIS may prevent formation of a binary complex by generating a 2- to 3-nt RNA–DNA duplex in the catalytic zone and thus preventing termination.
Regulation of pol II-like transcription fidelity by ribonuclease-enhancing elongation factors appears to be ubiquitous in that it has been observed from E. coli to yeast to mammals. In other transcriptional systems, the fidelity may be controlled by highly potent intrinsic ribonuclease activities. The intrinsic ribonuclease activities of vaccinia virus RNA polymerase (24) and eukaryotic RNA polymerase III (25) may function in this way, independent of factors.
In summary, we have demonstrated that synthetic RNA–DNA dumbbell constructs are versatile and effective templates for the investigation of transcription elongation. The ease with which dumbbells of any desired sequence can be prepared and with which their products can be analyzed by high-resolution methods makes them particularly attractive. From transcriptional analysis using dumbbell templates, it has been possible to demonstrate that the polymerase can misincorporate and elongate from a 3′ mismatched end. The pol II elongation from the mispaired end can be prevented by TFIIS, which enhances preferential removal of 3′ nucleotides from the mismatched transcripts by a factor of 4 × 104. Thus, TFIIS may control the fidelity of pol II transcription by a mechanism analogous to that of GreA in E. coli RNA polymerase-mediated transcription.
Acknowledgments
This work is dedicated to Professor H. Gobind Khorana on his 75th birthday. We thank L. Labeots for oligonucleotides; H. S. Yoon for recombinant TFIIS; A. Sitikov, E. Davydova, and T. Martin for discussions; M. Hochstrasser, T. Martin, T. Pan, M. Rosner, and U. Storb for critical comments on the manuscript; and J. Booker for preparation of the manuscript. This research was generously supported by the National Institute of Health Grant DK21901.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviation: pol II, RNA polymerase II.
References
- 1.Johnson K A. Annu Rev Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 2.Erie D A, Hajiseyedjavadi O, Young M C, von Hippel P H. Science. 1993;262:867–873. doi: 10.1126/science.8235608. [DOI] [PubMed] [Google Scholar]
- 3.Borukhov S, Sagitov V, Goldfarb A. Cell. 1993;72:459–466. doi: 10.1016/0092-8674(93)90121-6. [DOI] [PubMed] [Google Scholar]
- 4.Orlova M, Newlands J, Das A, Goldfarb A, Borukhov S. Proc Natl Acad Sci USA. 1995;92:4596–4600. doi: 10.1073/pnas.92.10.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bengal E, Flores O, Reinberg D, Aloni Y. Mol Cell Biol. 1991;11:1195–1206. doi: 10.1128/mcb.11.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kassavetis G A, Geiduschek E P. Science. 1993;259:944–945. doi: 10.1126/science.7679800. [DOI] [PubMed] [Google Scholar]
- 7.Kerppola T K, Kane C M. Mol Cell Biol. 1988;8:4389–4394. doi: 10.1128/mcb.8.10.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agarwal K, Baek K, Jeon C, Miyamoto K, Ueno A, Yoon H. Biochemistry. 1991;30:7842–7851. doi: 10.1021/bi00245a026. [DOI] [PubMed] [Google Scholar]
- 9.Reines D. J Biol Chem. 1992;267:3795–3800. [PMC free article] [PubMed] [Google Scholar]
- 10.Izban M G, Luse D S. J Biol Chem. 1993;268:12874–12885. [PubMed] [Google Scholar]
- 11.Marshall N F, Price D H. J Biol Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 12.Garret K P, Tan S, Bradsher J N, Lane W S, Conway J W, Conway R C. Proc Natl Acad Sci USA. 1994;91:5237–5241. doi: 10.1073/pnas.91.12.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Hawley D K. Proc Natl Acad Sci USA. 1993;90:843–847. doi: 10.1073/pnas.90.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeon C, Yoon H, Agarwal K. Proc Natl Acad Sci USA. 1994;91:9106–9110. doi: 10.1073/pnas.91.19.9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daube S S, von Hippel P H. Science. 1992;258:1320–1324. doi: 10.1126/science.1280856. [DOI] [PubMed] [Google Scholar]
- 16.Hodo H G, Blatti S P. Biochemistry. 1977;16:2334–2343. doi: 10.1021/bi00630a005. [DOI] [PubMed] [Google Scholar]
- 17.Jeon, C. J. (1993) Ph. D. thesis (University of Chicago, Chicago), p. 99.
- 18.Rice G, Chamberlin M J, Kane C M. Nucleic Acids Res. 1993;21:113–118. doi: 10.1093/nar/21.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doktycz M J, Goldstein R F, Paner T M, Gallo F J, Benight A S. Biopolymers. 1992;32:849–864. doi: 10.1002/bip.360320712. [DOI] [PubMed] [Google Scholar]
- 20.Martin F H, Tinoco I., Jr Nucleic Acids Res. 1980;8:2295–2299. doi: 10.1093/nar/8.10.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yager T D, von Hippel P H. Biochemistry. 1991;30:1097–1118. doi: 10.1021/bi00218a032. [DOI] [PubMed] [Google Scholar]
- 22.Chamberlin M. Harvey Lect. 1995;88:1–21. [PubMed] [Google Scholar]
- 23.Johnson T L, Chamberlin M J. Cell. 1994;77:217–224. doi: 10.1016/0092-8674(94)90314-x. [DOI] [PubMed] [Google Scholar]
- 24.Hagler J, Shuman S. J Biol Chem. 1993;268:2166–2173. [PubMed] [Google Scholar]
- 25.Whitehall S K, Bardeleben C, Kassavetis G A. J Biol Chem. 1994;269:2299–2306. [PubMed] [Google Scholar]