

Abstract
About one-third of breast cancers express a functional estrogen (β-estradiol [E2]) receptor (ER) and are initially dependent on E2 for growth and survival but eventually progress to hormone independence. We show here that ER+, E2-independent MCF-7/LCC1 cells derived from E2-dependent MCF-7 cells contain elevated basal NF-κB activity and elevated expression of the transcriptional coactivator Bcl-3 compared with the parental MCF-7 line. LCC1 NF-κB activity consists primarily of p50 dimers, although low levels of a p65/p50 complex are also present. The ER− breast cancer cell lines harbor abundant levels of both NF-κB complexes. In contrast, nuclear extracts from MCF-7 cells contain a significantly lower level of p50 and p65 than do LCC1 cells. Estrogen withdrawal increases both NF-κB DNA binding activity and expression of Bcl-3 in MCF-7 and LCC1 cells in vitro and in vivo. Tumors derived from MCF-7 cells ectopically expressing Bcl-3 remain E2 dependent but display a markedly higher tumor establishment and growth rate compared to controls. Expression of a stable form of IκBα in LCC1 cells severely reduced nuclear expression of p65 and the p65/p50 DNA binding heterodimer. Whereas LCC1 tumors in nude mice were stable or grew, LCC1(IκBα) tumors regressed after E2 withdrawal. Thus, both p50/Bcl-3- and p65/p50-associated NF-κB activities are activated early in progression and serve differential roles in growth and hormone independence, respectively. We propose that E2 withdrawal may initiate selection for hormone independence in breast cancer cells by activation of NF-κB and Bcl-3, which could then supplant E2 by providing both survival and growth signals.
About 60% of all diagnosed breast cancers express estrogen receptors (ERs), and about half of these are dependent on estrogen for growth and are initially responsive to endocrine therapy (15, 25, 48). These tumors eventually acquire resistance to hormonal manipulation as part of their progression toward a more malignant phenotype, and in many instances they cease to express ERs or express mutant forms of the ER (33, 34). The MCF-7 line is a widely used prototype for estrogen-dependent breast cancer. These cells form tumors in nude mice in the presence of circulating β-estradiol (E2), and the tumors regress rapidly through an apoptotic mechanism (21) when the source of E2 is removed (29, 44). In order to study the progression of breast cancer toward a hormone-independent phenotype, sublines derived from MCF-7 cells cultured in vivo and in vitro in the presence of subphysiological concentrations of estrogen have been isolated (13, 14). MCF-7/MIII cells were isolated from a small, slowly proliferating MCF-7 tumor that arose in an ovariectomized athymic mouse, and a second passage produced MCF-7/LCC1 cells, which form E2-independent tumors with a significantly reduced latency. Both cell lines retain the parental MCF-7 level of expression of the ER but display increased expression of some estrogen-regulated genes with a concomitant loss of E2 responsiveness in vitro. Although LCC1 cells can efficiently generate tumors in nude mice in the absence of estrogen, they grow more rapidly when estrogen is present and therefore retain a degree of estrogen responsiveness in vivo (9).
The transcription factor NF-κB is composed of a heterodimer of members of the Rel family of transcription factors, including p50 (NF-κB1), p65(RelA), c-Rel, RelB, and p52(NF-κB2). Transactivation domains are absent in p50 and p52, and thus they are active only as heterodimers with other members. This family of proteins contains Rel homology domains which mediate DNA binding, dimerization, and nuclear localization. Activation of NF-κB occurs following a wide variety of stimuli, including exposure to some cytokines and several kinds of stress. Inactive NF-κB is maintained in the cytoplasm as a result of interaction with an inhibitory subunit, IκB (4), of which there are four subtypes, α,β, γ, and ɛ (31). NF-κB activation follows phosphorylation of IκB by IκB kinases (α or β), which in turn are activated by an NF-κB-inducing kinase called NIK (24, 38). IκB phosphorylation results in its degradation and subsequent release, allowing NF-κB translocation to the nucleus, where it regulates a large number of genes involved in inflammation, immunity, cell adhesion, and apoptosis-regulatory molecules (2). Another member of the IκB family is the oncoprotein Bcl-3, which can disrupt the association between transcriptionally inactive p50 and p52 homodimers, allowing association of a transactivating partner. Bcl-3 can also directly activate transcriptional function in these complexes (reference 31 and references therein). Much of the information regarding the role of NF-κB in cell survival has come from the study of tumor necrosis factor alpha signaling in tumor cells. While the tumor necrosis factor alpha receptor activates a caspase cascade leading to apoptosis, in most cells a concomitant activation of NF-κB prevents cell death (3, 5). These observations led to the discovery that NF-κB regulates the activity of several survival genes, including genes for Bcl-x and inhibitors of apoptosis (IAPs) (reviewed in reference 42).
There is abundant evidence that NF-κB can promote tumorigenesis (32, 46). One of the earliest reports showed that antisense downregulation of the p65 subunit of NF-κB in fibrosarcoma cells could both inhibit tumorigenicity and cause tumor regression (22). More recently, inhibition of NF-κB activity by stable expression of a dominant negative inhibitory IκB kinase in mouse mammary tumor cells reduced their tumorigenic potential (7). Resistance to chemotherapeutic drugs is also impaired by NF-κB inhibition in tumor cells (32, 54). Studies have shown that breast cancer cell lines expressing the ER contain low levels of NF-κB DNA binding activity, while ER− breast cancer cells display constitutively high levels of NF-κB DNA binding and correspondingly high NF-κB transactivational activity (36, 50). NF-κB activity is also induced in rat mammary glands after treatment with carcinogens and appears to increase prior to malignant transformation of mammary epithelial cells (28).
In this study we have used the ER-positive, hormone-independent LCC1 and MCF-7 parental cells to determine if and at which stage during serial breast cancer cell progression toward hormone independence these cells begin to acquire elevated NF-κB activity. Using a tumor xenograft model, we show that (i) expression of the Bcl-3 protein in MCF-7 cells augments tumor establishment and growth but is insufficient to confer E2-independence and (ii) inhibition of p65-associated NF-κB activity with a dominant form of the NF-κB inhibitor IκBα reverts the E2-independent phenotype of LCC1 cells.
MATERIALS AND METHODS
Plasmids and antibodies.
Anti-p65 (A), anti-p50 (H-119), anti-p52 (K-27), anti-c-Rel (N), anti-Bax (N-20), and anti-Bcl-3 were obtained from Santa Cruz Biotechnology. Anti-Bcl-2 and anti-Bcl-xL were gifts from John Reed, La Jolla, Calif. Anti-FLAG M2 monoclonal antibody and anti-α-actin polyclonal antibody were purchased from Sigma. The 3X-NF-κB3-luciferase construct containing three copies of the NF-κB response element from the major histocompatibility complex class I gene and a mutated version of this element (19), pRC/CMV-FLAG-tagged IκBα S32A/S36A, the superrepressor form of IκBα (IκBαSR) (27), and CMV-2-FLAG-Bcl-3 were provided by A. S. Baldwin. Initial studies utilized the major histocompatibility complex reporter gene, and the κ light-chain reporter gene was obtained later since it produced higher overall enzyme values after transfection.
Cell culture and transfection.
MCF-7 cells were derived from several isolates. MCF-7(early) and MCF-7(late) cells are uncloned isolates of MCF-7 at controlled passages of 46 to 48 and 157 to 159, respectively. MCF-7/MIII and MCF-7/LCC1 are ER+, E2-independent cell lines. MIII cells were isolated from a slow-growing tumor resulting from inoculation of parental MCF-7 cells into an ovariectomized nude mouse. MIII cells were further passaged in ovariectomized nude mice and then reestablished in vitro as the continuous line MCF-7/LCC1, which forms tumors in ovariectomized mice with reduced latency (9). Both sublines were passaged fewer than 30 times after isolation. Other MCF-7 cells, not designated early or late passage, were obtained originally from L. Murphy (Winnipeg, Canada) and are of undetermined passage. MDA-MB-231 (ER−), MDA-MB-468 (ER−) and T47-D (ER+) cells were obtained from the American Type Culture Collection (Manassas, Va.). The tumorigenic characteristics of many of these breast cancer cell lines have been documented (45, 49). MCF-7(40F) is an MCF-7 derivative selected for resistance to adriamycin that is E2 independent and ER− (20). All MCF-7 derived cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (GIBCO-BRL) containing a high glucose concentration, 5% fetal bovine serum (GIBCO-BRL), and 2 μg of gentamicin sulfate per ml. T47-D, MDA-MB-468, and MDA-MB-231 cells were maintained in DMEM containing 5% serum and a low glucose concentration. SKBR-3 cells were maintained in McCoy's 5A medium with 10% fetal bovine serum. Incubation was at 37°C in a 5% CO2 humidified environment. In experiments requiring E2 depletion, cells were precultured for 7 days with several changes of phenol red-free DMEM containing 5% steroid-free fetal bovine serum that had been adsorbed to dextran-coated charcoal for 45 min at 45°C. E2 was added from a 1 mM stock in ethanol to a final concentration of 10−8 M for the indicated times. Transfections were performed with Lipofectamine according to the directions of the manufacturer (GIBCO-BRL). For stable transfection, pcDNA3-IκBαSR-FLAG or CMV-FLAG-Bcl-3 was introduced into MCF-7/LCC1 cells, and clones were selected in medium containing 50 μg of G418 per ml as previously described (52). Resistant clones were picked and expanded, and then lysates were subjected to immunoblot analysis with the anti-FLAG antibody. For transient cotransfections, expression-reporter constructs and pcDNA3-LacZ were introduced by using Superfect or Lipofectamine according to the manufacturer's directions. Cell extracts were harvested 48 h later and analyzed in a BioOrbit 1250 luminometer by using luciferase assay reagent (Promega). Reported values represent means ± standard errors (SE) from duplicate or triplicate experiments, normalized to LacZ activity determined by methylumbelliferyl-β-glucuronide assay (1), and are representative of those from at least three separate experiments.
EMSA analysis.
Electrophoretic mobility shift assays (EMSA) were performed with nuclear extracts from cultured cells or from tumors isolated as described by Osborn et al. (41). NF-κB site oligonucleotides were obtained from Promega (E3291) and end labeled with T4 polynucleotide kinase by using [γ-32P]ATP (Amersham). Five micrograms of nuclear extract was mixed with 5 μl of DNA binding buffer (20 mM HEPES [pH 7.9], 0.2 mM EDTA, 0.2 mM EGTA, and 2 mM dithiothreitol in 50% glycerol), 5 μg of poly(dI-dC), and 0.2 ng of labeled probe in a final volume of 20 μl and then incubated at room temperature for 25 min. Specific bands were verified with a 10 to 125 M excess of cold oligonucleotide 10 min prior to addition of the labeled probe, and equivalence of extract loading was demonstrated by EMSA with a DNA fragment containing the consensus Sp1 binding site (Promega). Samples were loaded on a 5% native polyacrylamide gel and run in nondenaturing Tris-glycine buffer. For supershift experiments, 2 μg of each antibody was added to extracts and left for 1 h prior to addition of the labeled probe.
Tumors in nude mice.
Six-week-old ovariectomized nude mice (nu/nu CD-1) were implanted subcutaneously with an estrogen release pellet (60-day-release pellet containing 0.72 mg of E2; Innovative Research of America, Sarasota, Fla.). Two days later, 2 × 106 cells derived from exponential cultures of wild-type MCF-7 cells were injected subcutaneously into the flank of the animal. For LCC1 experiments, three pooled clones of either LCC1(IκBαSR) or LCC1(pcDNA3) were injected subcutaneously into the contralateral flanks of 12 animals. Similarly, cell suspensions containing three pooled MCF-7(FLAG) or MCF-7(FLAG-Bcl-3) clones were injected subcutaneously into the contralateral flanks of 12 mice, while another 3 mice received only MCF-7(FLAG-Bcl-3) in one flank only. Tumors were allowed to form over a period of 4 to 6 weeks, and volumes were determined by caliper measurements as previously described (44). The E2 release pellet was then removed, and regression was monitored until the tumor reached 50% of its volume at pellet removal or over a time course as indicated, at which point the animal was sacrificed and the tumor was removed. Tumor protein lysates were prepared by snap freezing followed by pulverization under liquid N2. After the addition of radioimmunoprecipitation assay buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 0.1% sodium dodecyl sulfate [SDS], 0.5% sodium deoxycholate, 1% NP-40, 10 μg of phenylmethylsulfonyl fluoride per ml, 1 μg of aprotinin per ml, and 0.02% sodium azide), samples were sonicated and then incubated for 30 min on ice before centrifugation at 16,000 × g to remove insoluble material. Protein was measured with Bio-Rad reagent.
Immunoblot analysis.
Cell monolayers were washed twice with phosphate-buffered saline and lysed in 400 μl of RIPA buffer per 107 cells for 30 min on ice. Insoluble material was removed following centrifugation at 12,000 × g for 15 min, and soluble protein concentrations were determined with a Bio-Rad kit. Proteins (20 μg) were separated on SDS-7.5 or 10% polyacrylamide gels and transferred to polyvinylidene difluoride membranes. After exposure to primary antibody, proteins were detected with peroxidase-conjugated second antibody (Sigma) and chemiluminescent substrate (Dupont, NEN).
Immunocytochemistry and ISEL.
Seven-micrometer frozen sections were cut from LCC1(pcDNA3) and LCC1(IκBαSR) tumors. For Bax immunostaining, sections were fixed in formaldehyde for 30 min and incubated with polyclonal Bax NH2 terminus antibody followed by CY3-conjugated goat anti-rabbit immunoglobulin G (Jackson Laboratories). In situ end labeling (ISEL) was performed with terminal transferase and biotin-16-dUTP (Boehringer Mannheim) followed by CY2-labeled streptavidin (Amersham) as described previously (44). Sections were visualized and imaged with a Zeiss Axiophot fluorescence microscope equipped with Northern Eclipse software (EMPIX Imaging Inc., Mississauga, Ontario, Canada).
RESULTS
LCC1 cells display elevated NF-κB DNA binding activity.
Evidence shows that ER expression in breast cancer cell lines is associated with low baseline NF-κB activity, while breast cancer cell lines devoid of ER have high constitutive levels of NF-κB activity (36). Although LCC1 cells are E2 independent they express functional ER as determined by their increased growth rate in the presence of E2 (14). Table 1 presents a summary of the cell lines and isolates used in this study. In order to determine whether NF-κB activity correlates with E2 dependence, nuclear extracts from ER+ T47-D and ER+ LCC1 cells were assayed for NF-κB DNA binding, and the NF-κB activity was compared with that in MCF-7 cells selected for resistance to adriamycin [MCF-7ADR(40F) cells]. These cells have lost expression of the ER and are able to form tumors efficiently in ovariectomized nude mice (20). Figure 1A shows the results of an EMSA which demonstrates that, as predicted from the literature on NF-κB levels in ER− breast cancer cell lines, MCF-7(40F) cells also contain high levels of constitutive NF-κB DNA binding activity associated with fast- and slow-migrating complexes. Conversely, T47-D cells contained very low levels of NF-κB DNA binding activity, as previously reported (36). In contrast, LCC1 cells displayed intermediate levels of NF-κB activity associated primarily with the faster-migrating complex. In order to directly compare NF-κB DNA binding activities of cells derived from the same isolate of MCF-7 cells, we prepared nuclear extracts from the early passage of parental MCF-7 cells, MIII cells, and LCC1 cells and contrasted these NF-κB DNA binding complexes with those from ER− MDA-MB-231 cells. The results in Fig. 1B show again that LCC1 cells had high levels of NF-κB activity compared with the other MCF-7-derived cells. Similar to MCF-7(40F) cells, MDA-MB-231 cells contain a high level of constitutive NF-κB activity associated with two different complexes. The EMSA in Fig. 1C, in which an Sp1 binding site DNA fragment was used as a probe, contained equivalent amounts of the indicated nuclear extracts used for Fig. 1A and B and demonstrates that quantitative differences were not a function of extract loading. To determine the composition of the NF-κB complexes in LCC1 cells relative to ER− cells, we supershifted nuclear extracts from LCC1 and the ER− MCF-7(40F) and MDA-MB-231 cells with antibodies against NF-κB proteins. Figure 1D shows clearly that while p50 was present in complexes from all three cell lines, the ER− lines contained markedly higher levels of the slower-migrating p65/RelA complex. The p50 antibody also supershifted the p65-associated complex, thus indicating that the upper complex is a heterodimer of p50 and p65. Thus, NF-κB subunits appear to be differentially activated in breast cancer cell lines. Despite its commercial designation for supershift analysis, we were unable to supershift any of the complexes with this p52 antibody or with an antibody obtained from the laboratory of A. S. Baldwin which reportedly was capable of supershifting p52 but only on an erratic basis, and therefore we cannot formally rule out that the lower-migrating complex also contains heterodimers of p52/p50. Note that all of the cell lines were cultured with identical serum concentrations, thereby eliminating the influence of differential contributions of serum growth factors on NF-κB activity.
TABLE 1.
Cell lines
| Cell line | Descriptiona |
|---|---|
| MCF-7 (early or late) | Human mammary adenocarcinoma; well differentiated, ER positive, p53+/+ (39), controlled passages (early, 46 to 48 passages; late, 156 to 159 passages) |
| T47-D | Human mammary ductal carcinoma, differentiated epithelial, ER positive, p53−/+ (39) |
| MIII | MCF-7 (controlled passage) inoculated into ovariectomized nude mice; slow-growing tumor isolated 6 months after inoculation and reestablished in vitro, ER-positive and E2-responsive in vivo (14) |
| LCC1 | Isolated from rapidly growing tumors derived from second inoculation of MIII cells into ovariectomized nude mice and then reestablished in vitro; ER-positive and E2-responsive in vivo, increased constitutive expression of some E2-regulated genes (9) |
| MDA-MB-231 | Human mammary adenocarcinoma; poorly differentiated, ER negative, p53−/− (39) |
| MCF-7ADR(40F) | MCF-7 cells selected for 40-fold resistance to doxorubicin (adriamycin) [wild-type MCF-7 ED50, 14.5 nM; MCF-7ADR(40F) ED50, 474 nM (20)], p53mut (39), ER negative |
ED50, 50% effective dose.
FIG. 1.

NF-κB DNA binding activity increases with acquisition of E2 independence. (A and B NF-κB complexes in 5 μg of nuclear extract from T47-D, LCC1, and MCF-7(40F) cells (A) and from MCF-7(early passage), MIII, LCC1, and MDA-MB-231 cells (B) were subjected to EMSA as described in Materials and Methods, using an oligonucleotide containing a consensus NF-κB binding site. Arrows indicate fast- and slow-migrating complexes. A 10-fold excess of cold oligonucleotide (comp) was used for competition of specific bands in duplicate MCF-7(40F) and MDA-MB-231 lanes. (C) Five micrograms of the same nuclear extracts used for panels A and B was subjected to EMSA with the consensus Sp1 enhancer element as a probe. (D) Antibody supershift analysis of NF-κB complexes. NF-κB complexes from the indicated cell lines were incubated with antibodies against NF-κB proteins prior to DNA binding as described in Materials and Methods. Arrows indicate fast- and slow-migrating complexes; arrowheads identify antibody-shifted complexes. NS, normal serum; comp, a 10-fold excess of unlabeled probe was used to compete with specific bands. (E) Results from transient transfection of MCF-7(early) and LCC1 cells with the 3X-NF-κB3-luciferase and mutant-luciferase reporter constructs. The results represent values from triplicate experiments ± SE and were normalized to β-galactosidase activity from a cotransfected pCMV-LacZ plasmid. (F) Immunoblot analysis of nuclear p65, p50, and p52 in breast cancer cell lines. Ten micrograms of each nuclear extract was reacted with either anti-p65 antibody or an anti-p50 antibody which also cross-reacts with p52.
Not all DNA-bound NF-κB is transcriptionally active (56); therefore, NF-κB transactivational activity was tested by transient transfection of an NF-κB reporter gene. The graph in Fig. 1E shows that the basal level of NF-κB-luciferase reporter gene activity in LCC1 cells was about fivefold higher than that in the E2-dependent MCF-7(early) cells. Since our supershift analysis revealed major differences between the predominant NF-κB DNA binding complexes in ER+ cells and ER− cells, we performed immunoblot analysis on nuclear extracts from these cell lines to ascertain the relative levels of these proteins. The results in Fig. 1F indicate that MCF-7(early) nuclear extracts contain only trace levels of p65, while LCC1 nuclei contain a slightly higher level. In contrast, the ER−, E2-independent MCF-7(40F) and MDA-MB-231 cells harbor much higher nuclear levels of this protein. When the same extracts were assayed for p50 immunoreactivity, significantly less p50 was present in MCF-7(early) cells than in the other cells. The LCC1 and the ER− cell lines contained similar nuclear levels of p50. Given that p50 heterodimerizes with p65 in these cells, the nuclear level of p50 would affect both the fast- and slower-migrating NF-κB complexes. The relative nuclear content of p52 detected by this antibody was low and was essentially equivalent across all cell lines. Thus, nuclear NF-κB proteins and NF-κB DNA binding complexes are both qualitatively and quantitatively different in breast cancer cell lines and isolates grown in vitro under the same culture conditions. Progression appears to correlate with increased levels of NF-κB consisting predominantly of p50 dimers as well as with a discernible increase in the formation of p65/p50 DNA binding complexes.
E2 regulates NF-κB DNA binding activity in vivo and in vitro.
Hormone-dependent breast tumors undergo regression after E2 removal by ovariectomy or following antiestrogen therapy as the result of programmed cell death of a large majority of the cells (29, 44). A subset of these cells will escape apoptosis as a result of acquisition of hormone independence and/or antiestrogen resistance. Since steroid hormones are known to modulate the levels of NF-κB activity, we tested the effects of growth in E2-free medium on the NF-κB DNA binding activity of MCF-7 and LCC1 cells. Figure 2A shows that NF-κB activity began to increase in MCF-7 cells within 4 days after the medium was replaced with E2-free medium and continued to rise over the 12-day period studied. Thus, NF-κB activity in ER+ MCF-7 breast cancer cells is highly responsive to removal of E2 in vitro. As expected, LCC1 cells have a high constitutive level of NF-κB DNA binding activity, which underwent a slight increase within 12 days following culture in E2-free medium. In order to determine whether this increased level of NF-κB activity also occurs in MCF-7 tumors in vivo, we grew MCF-7 tumors in nude mice implanted with E2 release pellets and isolated nuclear extracts from a solid tumor every day for 7 days and again at 18 days after pellet removal. The EMSA in Fig. 2B shows that, compared with an that of an actively growing tumor in an animal implanted with an E2 release pellet, NF-κB activity began to increase within 3 days following E2 pellet removal and rose continuously to a maximum within the 7-day period. As the tumors regressed, MCF-7 NF-κB DNA binding activity remained elevated in nuclear extracts for 18 days after E2 pellet removal. To profile NF-κB complexes in vivo from E2-depleted MCF-7 xenografts, we similarly assayed DNA binding in nuclear extracts from regressing MCF-7 cell tumors. Supershift analysis of nuclear extracts from the MCF-7 tumor excised at day 6 following E2 release pellet removal (Fig. 2C) shows again that although the NF-κB activity was primarily p50/p50 (or possibly p50/p52), an anti-p65 reactive complex was also present. Thus, both NF-κB complexes are also detectable in regressing tumors and could potentially participate in the evolution of the E2-independent phenotype. Taken together, these results show that E2 removal both in vivo and in vitro is a potent stimulus in breast cancer cells of NF-κB activity, which might then contribute to E2 independence. One interpretation of this result is that E2 removal results in survival of cells with high endogenous levels of NF-κB rather than an induction of NF-κB activity. In order to address this issue, we tested the reversibility of the effect of E2 withdrawal on NF-κB DNA binding. MCF-7 cells were grown in E2-free medium for 10 days and then treated with vehicle or E2 for 72 h. The results in Fig. 2D demonstrate that, as expected, NF-κB activity was high in MCF-7 cells cultured without E2. In contrast, levels were strongly reduced in cells which had be reexposed to E2, thus indicating regulation of NF-κB binding rather than an altered composition of the cell population.
FIG. 2.

E2 regulates NF-κB DNA binding in vitro and in vivo. (A) Nuclear extracts (5 μg) from MCF-7 and MCF-7/LCC1 cells cultured in DMEM (lanes C) or in E2-free medium for the indicated number of days were subjected to EMSA with the NF-κB oligonucleotide. A 10-fold excess of cold competing oligonucleotide (comp) preferentially competes the specific NF-κB complexes. n.s., nonspecific band. The lower panel shows a gel shift of the Sp1 consensus oligonucleotide with the same extracts to control for extract loading. (B) MCF-7 tumors were grown in ovariectomized nude mice implanted with an E2 release pellet. Tumors were obtained prior to E2 pellet removal (control) and at the indicated times following pellet removal. Nuclear extracts were incubated with a labeled NF-κB oligonucleotide and subjected to EMSA as described in Materials and Methods. A 50-times concentration of cold oligonucleotide (comp) was used to compete specific binding. In the lower panel the same extracts were assayed for binding to the consensus Sp1 element. (C) Tumor extracts from day 6 after the E2 pellet removal described above were used for supershift analysis with antibodies (Ab) against p50 and p65. Arrowheads indicate supershifted complexes. Ns, normal serum. (D) MCF-7 cells cultured in stripped medium without E2 for 10 days were treated with E2 or vehicle (−) for 3 days. Nuclear extracts were isolated, and 5 μg was subjected to EMSA with the NF-κB site. Protein binding to the Sp1 element from the same extracts is shown in the panel on the right.
Expression of ER proteins in MCF-7 sublines.
Previous work by Nakshatri et al. (36) showed that loss of ER expression correlated with the acquisition of constitutive NF-κB activity. As LCC1 cells are E2 independent, it was possible that these cells had in fact begun to downregulate ER expression, thus resulting in higher levels of NF-κB activity. We (this work) and others (11, 17) have reported that E2 can modulate NF-κB activity; thus, alterations of ER levels in the presence or absence of E2 might indirectly affect NF-κB activity. In order to determine the relative expression levels of both of the ERs and to see whether E2 alters these levels, we performed immunoblot analysis of LCC1 cells, MIII cells, MCF-7(early) cells, and MCF-7 cells of undetermined passage treated with E2 or vehicle for 72 h. Figure 3 shows that, as expected, all cell lines express the 68-kDa ERα. Interestingly, LCC1 cells express a slightly higher level of ERα than the other cells. Similarly, immunoblotting for ERβ indicated that all of the cell lines expressed a 66-kDa protein corresponding to ERβ. Again, levels appeared elevated in LCC1 cells. There was no indication that either of the ERs was subject to E2 regulation in MIII or MCF-7 cells; however, E2 reproducibly increased both ERα and ERβ of MCF-7(early) cells and the level of ERβ in LCC1 cells above levels in vehicle-treated cultures. Thus, despite this marked increase in constitutive NF-κB activity in LCC1 cells, they retain high-level expression of ERα and ERβ.
FIG. 3.
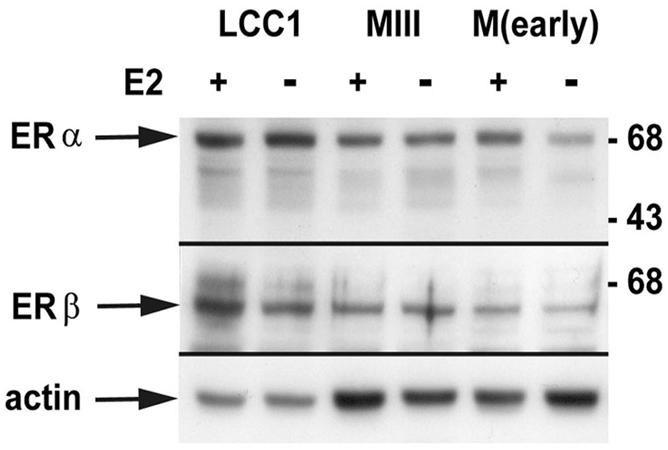
Immunoblot analysis of ER proteins. Twenty micrograms of whole-cell protein extract from MCF-7 and derived sublines treated with E2 or vehicle for 72 h was separated by SDS-polyacrylamide gel electrophoresis and immunoblotted with antibodies against ERα and ERβ. The expression of actin was used as an internal loading control. Numbers on the right indicated molecular masses in kilodaltons.
Estrogen regulates Bcl-3 protein expression.
The supershift experiments described above indicated that the NF-κB complex in LCC1 cells is composed primarily of p50, which in itself is transcriptionally inactive; however, activation of the transcriptional function of this complex has been shown to occur through complex formation with Bcl-3 (31). If p50 complexes are to be active in tumors following E2 removal, the accessory factor Bcl-3 might contribute to this activity. To investigate this, we subjected protein extracts from MCF-7 tumors to immunoblot analysis at intervals after removal of the E2 release pellet. The results in Fig. 4A show that control tumors contained low levels of Bcl-3, while the levels rose significantly within 1 day after E2 removal and remained elevated for the balance of the study. To assess whether Bcl-3 expression induced by E2 removal was principally due to the absence of E2 or was secondary to other effects on the tumor milieu, we analyzed extracts from LCC1 cells cultured in E2-free or E2-supplemented medium over several days. The results in Fig. 4B show that culture of LCC1cells in the absence of E2 increased the expression of Bcl-3 within 1 day compared with culture in E2-supplemented medium. The differential expression was already maximal by 2 days and remained so throughout the 4-day study period. Since NF-κB DNA binding is constitutively higher in E2-independent LCC1 cells and MDA-MB-231 cells than in MCF-7 cells and MIII cells in the early stage of E2 independence, we wished to determine whether basal levels of Bcl-3 might follow the same pattern. Figure 4C shows a Western blot analysis of Bcl-3 levels in extracts from early- and late-passage MCF-7 cells, MIII cells, LCC1 cells, and several ER− cells. The results indicate that levels of Bcl-3 are highest in the E2-independent lines, including LCC1 cells. A second comparison of Bcl-3 levels in MCF-7(late), MCF-7(40F) and MDA-MB-468 cells (Fig. 4D) shows that, unexpectedly, MCF-7(40F) cells express Bcl-3 at approximately the same level as MCF-7 cells. However, it is important to note that MCF-7(40F) cells were selected not for E2 independence but rather for adriamycin resistance and therefore do not necessarily represent a natural stage in hormone progression. Moreover, we cannot rule out that regulation of Bcl-3 expression in other ER− cell lines is accomplished by the same mechanism as in ER+ cell lines.
FIG. 4.

Bcl-3 is regulated by E2 in breast cancer cells in vivo and in vitro. (A) Bcl-3 expression in MCF-7 breast cancer tumors was determined by immunoblotting of 15 μg of tumor protein extracts on the indicated days after E2 release pellet removal. The molecular mass in kilodaltons is shown on the right. (B) Basal expression of Bcl-3 in E2-dependent and -independent breast cancer cells. Whole-cell protein extracts were isolated from the indicated breast cancer cell lines grown in appropriate phenol red-containing medium. In all experiments,15 μg of each extract was subjected to immunoblot analysis with the Bcl-3 antibody and either actin or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a protein loading control. (C) Immunoblot to detect Bcl-3 expression in vitro in the presence and absence of E2. LCC1 cells were cultured in DMEM or E2-free medium supplemented with E2 or vehicle as described in Materials and Methods for the indicated times. Lane c, control consisting of extracts from cells cultured in unstripped medium containing phenol red. (D) Comparison of Bcl-3 expression in the indicated cell lines as described for panel C.
Bcl-3 augments MCF-7 tumor growth but not E2 independence.
Bcl-3 has recently been shown to stimulate growth as well as provide a survival function in some cells (35, 47, 55). The results described above clearly show that p50/50 (and possibly p50/p52) complexes predominate in LCC1 cells, and the increased expression of Bcl-3 suggests that this protein could play a role in E2 independence. To assess the sufficiency of Bcl-3 expression in conferring E2 independence, we stably transfected MCF-7(early) cells with a FLAG-tagged Bcl-3 expression construct. Figure 5A shows an immunoblot of lysates from three pooled MCF-7(FLAG-Bcl-3) clones and three pooled MCF-7(FLAG) control clones reacted with anti-FLAG to detect transfected protein (upper panel) and anti-Bcl-3 (second panel) in order to compare the relative levels of Bcl-3 in these clones. Bcl-3 expression has been associated with induction of the cyclin D1 gene, and immunoblot analysis of the same nuclear extracts with an anti-cyclin D1 antibody showed that the constitutive level of cyclin D1 expression was increased in the transfected cells (third panel). Nuclear extracts from these pooled clones were also used for an EMSA to detect NF-κB DNA binding activity. Figure 5B shows that, consistent with its ability to interact with p50, the p50 complex was augmented in MCF-7(FLAG-Bcl-3) clones compared with controls when equal amounts of nuclear extract were assayed, suggesting a possible stabilization of the p50 DNA binding complex. We then used both MCF-7(FLAG-Bcl-3) and MCF-7(FLAG) clones to generate tumors in ovariectomized nude mice implanted with an E2 release pellet as described in Materials and Methods. The growth of control tumors [MCF-7(FLAG)]and MCF-7(FLAG-Bcl-3) tumors was monitored over 36 days, and the results are shown in Fig. 5C. Eleven of the 15 sites injected with MCF-7(FLAG-Bcl-3) cells produced tumors. On the other hand, only 4 of 12 control sites formed tumors over the same time period. Of the tumors that formed, the mean volumes (± SE) were 258 ± 45 mm3 for MCF-7(FLAG-Bcl-3) tumors and 67 ± 24 mm3 for MCF-7(FLAG) tumors. Tumor regression was monitored after E2 release pellet removal until the tumor was 50% of its original size at pellet removal. If Bcl-3 conferred E2 independence, we expected that MCF-7(Bcl-3) tumors would either remain stable or continue to grow. However, comparison with control MCF-7(FLAG) tumors showed virtually identical regression rates, requiring 14 days for the latter tumors and 15.5 days for MCF-7(FLAG-Bcl-3) tumors to regress to 50% of the original tumor volume. Thus, Bcl-3 alone does not render MCF-7 tumors stable after E2 withdrawal, suggesting that Bcl-3-mediated increases in p50 and/or p52 activity are either of insufficient magnitude or cannot confer an E2-independent phenotype. In contrast, the higher rate of tumor establishment and rapid tumor growth of MCF-7(FLAG-Bcl-3) cells compared with control cells shows that Bcl-3 can augment the growth and tumorigenicity of breast cancer cells.
FIG. 5.

Bcl-3 promotes E2-dependent MCF-7 tumor growth. (A) MCF-7 cells were transfected with either an empty FLAG vector or FLAG-Bcl-3. Shown is immunoblot analysis of cell extracts from three pooled clones of each transfectant with an anti-FLAG antibody. The lower panel shows a duplicate blot following incubation with anti-Bcl-3 to detect both endogenous and transfected protein. Sizes of molecular mass markers are shown on the left in kilodaltons. (B) EMSA to assess the effects of Bcl-3 overexpression on NF-κB DNA binding. Five micrograms of nuclear extract from pooled MCF-7(FLAG) and MCF-7(FLAG-Bcl-3) clones was used for gel shift analysis with either the NF-κB or Sp1 sequence as a probe. Samples were assessed simultaneously on the same gel. (C) Tumor volumes derived from MCF-7(FLAG) and MCF-7(FLAG-Bcl-3) cells 36 days after cell inoculation. Mice 1 to 12 were injected subcutaneously with pooled clones of control or MCF-7(FLAG-Bcl-3) on either flank. Mice 13 to 15 received only an MCF-7(FLAG-Bcl-3) inoculation.
Induction of both p65- and p50-associated activity after E2 withdrawal in vitro and in vivo.
The relative levels of p65 and p50 in nuclei from MCF-7(early) and LCC1 cells and the results in Fig. 1B suggested that there were both qualitative and quantitative differences between NF-κB complexes in the hormone-dependent and hormone-independent cells. To characterize the NF-κB complexes in these two isolates, it was necessary to use MCF-7(early) cells which were cultured in E2-free medium, since without E2 withdrawal there was insufficient NF-κB DNA binding activity to evaluate. We performed an EMSA and supershift analysis of nuclear extracts from MCF-7(early) and LCC1 cells which, for consistency, were both derived from culture in E2-depleted medium for 12 days (Fig. 6). Supershift analysis with the p50 antibody again revealed the presence of a major complex containing p50, while the p52 antibody was unable to supershift any complex. Importantly, the p65 antibody identified a strong p65-associated complex in the LCC1 cells. By contrast, the level of p65 DNA binding activity was significantly lower in the MCF-7(early) nuclear extracts. Taken together with the nuclear expression of p65 and p50 in MCF-7 and LCC1 cells, these results show that after E2 withdrawal, LCC1 cells contain significantly more p65-associated activity than do their E2-dependent counterparts.
FIG. 6.

Induction of both p65- and p50-associated NF-κB activity following E2 withdrawal. Nuclear extracts were collected from MCF-7(early) and LCC1 cells cultured for 12 days in phenol red-free medium with charcoal-stripped serum to induce NF-κB activity. Five micrograms of each extract was used for EMSA with the NF-κB probe and antibody supershift analysis with the indicated antibodies (Ab). The same quantity of extract was also subjected to EMSA with the Sp1 probe as a control for extract loading.
LCC1 cells constitutively expressing IκBα revert to an estrogen-dependent tumor phenotype.
In order to test the hypothesis that NF-κB activity contributes significantly to the E2-independent phenotype, we stably transfected LCC1 cells with a FLAG-tagged degradation-resistant form of the NF-κB inhibitor IκBα called IκBαSR (27). IκBα most effectively inhibits p65-containing complexes (26), although some minimal inhibition of p50/p50 activity has also be documented (30). Figure 7A shows immunoblot analysis with the anti-FLAG monoclonal antibody of four positive clones and control extracts from pcDNA3-transfected LCC1 cells. The effects of IκBαSR expression on relative levels of nuclear NF-κB complexes were tested by supershift analysis. Figure 7B shows that IκBαSR expression had little effect on the abundant, fast-migrating p50-containing complex. In contrast, the p65 complex was virtually absent from the LCC1(IκBαSR) cells. To confirm that any reversion of LCC1(IκBαSR) tumors to E2 dependence was associated with decreased nuclear levels of p65, we performed immunoblot analysis of nuclear extracts from LCC1(pcDNA3) and LCC1(IκBαSR) tumors. Figure 7C shows that p65 levels are markedly reduced in LCC1(IκBαSR) tumors compared with controls. On the other hand, both the p50 and p52 nuclear contents remained unaltered in these tumor types. The p50 antibody, which simultaneously detects p52 in extracts from cultured cells, did not detect p52 in the tumor extracts. To assess p52, we utilized a separate anti-p52 antibody, which indicated that the levels of nuclear p52 were unchanged in IκBαSR-expressing tumors. Evaluation of the effects of IκBαSR on NF-κB transcriptional activity following transient transfection of the three highest-expressing clones with the 3X-NF-κB reporter gene or the same reporter gene containing a mutant NF-κB response element showed that transcriptional activity in the three LCC1(IκBαSR) clones ranged from 20- to 100-fold less than that in the control LCC1(pcDNA3) clones (Fig. 7D). We then inoculated three pooled LCC1(pcDNA3) clones and pooled LCC1(IκBαSR) clones 1, 3, and 4 into the contralateral sides of ovariectomized nude mice implanted with an E2 release pellet. Tumors from both groups grew at various rates under these conditions of hormone replacement, and there was no significant difference in the final tumor volumes [mean ± standard deviation, 183 ± 88 mm3 for LCC1(pcDNA3) tumors and 160 ± 88 mm3 for LCC1(IκBαSR) tumors]. Tumor volumes were measured 2 weeks after E2 pellet removal, and the percent regression was calculated. Figure 7E is a histogram showing the results of this analysis. While LCC1(pcDNA3) tumor volumes either remained stable or continued to grow after pellet removal, LCC1 (IκBαSR) tumors underwent significant regression. In cells engaged in apoptosis through the mitochondrial death pathway, the Bax protein undergoes a conformational change exposing its otherwise buried N terminus associated with mitochondrial translocation (37). Figure 7F depicts sections from LCC1(pcDNA3) and LCC1(IκBαSR) tumors immunostained with an antibody against the N terminus of Bax, showing high levels of translocated Bax in the LCC1(IκBαSR) tumors but not in control tumors. Moreover, detection of DNA fragmentation in apoptotic cells by using ISEL showed that LCC1(IκBαSR) tumors contained large numbers of apoptotic nuclei, while LCC1(pcDNA3) tumors had almost none. Thus, NF-κB inhibition is sufficient to restore an E2-dependent phenotype to LCC1 tumors, rendering them apoptotic after E2 removal through a mitochondrial pathway of cell death involving Bax translocation.
FIG. 7.

MCF-7/LCC1 cells constitutively expressing IκBαSR lose p65/p50 activity and revert to an E2-dependent tumor phenotype. (A) Immunoblot analysis to detect FLAG-tagged IκBαSR expression in stable LCC1 clones with an anti-FLAG monoclonal antibody. Clones are designated 1 to 4. CON, control. (B) Five micrograms of nuclear extract from LCC1(pcDNA3) and LCC1(IκBαSR) cell cultures was subjected to EMSA and antibody (Ab) supershift analysis as described in Materials and Methods. NS, normal serum. (C) Nuclear extracts (10 μg) from LCC1(IκBαSR) and LCC1(pcDNA3) tumors were analyzed by immunoblotting for expression of p65, p50, and p52. The p50 antibody did not identify a p52 band from these extracts, and therefore a p52-specific antibody was used to assess changes in expression between clones. (D) NF-κB activity was evaluated in LCC1(IκBαSR) clones 1, 3, and 4 (from left to right) following transient cotransfection of the NF-κB-luciferase and LacZ reporter genes. The results presented are the averages from two experiments, expressed in arbitrary units, and are normalized to β-galactosidase activity. (E) Three pooled clones each of LCC1(pcDNA3) and LCC1(IκBαSR) cells were inoculated into ovariectomized nude mice implanted with an E2 release pellet. The histogram shows the percent regression of LCC1 (n = 11) and LCC1(IκBαSR) (n = 9) tumors following removal of the release pellet. Bars represent standard errors. (F) Sections of LCC1 and LCC1(IκBαSR) tumors were subjected to ISEL to detect free 3′-OH DNA ends in apoptotic nuclei or immunostained with an antibody against the NH2 terminus of Bax to detect activated mitochondrial Bax.
Effect of NF-κB inhibition on E2-regulated genes.
Recent work has shown that expression of both of the antiapoptotic proteins Bcl-2 and Bcl-x is induced by NF-κB in neurons (10, 51). Since LCC1(I-κBαSR) tumors undergo apoptotic regression following E2 withdrawal, we tested whether the reduced NF-κB in these cells was associated with a reduction in Bcl-2 and/or Bcl-x. We first considered the expression of these proteins in the respective tumors on various days following E2 release pellet removal. The results in Fig. 8A show that Bcl-2 protein levels were strongly reduced in LCC1(IκBαSR) cells compared with LCC1(pcDNA3) cells for the entire period after withdrawal of E2. In contrast, Bcl-x expression in the two tumor types was indistinguishable. During the initial characterization of MIII and LCC1 cells, it was found that the expression of a number of normally E2-responsive genes was constitutively upregulated and no longer responsive to E2 (9). This was also true of another independent MCF-7 subline selected for E2-independent growth (13). E2 can also regulate expression of Bcl-2 (43, 52), although the promoter for this gene is complex and may also be regulated by mutant p53. A comparison of the expression of Bcl-2 and its regulation by E2 in vitro is shown in Fig. 8B. Unlike for several other E2-regulated gene products, LCC1 cells contain lower levels of Bcl-2 than do parental MCF-7 cells. This expression is refractory to E2 regulation. As observed in the tumors, stable expression of IκBαSR further reduced Bcl-2 expression in vitro, and this was accompanied by a weak reinstatement of E2 regulation. Importantly, we have previously shown that constitutive Bcl-2 expression is sufficient to prevent regression of MCF-7 tumors in nude mouse xenografts (44). Taken together, these results suggest that the reduction in Bcl-2 expression in LCC1(IκBαSR) tumors could contribute to the E2 withdrawal-induced apoptosis and subsequent regression.
FIG. 8.

Expression of IκBαSR downregulates Bcl-2 expression. (A) Whole-cell extracts from LCC1(pcDNA3) (lanes P) and LCC1(IκBαSR) (lanes I) cell tumors at different days following E2 pellet removal were analyzed by immunoblotting for expression of Bcl-2. Blots were stripped and reacted with antibody against Bcl-x. Loading of the gel was controlled for by reactivity with an actin antibody. (B) LCC1 cells stably transfected with pcDNA3 or IκBαSR together with MIII and M(late) cells were cultured in E2-free medium for 10 days and then treated with E2 or vehicle for 48 h. E2 regulation of Bcl-2 expression in vitro in the MCF-7-derived cells and clones was assessed by immunoblotting of 15 μg of cell extract. Actin was used as an internal loading control for all blots. Molecular masses in kilodaltons are shown on the right.
DISCUSSION
The events which precede the loss of E2 dependence and a more tumorigenic phenotype in breast cancer cells are not understood. The LCC1 subline of the E2-dependent human breast cancer cell line MCF-7 was selected under conditions of E2 depletion, resulting in an experimental model of progression in that these cells grow in a hormone-independent manner but retain wild-type levels of ER expression. Apart from the observation that some E2-regulated genes are constitutively expressed in these cells, little is known about the cellular events that contribute to the loss of E2 dependence. While it is clear that NF-κB activity rises in the context of carcinogen-induced mammary epithelial cell transformation (28), in general there is also a marked increase in constitutive NF-κB activity in ER− cells compared with ER+ cells (36). Using E2-independent LCC1 cells, the work described here shows that an increase in constitutive NF-κB activity occurs in cells selected for E2 independence prior to loss of ER expression. Moreover, inhibition of this NF-κB activity is sufficient to confer sensitivity to estrogen removal on LCC1 tumors through a mitochondrial death pathway. This observation also clearly indicates that LCC1 cells have not sustained damage to their apoptotic machinery during progression toward E2 independence. In contrast to K-BALB murine fibrosarcoma cells and mouse mammary tumor cells, in which tumorigenicity was completely blocked by inhibition of NF-κB activity (7, 22), at least in animals supplemented with E2, LCC1(IκBαSR) cells were able to form tumors as well as control cells. Thus, inhibition of NF-κB activity through IκBα does not interfere with E2-driven tumorigenicity.
The process of selection requires epigenetic changes which render some cells resistant to the selective pressure. Therefore, to investigate the mechanism by which this occurs, we have analyzed the consequence of E2 removal on NF-κB activity both in vitro and in tumors. The reduction in E2 levels following removal of the E2 pellet in ovariectomized athymic mice and culture in E2-free medium results in a rapid increase in NF-κB levels in MCF-7 cells. The kinetics were fastest in vivo, which may reflect E2 clearance through metabolism. Additionally, cells in culture were exposed to estrogenic phenol red prior to removal to phenol red-free stripped medium, and the clearance rate of phenol red in these cells. Several reports have previously shown that the ligand-bound ER is involved in reciprocal regulation with NF-κB (reference 17 and references therein). Both ERα and the ERβ can inhibit NF-κB-dependent transcription in a manner involving the ligand binding domain of the receptor but not the squelching of coactivator proteins (11). Discreet parts of the ER ligand binding domain surface are required for this transrepression of NF-κB activity, which are separate from those involved in transactivation (53). Estrogen has been clearly demonstrated to repress NF-κB activity, and correspondingly, NF-κB induction follows E2 withdrawal. Estrogen removal over the time course of our experiments in vitro is only cytostatic for MCF-7 cells. Given this and the fact that NF-κB activation is reversed by reintroduction of E2 in vitro, it is unlikely that the induction of NF-κB DNA binding seen after removal of E2 in vivo is the result of selection for cells with higher basal levels of NF-κB. Moreover, NF-κB is a positive regulator of cell growth (12), thus arguing against subpopulations of MCF-7 cells containing constitutively elevated NF-κB activity.
Since the predominant NF-κB complex in MCF-7 and LCC1 cells is either a homodimeric complex of p50 or a heteromeric complex of p50 and p52, activation after hormone removal would not be expected to yield a strong transcriptional response. Our finding that the level of the p50-coactivating protein Bcl-3 is increased after E2 removal provides a mechanism by which E2 withdrawal induces p50-associated NF-κB activity in breast cancer cells that do not contain high levels of the alternate transcriptionally active partner, p65. The observation that Bcl-3 increases the E2-dependent growth of MCF-7 cells may be due in part to the reported ability of Bcl-3/p50/p52 complexes to induce cyclin D1 (23, 55) as well as stimulate AP-1-mediated transactivation and cellular proliferation (35). Although the Bcl-3 regulatory regions contain NF-κB enhancer elements (40) that are responsive to NF-κB (8, 10), this mechanism cannot account for the rapid increase in Bcl-3 protein expression observed both in vitro and in vivo after E2 withdrawal, suggesting that Bcl-3 expression may be more directly under control of the ER.
The present data indicate that p50 and p65 are differentially expressed during progression and that early in this process NF-κB activity increases and is composed primarily of p50/p50 (and possibly p50/p52), along with a smaller increase in p50/p65. Highly malignant ER− cells have both p65/p50 and p50 (p50/p52)-associated NF-κB DNA binding activity. Evidence suggests that the p65 protein plays a critical role in tumorigenesis (22). Nuclear extracts from ER+ MCF-7 cells of limited passage express only trace levels of p65 and comparatively low levels of p50. LCC1 nuclei contain slightly elevated p65, although the level remains significantly lower than that in the ER− cell lines. However, LCC1 p50 nuclear expression is essentially similar to that in the E2-independent breast cancer cell lines. Together with the finding that basal Bcl-3 expression in LCC1 cells is also higher than that in the parental MCF-7 isolate, these results can account for the higher constitutive level of both NF-κB complexes in LCC1 cells. The expression pattern of p65 and p50/p52 in breast tumors is unclear. It has been suggested that breast cancer cell lines but not tumors express p65. While Cogswell et al. (16) detected only low levels of p65 protein in a panel of four breast tumor samples, only one of these was ER−. Supershift analysis was done on an ER+ tumor which, in agreement with our results, showed primarily p50-associated NF-κB DNA binding activity. Sovak et al. (50) detected nuclear p65 in 15 of 23 tumor samples, although that study did not differentiate on the basis of ER status. Thus, it appears that NF-κB proteins and complexes are differentially present in breast cancer cells, which, at least in cell lines, correlates with both ER expression and the stage of progression. One of the most important outcomes of this study is that it describes an underlying reason for why primary breast tumors with functional ERs may be marginally or not at all E2 dependent. With respect to both Bcl-3 expression and NF-κB activity, like LCC1 cells and MDA-MB-231 cells, tumors that display elevated Bcl-3 could be either ER+ or ER−. Another variable that could affect these parameters in tumors that are either truly E2 dependent or just E2 responsive is the level of circulating E2 in a patient at the time of biopsy. Based on our results, Bcl-3 and NF-κB activity would not be predicted to correlate simply with ER status in the absence of any other information about the tumor or patient. Instead, ER expression combined with Bcl-3 and differential NF-κB activity might predict the actual hormone-dependent status of the tumor and prove to be a useful marker for progression.
The targets of NF-κB are numerous and varied, ranging from cytokines in the immune system to cell adhesion molecules, transcription factors, a variety of enzymes, and certain survival proteins (42). The last group includes the antiapoptotic protein Bcl-xL as well as Bcl-2, whose expression has been shown to be activated by NF-κB in neuronal cells (10, 51). We have previously demonstrated that Bcl-2 is positively regulated by E2 in MCF-7 cells (52), although this regulation appears to be absent in LCC1 cells and basal levels of Bcl-2 are lower than in related MCF-7 cells. We have also shown that Bcl-2 expression is sufficient to prevent E2 withdrawal regression of MCF-7 tumors in nude mice (44). Thus, a further decrease in expression of Bcl-2 in LCC1(IκBαSR) cells would likely contribute to the acquired E2 sensitivity of LCC1(IκBαSR) tumors by facilitating the apoptotic response following the death signal constituted by E2 release pellet removal. The fact that Bcl-2 but not Bcl-x was reduced by IκBαSR expression suggests that p65/p50 complexes may positively regulate Bcl-2 expression while Bcl-x is regulated either by different NF-κB complexes or by other means in these cells. The IAP protein hIAP1 (18) has been shown to be induced by NF-κB (reference 42 and references therein), and while our preliminary findings indicate higher levels of both hIAP1 and hIAP2 in those breast cancer cells containing elevated constitutive NF-κB activity (data not shown), the role of these proteins in hormone independence requires further investigation.
The work presented here demonstrates that NF-κB complexes play distinct roles in E2-dependent and -independent growth and survival of breast cancer cells. Complexes of p50 (and possibly p52) and Bcl-3 in LCC1 cells promote the growth of LCC1 cells in an E2-dependent manner and, although this was not directly tested here, likely contribute to growth promotion in the presence of an appropriate survival signal in E2-independent cells as well. The majority of NF-κB DNA binding activity is composed of p50 dimers; however, expression of Bcl-3, which is required for the transcriptional activity of these complexes, is insufficient to confer E2 independence to MCF-7 cells. Thus, p65-containing complexes in LCC1 cells, either alone or in concert with p50 dimer/Bcl-3 complexes, clearly have a critical role in conferring the E2-independent phenotype. Supporting this hypothesis is the observation that expression of IκBαSR protein clearly reduced nuclear p65 levels in LCC1 tumors but had little effect on p50 or p52. Moreover p65 complexes were virtually undetectable by supershift analysis in LCC1(IκBαSR) cells. The crystal structure of the IκBα:NF-κB complex has been resolved, revealing a stable and extensive interaction between IκBα and p65. The outcome of this interaction is a critical change in the conformation of p65, resulting in allosteric inhibition of DNA binding as well as masking of the nuclear localization signal in p65 (26). Although IκBα can bind p50 homodimers, it does so with a 60-fold-lower affinity than in its interaction with p65/p50 (30). Thus, the data presented here, as well as the fact that all ER− or E2-independent cell lines have high levels of p65/p50 NF-κB activity, support the hypothesis that breast cancer cell progression requires increased p65/p50 activity.
Our results together with those in the literature suggest the model in Fig. 9, which indicates pathways of growth and survival in ER+ breast cancer cells in both the presence and absence of E2. E2 provides dual growth and survival signals at least in part by increasing cyclin D1 and Bcl-2 protein levels. We surmise that the increase in Bcl-3 after E2 withdrawal could also contribute to increased proliferative rates, based on the increased rate of tumor growth generated by Bcl-3-overexpressing cells. This increased proliferation is insufficient, however, to prevent the overall regression of the tumor over time. Thus, our model suggests that while most E2-dependent cells will undergo cell death after E2 withdrawal in vivo, a subset with adequate levels of p50/p52/Bcl-3 and p50/p65 will both grow and survive, thus establishing the E2-independent phenotype. It is possible that these two events may not occur simultaneously in tumor cells and that the immediate induction of Bcl-3 after E2 withdrawal may be sufficient over the short term to maintain the tumor while the proliferative rate exceeds the rate of apoptosis. This could provide adequate time for other signaling pathways, such as ErbB2, which is also induced after E2 withdrawal in MCF-7 cells (data not shown), to induce p50/p65-associated activity (6, 57). Studies to investigate the possible role of ErbB receptors in this process are under way.
FIG. 9.

Proposed model of induction of NF-κB-mediated estrogen-independent growth and survival following estrogen withdrawal from ER+ breast cancer cells. E2 provides growth and survival signals to ER+ breast cancer cells. E2 withdrawal induces NF-κB activity which in most cells is not sufficient to support E2-independent growth, and the tumor regresses. However, some cells could persist as E2-independent variants as a result of adequate expression of differential NF-κB activity. ER− breast cancer cells have constitutive p65 and p50 activity. See text for details.
The ability of IκBα to inhibit E2-independent growth in this study shows that NF-κB activation may be the most critical early event following E2 withdrawal resulting in breast cancer progression. Therefore, in addition to representing a potential therapeutic target in ER− breast cancer as proposed by Biswas et al. (7), NF-κB inhibition may well prove to be effective in obviating progression. Ironically, a reduction in circulating E2 levels (or spontaneous loss of ER expression) in individuals with hormone-dependent breast cancer would directly precipitate the early induction of NF-κB activity, which could confer E2-independent growth and survival to these cells.
Acknowledgments
This work was supported in part by grant 13266 from the National Cancer Institute of Canada/Canadian Institutes for Health Research Canadian Breast Cancer Research Initiative to M.A.C.P.
REFERENCES
- 1.Ausubel, F. M., R. Brent, R. E Kingston, D. D. Moore, J. G. Seidman, J. Smith, and K. Struhl. 1989. Short protocols in molecular biology, 3rd ed. John Wiley & Sons, New York, N.Y.
- 2.Baeuerle, P. A., and D. Baltimore. 1996. NF-κB: ten years after. Cell 87:13-20. [DOI] [PubMed] [Google Scholar]
- 3.Baichwal, V. R., and P. A. Baeuerle. 1997. Apoptosis: activate NF-κB or die? Curr. Biol. 7:R94-R96. [DOI] [PubMed] [Google Scholar]
- 4.Beg, A. A., S. M. Ruben, R. I. Scheinman, S. Haskill, C. A. Rosen, and A. S. Baldwin, Jr. 1992. IκB interacts with the nuclear localization sequences of the subunits of NF-κB: a mechanism for cytoplasmic retention. Genes Dev. 6:1899-1923. [DOI] [PubMed] [Google Scholar]
- 5.Beg, A. A., and D. Baltimore. 1996. An essential role for NF-κB in preventing TNFα-induced death. Science 274:782-784. [DOI] [PubMed] [Google Scholar]
- 6.Beraud, C., W. J. Henzel, and P. A. Baeuerle. 1999. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-kappaB activation. Proc. Natl. Acad. Sci. USA 96:429-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biswas, D. K., S.-C. Dai, A. Cruz, B. Weiser, E. Graner, and A. B. Pardee. 2001. The nuclear factor kappa B (NF-κB): a potential therapeutic target for estrogen receptor negative breast cancers. Proc. Natl. Acad. Sci. USA 98:10386-10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braiser, A. R., M. Lu, T. Hai, Y. Lu, and I. Boldogh. 2001. NF-κB-inducible Bcl-3 expression is an autoregulatory loop controlling nuclear p50/NF-κB1 residence. J. Biol. Chem. 276:32080-32093. [DOI] [PubMed] [Google Scholar]
- 9.Brunner, N., V. Boulay, A. Fojo, C. E. Freter, M. E. Lippman, and R. Clarke. 1993. Acquisition of hormone-independent growth in MCF-7 cells is accompanied by increased expression of estrogen-regulated genes but without detectable DNA amplifications. Cancer Res. 53:283-290. [PubMed] [Google Scholar]
- 10.Bui, N. T., A. Livolsi, J.-F. Peyron, and J. H. M Prehn. 2001. Activation of nuclear factor κB and bcl-x survival gene expression by nerve growth factor requires tyrosine phosphorylation of IκBα. J. Cell Biol. 152:753-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerillo, G., A. Rees, N. Manchanda, C. Reilly, I. Brogan, A. White, and M. Needham. 1998. The oestrogen receptor regulates NF-κB and AP-1 activity in a cell-specific manner. J. Steroid Biochem. Mol. Biol. 67:79-88. [DOI] [PubMed] [Google Scholar]
- 12.Chen, F., V. Castronova, and X. Shi. 2001. New insights into the role of nuclear factor-κB in cell growth regulation. Am. J. Pathol. 159:387-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho, H., P. A. Ng, and B. S. Katzenellenbogen. 1991. Differential regulation of gene expression by estrogen in estrogen growth-independent and -dependent MCF-7 human breast cancer cell sublines. Mol. Endocrinol. 5:1323-1330. [DOI] [PubMed] [Google Scholar]
- 14.Clarke, R., N. Brunner, B. S. Katzenellenbogen, E. W. Thompson, M. J. Norman, C. Koppi, S. Paik, M. E. Lippman, and R. B. Dickson. 1989. Progression of human breast cancer cells from hormone-dependent to hormone-independent growth both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 86:3649-3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarke, R., E. W. Thompson, F. Leonessa, M. Lippman, M. McGarvey, T. L. Frandsen, and N. Brunner. 1993. Hormone resistance, invasiveness and metastatic potential in breast cancer. Breast Cancer Res. Treat. 24:227-239. [DOI] [PubMed] [Google Scholar]
- 16.Cogswell, P. C., D. C. Guttridge, W. K. Funkhouser, and A. S. Baldwin. 2000. Selective activation of NF-κB subunits in human breast cancer: potential roles for NF-κB/p52 and for Bcl-3. Oncogene 19:1123-1131. [DOI] [PubMed] [Google Scholar]
- 17.Delfino, F., and W. H. Walker. 1999. Hormonal regulation of the NF-κB signaling pathway. Mol. Cell. Endocrinol. 157:1-9. [DOI] [PubMed] [Google Scholar]
- 18.Deveraux, Q. L., S. L. Schendel, and J. C. Reed. 2001. Anti-apoptotic proteins. The bcl-2 and inhibitor of apoptosis protein families. Cardiol. Clin. 19:57-74. [DOI] [PubMed] [Google Scholar]
- 19.Finco, T. S., and A. S. Baldwin, Jr. 1993. Kappa B site-dependent induction of gene expression by diverse inducers of nuclear factor B requires Raf-1. J. Biol. Chem. 268:17676-17679. [PubMed] [Google Scholar]
- 20.Fruehauf, J. P., E. G. Mimnaugh, and B. K. Sinha. 1991. Doxorubicin-induced cross-resistance to tumor necrosis factor (TNF) related to differential TNF processing. J. Immunother. 10:165-173. [DOI] [PubMed] [Google Scholar]
- 21.Gross, A., J. M. McDonnell, and S. J. Korsmeyer. 1999. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13:1899-1911. [DOI] [PubMed] [Google Scholar]
- 22.Higgins, K. A., J. R. Perez, T. A. Coleman, K. Dorshkind, W. A. McComas, U. M. Sarmiento, C. A. Rosen, and R. Narayanan. 1993. Antisense inhibition of the p65 subunits of NF-κB blocks tumorigenicity and causes tumor regression. Proc. Natl. Acad. Sci. USA 90:9901-9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinz, M., D. Krappmann, A. Eichten, A. Heder, C. Scheidereit, and M. Strauss. 1999. NF-κB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell. Biol. 19:2690-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiroasu, N., M. Shindo, S. Sakon, S. Nishinaka, M. Mihara, H. Yagita, and K. Okumura. 1998. Differential regulation of IκB kinase α and β by two upstream kinases, NF-κB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc. Natl. Acad. Sci. USA 95:3537-3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horwitz, K. B. 1993. Mechanisms of hormone resistance in breast cancer. Breast Cancer Res. Treat. 26:119-130. [DOI] [PubMed] [Google Scholar]
- 26.Huxford, T., D.-B. Huang, S. Malek, and G. Ghosh. 1998. The crystal structure of the IκBα/NF-κB complex reveals mechanisms of NF-κB inactivation. Cell 95:759-770. [DOI] [PubMed] [Google Scholar]
- 27.Iimuro Y., T. Nishiura, C. Hellerbrand, K. E. Behrns, R. Schoonhoven, J. W. Grisham, and D. A. Brenner. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J. Clin. Invest. 101:802-811. [DOI] [PMC free article] [PubMed]
- 28.Kim, D. W., M. A. Sovak, G. Zanieski, G. Nonet, R. Romieu-Mourez, A. W. Lau, L. J. Hafer, P. Yaswen, M. Stampfer, A. E. Rogers, J. Russo, and G. E. Sonenshein. 2000. Activation of NF-κB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis 21:871-879. [DOI] [PubMed] [Google Scholar]
- 29.Kyprianou, N., H. F. English, N. E. Davidson, and J. T. Isaacs. 1991. Programmed cell death during regression of the MCF-7 human breast cancer following estrogen ablation. Cancer Res. 51:162-166. [PubMed] [Google Scholar]
- 30.Malek, S., T. Huxford, and S. Ghosh. 1996. IκBα functions through direct contacts with the nuclear localization signals and DNA binding sequences of NF-κB. J. Biol. Chem. 273:25427-25435. [DOI] [PubMed] [Google Scholar]
- 31.May, M. J., and S. Ghosh. 1997. Rel/NF-κB proteins: an overview. Semin. Cancer Biol. 8:63-73. [DOI] [PubMed] [Google Scholar]
- 32.Mayo, M. W., and A. S. Baldwin. 2000. The transcription factor NF-kappa B: control of oncogenesis and cancer chemotherapy resistance. Biochem. Biophys. Acta 1470:M55-M62. [DOI] [PubMed] [Google Scholar]
- 33.McGuire, W. L., G. C. Chamness, and S. A. W. Fuqua. 1991. Estrogen receptor variants in clinical breast cancer. Mol. Endocrinol. 5:1571-1577. [DOI] [PubMed] [Google Scholar]
- 34.Murphy, L. C., and H. Dotzlaw. 1989. Variant estrogen receptor mRNA species detected in human breast cancer biopsy samples. Mol. Endocrinol. 3:687-693. [DOI] [PubMed] [Google Scholar]
- 35.Na, S.-Y., J.-E. Choi, H.-J. Kim, B. H. Jhun, Y.-C. Lee, and J. W. Lee. 1999. Bcl-3, an IκB protein, stimulates activating protein-1 transactivation and cellular proliferation. J. Biol. Chem. 274:28491-28496. [DOI] [PubMed] [Google Scholar]
- 36.Nakshatri, H., P. Bhat-Nakshatri, D. A. Martin, R. J. Goulet, and G. W. Sledge. 1997. Constitutive activation of NF-κB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 17:3629-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nechushtan, A., C. L. Smith, Y.-T. Hsu, and R. J. Youle. 1999. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 18:2330-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemoto, S., J. A. Di Donato, and A. Lin. 1998. Coordinate regulation of IκB kinases by mitogen-activated protein kinase kinase kinase-1 and NF-κB-inducing kinase. Mol. Cell. Biol. 18:7336-7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Connor, P. M., J. Jackman, I. Bae, T. G. Myers, S. Fan, M. Mutoh, D. A. Scudiero, A. Monks, E. A. Sausville, J. N. Weinstein, S. Friend, A. J. Fornace, and K. W. Kohn. 1997. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute Anticancer Drug Screen and correlation with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 57:4285-4300. [PubMed] [Google Scholar]
- 40.Ohno, H., G. Takimoto, and T. W. McKeithan. 1990. The candidate protooncogene Bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell 60:991-999. [DOI] [PubMed] [Google Scholar]
- 41.Osborn, L., S. Kunkel, and G. J. Nabel. 1989. Tumor necrosis factor alpha and interleukin 1 stimulate human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. USA 86:2336-2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pahl, H. L. 1999. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 18:6853-6866. [DOI] [PubMed] [Google Scholar]
- 43.Perillo, B., A. Sasso, C. Abbondanza, and G. Palumbo. 2000. 17β-Estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements in the coding sequence. Mol. Cell. Biol. 20:2890-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pratt, M. A. C., S. Krajewski, M. Menard, M. Krajewska, H. Macleod, and J. C. Reed. 1998. Estrogen withdrawal-induced human breast cancer tumour regression in nude mice is prevented by Bcl-2. FEBS Lett. 440:403-408. [DOI] [PubMed] [Google Scholar]
- 45.Price, J. E., A. Polyzos, R. D. Zhang, and L. M. Daniels. 1990. Tumorigenicity and metastasis of human breast carcinoma cell lines in nude mice. Cancer Res. 50:717-721. [PubMed] [Google Scholar]
- 46.Rayet, B., and C. Gelinas. 1999. Aberrant rel/nfkb genes and activity in human cancer. Oncogene 18:6938-6947. [DOI] [PubMed] [Google Scholar]
- 47.Rebollo, A., L. Dumoutier, J.-C. Renauld, A. Zaballos, V. Ayllón, and C. Martínez-A. 2000. Bcl-3 expression promotes cell survival following interleukin-4 deprivation and is controlled by AP1 and AP1-like transcription factors. Mol. Cell. Biol. 20:3407-3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santen, R., A. Manni, H. Harvey, and C. Redmond. 1990. Endocrine treatment of breast cancer in women. Endocrine Rev. 11:221-265. [DOI] [PubMed] [Google Scholar]
- 49.Sommers, C. L., S. W. Byers, E. W. Thompson, J. A. Torri, and E. P. Gelmann. 1994. Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res. Treat. 31:325-335. [DOI] [PubMed] [Google Scholar]
- 50.Sovak, M. A., R. E. Bellas, D. W. Kim, G. J. Zanieski, A. E. Rogers, A. M. Traish, and G. E. Sonenshein. 1997. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J. Clin. Investig. 100:2952-2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tamatani, M., Y. H. Che, H. Matsuzaki, S. Ogawa, H. Okado, S. Miyake, T. Mizuno, and M. Tohyama. 1999. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J. Biol. Chem. 274:8531-8538. [DOI] [PubMed] [Google Scholar]
- 52.Teixeira, C., J. C. Reed, and M. A. C. Pratt. 1995. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 protooncogene expression in human breast cancer cells. Cancer Res. 55:3902-3907. [PubMed] [Google Scholar]
- 53.Valentine, J. E., E. Kalkhoven, R. White, S. Hoare, and M. G. Parker. 2000. Mutations in the estrogen receptor ligand binding domain discriminate between hormone-dependent transactivation and transrepression. J. Biol. Chem. 275:25322-25329. [DOI] [PubMed] [Google Scholar]
- 54.Wang, C.-Y., J. C. Cusack, R. Liu, and A. S. Baldwin. 1999. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-κB. Nat. Med. 5:412-417. [DOI] [PubMed] [Google Scholar]
- 55.Westerheide, S. D., M. W. Mayo, V. Anest, J. L. Hanson, and A. S. Baldwin, Jr. 2001. The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G1 transition. Mol. Cell. Biol. 21:8428-8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoza, B. K., J. Y. Q. Hu, and C. E. McCall. 1996. Protein tyrosine-kinase activation is required for lipopolysaccharide induction of interleukin 1β and NF-κB activation, but not NF-κB nuclear translocation. J. Biol. Chem. 271:18306-18309. [DOI] [PubMed] [Google Scholar]
- 57.Zhou, P. B., M. C.-T. Hu, S. A. Miller, Z. Yu, W. Xia, S.-Y. Lin, and M.-C. Hung. 2000. Her-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-κB pathway. J. Biol. Chem. 275:8027-8031. [DOI] [PubMed] [Google Scholar]