

Abstract
It has been known for many years that fluoroacetate and fluorocitrate when metabolized are highly toxic, and that at least one effect of fluorocitrate is to inactivate aconitase. In this paper we present evidence supporting the hypothesis that the (−)-erythro diastereomer of 2-fluorocitrate acts as a mechanism based inhibitor of aconitase by first being converted to fluoro-cis-aconitate, followed by addition of hydroxide and with loss of fluoride to form 4-hydroxy-trans-aconitate (HTn), which binds very tightly, but not covalently, to the enzyme. Formation of HTn by these reactions is in accord with the working model for the enzyme mechanism. That HTn is the product of fluorocitrate inhibition is supported by the crystal structure of the enzyme-inhibitor complex at 2.05-Å resolution, release of fluoride stoichiometric with total enzyme when (−)-erythro-2-fluorocitrate is added, HPLC analysis of the product, slow displacement of HTn by 106-fold excess of isocitrate, and previously published Mössbauer experiments. When (+)-erythro-2-fluorocitrate is added to aconitase, the release of fluoride is stoichiometric with total substrate added, and HPLC analysis of the products indicates the formation of oxalosuccinate, and its derivative α-ketoglutarate. This is consistent with the proposed mechanism, as is the formation of HTn from (−)-erythro-2-fluorocitrate. The structure of the inhibited complex reveals that HTn binds like the inhibitor trans-aconitate while providing all the interactions of the natural substrate, isocitrate. The structure exhibits four hydrogen bonds <2.7 Å in length involving HTn, H2O bound to the [4Fe–4S] cluster, Asp-165 and His-167, as well as low temperature factors for these moieties, consistent with the observed very tight binding of the inhibitor.
Keywords: [Fe–S] enzyme, mechanism based inhibitor, low barrier hydrogen bonds
The mechanism of the inhibitory effects of fluorocitrate on the enzyme aconitase [citrate(isocitrate)hydrolyase, EC 4.2.1.3] has been a long-standing problem in biochemistry. The toxic nature of fluoroacetate was discovered over 50 years ago (1, 2) and citrate was found to accumulate in tissues poisoned with compounds that could provide the fluoroacetyl residue. On this basis Peters (3) and Martius (4) proposed that the inhibitory substance was a fluorotricarboxylic acid. Subsequently it was shown that 2-fluorocitrate is indeed produced metabolically via the citrate synthase reaction (5) and that in the presence of this substance the enzyme aconitase is inhibited (6). Aconitase catalyzes the conversion of citrate to isocitrate (Iso) via the obligatory intermediate cis-aconitate (Scheme SI).
Scheme I.
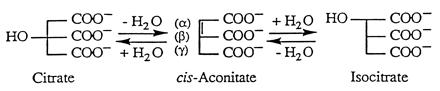
The stereochemistry of the dehydration/rehydration reactions requires that cis-aconitate bind to the enzyme in two different modes related by 180° rotation about the Cα–Cβ bond (7). It has since been shown that the enzyme contains a [4Fe–4S] cluster and that substrate binds to a specific iron of the cluster, Fea, during catalytic turnover (8).
The synthesis and purification of the four different diastereoisomers of 2-fluorocitrate by Kun and Dummel (9) led to the identification of (−)-erythro-2-fluorocitrate, (2R,3R), as the inhibitory isomer of the enzyme (10). Kinetic studies of this compound with aconitase have yielded a range of KI values and have demonstrated that depending on which substrate was used in the studies, inhibition may be either competitive or noncompetitive and that this competition is reversible (11, 12, 13). Villafranca and Platus (11) further showed that aconitase is rapidly inactivated by (−)-erythro-2-fluorocitrate. The availability of these isomers also made it possible to demonstrate that fluoride ion is released from both the (−)-erythro- (11, 14, 15) and the (+)-erythro-, (2S,3S), diastereoisomers when they are incubated with enzyme. However, for the former, the amount of fluoride released is approximately stoichiometric to the enzyme concentration present in the assay (11, 14) whereas for the latter, the loss is limited only by the original concentration of the fluorocitrate used (M.H.E., M.C.K., and H.B., unpublished results).
More recent spectroscopic data have indicated that the product of the defluorination reaction of (−)-erythro-2-fluorocitrate with aconitase is a very tight binding inhibitor. This comes from Mössbauer studies where enzyme that had been incubated with fluorocitrate and desalted to remove small nonprotein bound molecules yielded a spectrum virtually identical to that of the original sample (14). These spectra indicated that a single species was bound to ≈80% of Fea of the Fe–S cluster. Incubation of this sample with a 10- to 20-fold molar excess of citrate did not displace this species (it is difficult to reach a very high excess of substrate in the concentrated protein solutions used for Mössbauer spectroscopy). However, in an assay for enzymatic activity on a dilute aliquot of that sample, to which a 106-fold excess of Iso over the enzyme-inhibitor complex had been added, activity slowly developed (lag kinetics), indicating that the compound was tightly but not irreversibly bound to the enzyme. Based on these observations it has been proposed that the inhibitory product formed by the defluorination of (−)-erythro-2-fluorocitrate is 4-hydroxy-trans-aconitate (HTn) (14).
In this paper we present x-ray crystallographic data and chemical evidence to support this conclusion. We argue that these results provide a useful test for the proposed model of the catalytic mechanism of aconitase. In the light of present thinking, the metabolic conversion of fluoroacetate to (−)-erythro-2-fluorocitrate, in the past referred to as the “lethal synthesis” (16), can now well be viewed as the biosynthesis of a mechanism based inactivator (17).
EXPERIMENTAL METHODS
Materials.
Bovine mitochondrial aconitase was prepared and enzymatic assays were performed as described (18). The cyclohexylammonium salts of (−)-erythro-2-fluorocitrate and (+)-erythro-2-fluorocitrate were the kind gift of E. Kun (San Francisco State University). All other chemicals and enzymes were obtained from Sigma.
HPLC.
The products of the reaction of aconitase with the fluorocitrates were separated on an Organic Acid column obtained from Benson Polymeric (Reno, NV) and monitored at 210 nm following isocratic elution with 0.003 M H2SO4. The products of the reaction with the (+)-erythro-isomer were separated from enzyme using a Centricon-10 ultrafiltration unit. In the case of the (−)-erythro-isomer, the tight binding product was released by heat denaturation. The enzymatic determination of α-ketoglutarate using glutamate dehydrogenase was performed as described (19).
Crystals.
The crystals of the complex formed on addition of fluorocitrate to aconitase (enzyme-inhibitor complex) were obtained by the vapor diffusion method under anaerobic conditions at room temperature as described (20). The enzyme solution was incubated for 18 h at room temperature before being used for crystallization. The hanging drops were prepared by mixing 12 μl of a 16.4 mg/ml activated enzyme stock solution in 1.5 mM fluorocitrate, 25 mM Hepes (pH 7.5), with 18 μl of saturated (NH4)SO4 in 15 mM tricarballylate, 0.35 M NaCl, 0.25 M bis-Tris·HCl (pH 7.0). The drops were microseeded with monoclinic crystals of the aconitase–nitroisocitrate complex (20) and equilibrated against 1 ml of 2.2 M (NH4)SO4 in 5 mM tricarballylate, 0.35 M NaCl, 0.25 M bis-Tris·HCl (pH 7.0). The crystals grew within 8 weeks to a size of 1.0 × 0.8 × 0.3 mm and were characterized by precession photography. The crystals are monoclinic, space group B2, with unit cell dimensions of a = 185.9, b = 71.8, c = 72.2 Å, γ = 77.7°, and one molecule per asymmetric unit.
Data Collection.
Diffraction data were collected at 18°C using CuKα radiation from a GX-21 rotating anode generator operated at 40 kW, 50 mA with 0.2 mm focal spot, and equipped with focusing mirrors. Data were recorded with a Siemens multiwire area detector and processed with the xengen suite of programs (21). The detector was set at 15 cm from the crystal and was equipped with a helium cone. Data were collected in ω runs of 60–80° with steps of 0.25°/frame using alternate 2θ settings of 25° and 0° and 8-min and 3-min exposures, respectively. φ was advanced 30° between ω runs. The crystals were translated in the beam to minimize the effect of decay, and two crystals were used to complete the data set. For 267,668 total observations in the resolution range 20.0–2.05 Å there are 52,746 unique reflections (93.5% complete overall; 47.0% complete in the shell 2.23–2.05 Å) with Rsymm(F) of 6.0% for all data (I/σ(I) 13.9 overall; 3.2 in the shell 2.23–2.05 Å).
Structure Determination and Refinement.
The structure was solved by substituting the model for the protein from the isomorphous trans-aconitate–aconitase complex (22) and refined using xplor (23). The inhibitor and all H2O molecules, including one bound to Fe4(Fea) of the [4Fe–4S] cluster, were omitted from this model. The model was refined as a rigid body in two segments comprising residues 2–520 and 521–754 against data in the range 8.0-Å to 4.0-Å resolution. The resolution was extended in four steps with positional refinement and the refinement converged to an R-factor of 16.2% for all data in the range 8.0 to 2.5 Å. Individual temperature factor refinement reduced the R-factor to 15.4%. Side chains of all residues and water molecules were checked and repositioned in 2|Fo| − |Fc| maps using xtalview (24). Positional refinement was repeated in four more steps until all data were included to 2.2 Å (R-factor, 16.5%).
At this stage a 2|Fo| − |Fc| map revealed unambiguously defined electron density in the active site and adjacent to Fe4 consistent with the α, β, γ-carboxyl, and γ-hydroxyl groups of HTn (Fig. 1). A model for this compound was obtained by positioning trans-aconitate (Tn) into the electron density as observed for the trans-aconitate–aconitase complex (22). Only minor adjustment of the model was necessary. The hydroxyl group was fit to the density and modeled to coordinate to Fe4 in the manner observed for the Iso and nitroisocitrate (Nis) complexes of aconitase (20). Positional refinement was continued in three more steps until all the data to 2.05 Å were included. Inspection of a 2|Fo| − |Fc| map indicated that no further adjustment of the inhibitor or the protein was necessary. A |Fo| − |Fc| map calculated with data from 20.0 Å to 2.05 Å showed one large peak adjacent to Fe4 of the [4Fe–4S] cluster. This peak was modeled as a water molecule bound to Fe4 as observed in four other aconitase complexes (20, 22). The stereochemistry of the [4Fe–4S] cluster, bound H2O, and HTn was restrained using the parameters employed for refinement of the structures of the Tn and Iso complexes (20, 22).
Figure 1.
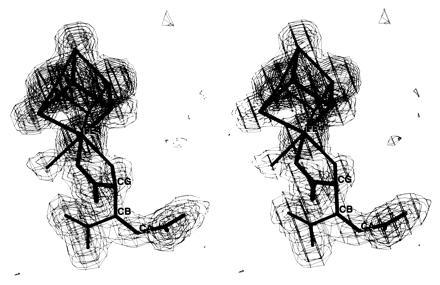
Stereo figure of the electron density for the HTn complex of aconitase. The Fourier map is computed with coefficients |Fo| − |Fc|, all observed data in the resolution range 20.0–2.05 Å, and phases derived from the refined model with the [4Fe–4S] cluster, H2O bound to Fe4 and HTn omitted. The map is contoured at 3, 6, 9, 12, and 15 σ. CA, CB, and CG atoms correspond to Cα, Cβ, and Cγ, respectively, of the inhibitor. Fe4 is the spectroscopically unique cluster subsite also denoted as Fea.
Positional and B-factor refinement converged to an R-factor of 0.177 for all reflections with |Fo| > 0 in the resolution range of 8.0 to 2.05 Å. The final model has 5838 nonhydrogen atoms and 256 water molecules. The rms deviations from ideality are 0.012 Å for bond lengths and 3.01° for bond angles. The Ramachandran plot shows six nonglycyl residues outside of allowed ranges (data not shown); these residues are on the surface of the molecule and have high B-factors.
RESULTS
When aconitase is incubated with (−)-erythro-2-fluorocitrate, fluoride ion stoichiometric to the enzyme concentration is lost (11, 14). The tightly bound defluorinated product can only be released by a large excess of substrate (≥104-fold) or by denaturation of the protein. Separation by HPLC of the product obtained by heat denaturation yielded a substance with a retention time of 9.3 min compared with 7.6 min for the 2-fluorocitrate. When the isolated 9.3-min fraction was added to fresh active enzyme and enzymatic activity with Iso was then determined, lag kinetics identical to those seen with the original sample prior to denaturation were observed. Unfortunately we were never able to obtain a sufficient quantity of pure material to identify this substance by physical chemical means. In contrast to the (−)-erythro- isomer the (+)-erythro- compound upon treatment with aconitase loses fluoride stoichiometric to the amount of fluorocitrate added. Furthermore, lag kinetics are not observed with enzyme so treated, and the products formed can be separated from protein by ultrafiltration. Analysis of the ultrafiltrate by HPLC gave two new peaks positioned at 8.1 and 9.2 min. This pattern was identical to one given by an authentic sample of oxalosuccinate. Furthermore, when either the products of the reaction or oxalosuccinate itself were treated with acid and heated for 15 min at 95°C and rechromatographed there was a decrease in the 8.1-min peak with a concomitant increase in the 9.2-min peak. This latter peak was identified as α-ketoglutarate both by comparison of the HPLC pattern of the pure compound and by enzymatic assay with glutamate dehydrogenase (19). The implications of these findings in regard to the enzyme mechanism will be discussed below.
Fig. 1 shows the 2.05-Å resolution electron density map for the fluorocitrate inhibited complex of aconitase. The density is clear for trans coplanar carboxyl groups bonded to Cα and Cβ and carboxyl and hydroxyl groups tetrahedrally bonded to the same carbon (Cγ). These features are consistent with the bound molecule being HTn. The appearance of the electron density is very similar to that for the Tn complex (22) with the addition of density for a hydroxyl group adjacent to Fe4. Electron density for a small molecule bound to Fe4 is also like that observed for the Tn complex and, as in that structure, has been modeled as H2O (25).
Fig. 2 shows the structure of HTn, the [4Fe–4S] cluster and four residues implicated in the catalytic mechanism (8). HTn binds in a nearly identical fashion as Tn except that the hydroxyl replaces a hydrogen in Tn (22). For both HTn and Tn the binding mode mimics that of Iso and the reaction intermediate analog, Nis (20). However, HTn and Tn bind in an inverted manner with respect to Iso and Nis with the Cα and Cγ carbon positions interchanged. Consequently, the double bond is removed by one carbon–carbon bond from Fe4. In the Iso, Nis, and HTn complexes Fe4 is six-coordinate with a H2O molecule also being bound. In the Tn complex Fe4 is five-coordinate but the geometry is very similar with the position of the hydroxyl group being vacant. Least squares superposition of the structures of the Iso, Nis, HTn, and Tn complexes in all pairwise combinations results in rms differences in the range 0.108 to 0.226 Å for 753 Cα atoms and 0.06 to 0.39 Å at Fe4. By both criteria the structures of the complexes with Nis and HTn are the two most similar. Bond lengths to Fe4 and average B-factors for the bound species are listed in Table 1. The HTn complex exhibits the lowest B-factors, the shortest Fe4-carboxyl distance, and a Fe4-hydroxyl distance as short as in the Nis complex.
Figure 2.
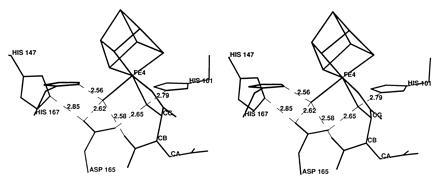
Stereo figure of the structure of the HTn complex of aconitase showing the environment of three oxygen atoms coordinated to Fe4 of the [4Fe–4S] cluster in the active site. In the complex the hydroxyl oxygen and one Cγ-carboxyl oxygen of HTn are bonded to Fe4 in addition to a H2O molecule. The six hydrogen bonds indicated (in Å) are conserved in the structures of the Iso, Nis, and Tn complexes except that Tn lacks a hydroxyl.
Table 1.
Bond lengths and B-factors in four aconitase structures
| Species bound | Iso | Nis | Tn | HTn |
|---|---|---|---|---|
| Bond length, Å | ||||
| Fe*–carboxyl† | 2.49 | 2.41 | 2.61 | 2.27 |
| Fe-hydroxyl‡ | 2.15 | 2.35 | — | 2.15 |
| Fe-H2O§ | 2.21 | 2.44 | 2.24 | 2.48 |
| Average B, Å2 | ||||
| [4Fe–4S] | 14.0 | 10.8 | 15.3 | 6.9 |
| H2O§ | 15.9 | 11.9 | 17.3 | 10.5 |
| Substrate/inhibitor | 15.7 | 15.7 | 14.3 | 8.3 |
Fe4 of the [4Fe–4S] cluster.
Cα carboxyl oxygen for Iso and Nis, Cγ-carboxyl oxygen for Tn and HTn.
Hydroxyl oxygen of substrate or inhibitor.
Solvent species modeled as H2O.
A network of hydrogen bonds is formed in the HTn complex involving the inhibitor, bound H2O and active site residues (Fig. 2). Three histidines interact with carboxylates (His-101·Asp-100, His-147·Asp-165, His-167·Glu-262) so that each is expected to be protonated. If this is the case, His-101 can then donate a hydrogen bond to the inhibitor hydroxyl, and the hydroxyl can donate a hydrogen bond to Asp-165. At the same time the bound H2O on Fe4 can donate hydrogen bonds to the inhibitor Cβ-carboxyl and Asp-165 while accepting a hydrogen bond from His-167. If both bonding and hydrogen bonding interactions are considered, the geometry at the inhibitor hydroxyl and bound water is essentially tetrahedral. A similar arrangement is observed in the structures of Iso and Nis complexes. Hydrogen bonds involving these two oxygen atoms are expected to play a key role in promoting the proton transfers required for substrate binding and catalysis (20, 22).
DISCUSSION
The stoichiometric loss of fluoride ion from (−)-erythro-2-fluorocitrate with the formation of HTn can be explained very nicely in terms of the proposed mechanism for aconitase action. As shown in Fig. 3 the cis-aconitate formed by the dehydration of citrate must rotate 180° about the Cα–Cβ bond—i.e., “flip” before hydration to the correct Iso stereoisomer can occur (8, 22). To account for the formation of HTn, the enzyme must first convert the 2-fluorocitrate to fluoro-cis-aconitate which must then flip to bind in the alternate mode. The addition of OH− results in the loss of fluoride with the formation of HTn which remains tightly bound to the enzyme (Fig. 4). Similarly, the (+)-erythro-isomer can serve as a pseudo-substrate with the proposed formation of 2-fluoroisocitrate (Fig. 5). However, in this case loss of fluoride leads to the formation of oxalosuccinate which is released from the enzyme. This allows for turnover to continue until the pseudo-substrate is depleted and also accounts for the complete loss of fluoride by this isomer. It should be pointed out that we have no chemical evidence to date for the formation of the proposed fluoroisocitrate intermediates. Also, to date repeated attempts to synthesize HTn have been unsuccessful (K. B. Sharpless, personal communication). The enzymatic synthesis of this compound prevents all the unwanted side reactions that can occur in the chemical synthesis of this highly oxygenated molecule. However, since production of HTn by aconitase occurs as a single turnover event, it would take unreasonable quantities of enzyme to produce sufficient quantities of the compound for separation, purification and characterization and for use in further studies.
Figure 3.
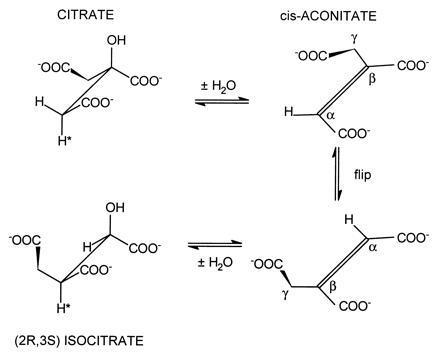
Proposed mechanism for the reactions catalyzed by aconitase. The “flip” step indicates that cis-aconitate must be released (displaced) and rebind in the alternate binding mode.
Figure 4.
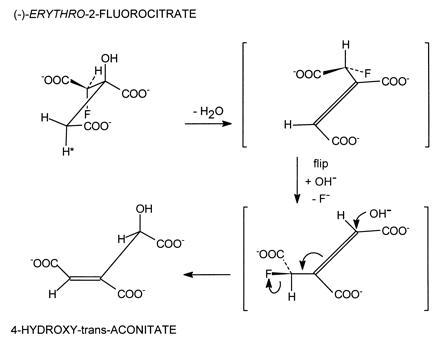
Proposed steps in the reaction leading to the synthesis of HTn by aconitase. The (−)-erythro isomer of 2-fluorocitrate loses fluoride stoichiometric with total enzyme concentration.
Figure 5.
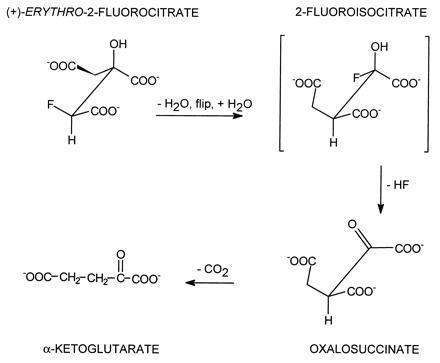
Proposed steps and products in the reaction of the (+)-erythro isomer of 2-fluorocitrate with aconitase. The oxalosuccinate formed is unstable and loses CO2 to form α-ketoglutarate. In this case release of fluoride is stoichiometric with total substrate added.
The crystallographic data are fully consistent with the tightly bound product of 2-fluorocitrate inhibition being HTn. Although the electron density is only at 2.05-Å resolution for this small molecule, two essential features of HTn are well resolved: coplanar carboxyl group carbon atoms trans to Cα and Cβ; and a tetrahedrally bonded carbon on Cγ. The structure explains why HTn is a potent inhibitor. HTn, like Tn, mimics the natural substrate, Iso, by placing all three carboxyl groups at equivalent positions in the active site while displacing the double bond from the carbon closest to Fe4. Consequently, the enzyme cannot hydrate the double bond. In addition, HTn provides the interaction of a hydroxyl group with Fe4, His-101, and Asp-165 in a manner very similar to that in the Iso and Nis complexes. HTn therefore is a nonhydratable compound with all the interactions of Iso. In this regard, HTn appears to be a reaction intermediate analog like Nis (26). The virtual identity of the structures of the HTn and Nis complexes, when superposed, supports this interpretation. The very low B-factors and short Fe–oxygen distances are also consistent with this interpretation (Table 1).
The structure of the HTn·aconitase complex exhibits short (<2.7 Å) hydrogen bonds (Fig. 2), which is consistent with the interpretation that HTn acts as a reaction intermediate analog. One of these hydrogen bonds, between H2O bound to Fe4 and the Cβ-carboxyl group of the substrate or inhibitor, may be a low-barrier hydrogen bond (27). The Cβ carboxyl group is expected to carry extra negative charge in the carbanion transition state (20, 27). Very short hydrogen bonds between donor and acceptor atoms with similar pKa values are proposed to stabilize transition states by up to 20 kcal/mol, accounting for the rate enhancement observed in enzymic reactions (27, 28, 29). In the structures of the Iso, Nis, and HTn complexes this distance is 2.73, 2.52, and 2.58 Å, respectively (Fig. 2), in agreement with the hypothesis. However, it has also been argued that multiple hydrogen bonds can have an additive effect, and that this accounts for the stabilization of the transition state (30, 31). In this regard the structures of the Iso, Nis, and HTn complexes have one, two, and four short hydrogen bonds, respectively, consistent with the very tight binding of HTn. It is interesting to note that all of the short interactions observed with HTn involve the bound H2O or Asp-165; the distance between these two particular moieties (2.62 Å) is also <2.7 Å in the structures of the Nis and Iso complexes.
Finally, a comment regarding the toxicity of fluoroacetate/fluorocitrate should be made. Whether or not the inhibition of aconitase by HTn accounts for the neurotoxic effects of these compounds cannot be answered by our studies despite the fact that HTn has been shown to be an extremely tight binding inhibitor of aconitase. It has been suggested that the accumulation of citrate with its ability to bind calcium may be the cause of the convulsive seizures in animals to whom fluoroacetate or 2-fluorocitrate has been administered (32). Another hypothesis that has been proposed is that 2-fluorocitrate interferes with membrane processes, including the transport of tricarboxylic acids (33). The elucidation of the actual mechanism(s) of toxicity awaits among other things, the successful synthesis and purification of HTn.
Acknowledgments
We acknowledge discussions with Drs. W. W. Cleland and J. V. Schloss who suggested that HTn may be formed in the reaction of (−)-erythro-2-fluorocitrate with aconitase and also comments of Dr. Cleland and Dr. J.-J. Kim on the manuscript. We also acknowledge support of this work by National Institutes of Health Grants GM38412 to H.B., GM51831 to M.C.K., and GM36325 to C.D.S.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: HTn, 4-hydroxy-trans-aconitate; Tn, trans-aconitate; Iso, isocitrate; Nis, nitroisocitrate.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY 11973 (reference 1FGH).
References
- 1.Marais J S C. Onderstepoort J Vet Sci Anim Ind. 1944;20:67–73. [Google Scholar]
- 2.McCombie H, Saunders B C. Nature (London) 1946;158:382–385. [Google Scholar]
- 3.Liebecq C, Peters R A. Biochim Biophys Acta. 1949;3:215–230. [PubMed] [Google Scholar]
- 4.Martius C. Ann Chem. 1949;561:227–232. [Google Scholar]
- 5.Peters R A, Wakelin R W, Buffa P, Thomas L C. Proc R Soc London Ser B. 1953;140:497–507. doi: 10.1098/rspb.1953.0004. [DOI] [PubMed] [Google Scholar]
- 6.Morrison J F, Peters R A. Biochem J. 1954;56:473–479. doi: 10.1042/bj0580473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gawron O, Mahajan K P. Biochemistry. 1966;5:2343–2350. doi: 10.1021/bi00871a024. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy M C, Stout C D. Adv Inorg Chem. 1992;38:323–339. [Google Scholar]
- 9.Kun E, Dummel R J. Methods Enzymol. 1969;13:623–672. [Google Scholar]
- 10.Carrell H L, Glusker J P, Villafranca J J, Mildvan A S, Dummel R J, Kun E. Science. 1970;170:1412–1414. doi: 10.1126/science.170.3965.1412. [DOI] [PubMed] [Google Scholar]
- 11.Villafranca J J, Platus E. Biochem Biophys Res Commun. 1973;55:1197–1207. doi: 10.1016/s0006-291x(73)80021-x. [DOI] [PubMed] [Google Scholar]
- 12.Brandt M D, Evans S M, Mendes-Morao J, Chappell J B. Biochem J. 1973;134:217–224. doi: 10.1042/bj1340217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eanes R Z, Kun E. Mol Pharmacol. 1974;10:130–139. [PubMed] [Google Scholar]
- 14.Kent T A, Emptage M H, Merkle H, Kennedy M C, Beinert H, Münck E. J Biol Chem. 1985;260:6871–6881. [PubMed] [Google Scholar]
- 15.Tecle B, Casida J E. Chem Res Toxicol. 1989;2:429–435. doi: 10.1021/tx00012a012. [DOI] [PubMed] [Google Scholar]
- 16.Peters R A. Adv Enzymol. 1957;18:113–159. doi: 10.1002/9780470122631.ch3. [DOI] [PubMed] [Google Scholar]
- 17.Silverman R B. Methods Enzymol. 1995;249:240–283. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- 18.Kennedy M C, Emptage M H, Dreyer J-L, Beinert H. J Biol Chem. 1983;258:11098–11105. [PubMed] [Google Scholar]
- 19.Bergmeyer H U. Methods Enzym Anal. 1985;3:20–24. [Google Scholar]
- 20.Lauble H, Kennedy M C, Beinert H, Stout C D. Biochemistry. 1992;31:2735–2748. doi: 10.1021/bi00125a014. [DOI] [PubMed] [Google Scholar]
- 21.Howard A J, Nielsen C, Xuong N H. Methods Enzymol. 1985;114:452–471. doi: 10.1016/0076-6879(85)14030-9. [DOI] [PubMed] [Google Scholar]
- 22.Lauble H, Kennedy M C, Beinert H, Stout C D. J Mol Biol. 1994;237:437–451. doi: 10.1006/jmbi.1994.1246. [DOI] [PubMed] [Google Scholar]
- 23.Brünger A T, Karplus M, Petsko G A. Acta Crystallogr. 1989;45:50–61. [Google Scholar]
- 24.McRee D E. J Mol Graphics. 1992;10:44–47. [Google Scholar]
- 25.Werst M M, Kennedy M C, Beinert H, Hoffman B M. Biochemistry. 1990;29:10526–10532. doi: 10.1021/bi00498a015. [DOI] [PubMed] [Google Scholar]
- 26.Schloss J V, Porter D J T, Bright H J, Cleland W W. Biochemistry. 1980;19:2358–2362. doi: 10.1021/bi00552a012. [DOI] [PubMed] [Google Scholar]
- 27.Cleland W W, Kreevoy M M. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 28.Cleland W W. Biochemistry. 1992;31:317–319. doi: 10.1021/bi00117a001. [DOI] [PubMed] [Google Scholar]
- 29.Frey P A, Whitt S A, Tobin J B. Science. 1994;264:1927–1930. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 30.Shan S, Loh S, Herschlag D. Science. 1996;272:97–101. doi: 10.1126/science.272.5258.97. [DOI] [PubMed] [Google Scholar]
- 31.Shan S, Herschlag D. J Am Chem Soc. 1996;118:5515–5518. [Google Scholar]
- 32.Clarke D D. Neurochem Res. 1991;16:1055–1058. doi: 10.1007/BF00965850. [DOI] [PubMed] [Google Scholar]
- 33.Kun E, Kirsten E, Sharma M L. Proc Natl Acad Sci USA. 1977;74:4942–4946. doi: 10.1073/pnas.74.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]