

Abstract
Purpose
Our previous studies have shown tissue hypoxia in the lung after irradiation contributing to radiation-induced injury. The purpose of this study was to determine the temporal onset of hypoxia following irradiation and show how it relates to pulmonary vascular damage, macrophage accumulation, production of reactive oxygen species and cytokines.
Methods and Materials
Female Fisher 344 rats were irradiated to the right hemithorax with a single dose of 28Gy. Serial studies were performed up to 20 weeks following irradiation. Radionuclide lung perfusion studies were carried out to detect changes in the pulmonary vasculature. Immunohistochemical studies were conducted to determine macrophages, tissue hypoxia (CA-9 marker), oxidative stress (8-OHdG), as well as expression of profibrogenic (TGF-β) and proangiogenic (VEGF) cytokines.
Results
Significant changes in lung perfusion along with tissue hypoxia were observed at three days after irradiation. Significant oxidative stress was detected one week post radiation, whereas macrophages started to accumulate at 4 weeks. Significant increase in TGF-β expression was seen within one day after radiation and for VEGF at 2 weeks post radiation. Hypoxia, oxidative stress, and both cytokines continued to rise with time after irradiation. The steepest increase correlated with vast macrophage accumulation.
Conclusions
Early changes in lung perfusion initiate among other factors the development of hypoxia and chronic oxidative stress after irradiation. Tissue hypoxia is associated with a significant increase in activation of macrophages and their continuous production of ROS stimulating production of fibrogenic and angiogenic cytokines and maintaining the development of chronic radiation-induced lung injury.
Keywords: radiotherapy, radiation pneumonitis, lung fibrosis, hypoxia, oxidative stress
Introduction
Radiation induced pulmonary toxicity is a significant cause of morbidity and mortality in patients treated for tumors in the thoracic region 1, 2. Apparent clinical, radiographic and histologic changes caused by radiation usually develop within weeks to months following irradiation and have been well documented 3, 4. However, the mechanisms underlying the pathogenesis are still uncertain. The classical linear “target cell” concept, referring exclusively to the depletion of clonogenic cells by irradiation, is insufficient to explain the pathogenesis of radiation induced lung injury. In the last decade, it has been well established that a cascade of events on the cellular and molecular level begins immediately after exposure to radiation and proceeds during a period of clinically occult pulmonary injury 5, 6. Radiation activates various cellular signaling pathways that lead to expression and activation of proinflammatory and profibrotic cytokines, vascular injury, and activation of the coagulation cascade leading to the development of edema, inflammatory responses, and the initiation of wound-healing processes 7–11. Multiple factors in this process have been investigated, but the main initiating events and driving forces in the perpetuation of radiation-induced lung injury are unknown, hampering the development of protection strategies and causal treatment of late radiation sequelae as well as the search for predictive markers.
Our previous studies have shown that hypoxia develops in rodent lung tissue after radiation and is associated with increased macrophage accumulation and activation, oxidative stress, and pro-fibrogenic/pro-angiogenic cytokine activity contributing to radiation-induced pulmonary injury 12–14. In general, tissue hypoxia is considered a major signal in wound healing and tissue remodeling processes 15, 16. Another important element in tissue remodeling processes and specifically in fibrogenesis is the balance of reactive oxygen (ROS) and nitrogen species (RNS) 17. In our in vitro studies we have demonstrated that hypoxia elicits macrophages to produce higher levels of TGF-β (transforming growth factor-beta), VEGF (vascular endothelial growth factor) and superoxide leading to enhanced oxidative stress 14. In addition, superoxide contributed to increased macrophage cytokine production, which was diminished by applying an antioxidant superoxide dismutase mimetic14.
Hence we hypothesize that hypoxia (and consequently oxidative stress) is one of the driving forces in initiating and perpetuating radiation induced lung injury. Therefore, the goal of this study is to determine the temporal onset and course of hypoxia after irradiation of the right hemithorax of rats and to determine how it correlates to vascular damage, macrophage accumulation, reactive oxygen species and cytokines which are known to be involved in processes leading to both immediate and chronic lung tissue injury.
Methods and Materials
Animals
Experiments were performed using 70 female Fisher-344 rats with prior approval from the Duke University Institutional Animal Care and Use Committee. The animals were housed 3 per cage and maintained under identical standard laboratory conditions. Food and water were provided ad libitum. Serial studies were performed before and at 1, 3, and 7 days, and 2, 4, 6, 8, 10, 14 and 20 weeks after irradiation.
Irradiation
At the time of irradiation all rats weighed between 160 and 170 g to minimize possible variations in lung size. The animals were anesthetized before irradiation with intraperitoneal injection of ketamine (65 mg/kg) and xylazine (4.5 mg/kg) and placed in a prone position. Hemithoracic radiation was delivered to the right lung with a single dose of 28 Gy using 150 kV x-rays with a dose rate of 0.71 Gy/min (Therapax 320, Pantak Inc., East Haven, CT). 12 mm lead blocks were used to protect the left thorax and the rest of the body.
Radionuclide Lung Perfusion Assay
Ten animals were followed up for 10 weeks using lung perfusion assays performed prior to and longitudinally in time at 1, 3, 5, and 7 days, and 2, 4, 6, 8, and 10 weeks after irradiation. For the assay, the animals were anesthetized with intraperitoneal injection of pentobarbitol (35 mg/kg) and placed in a prone body position on a flat surface in order to be imaged from the posterior aspect by scintigraphy (Picker Dyna Camera). For scintigraphic imaging the gamma camera was collimated with a pin hole collimator and positioned 25 mm above the animal. 99mTc-labeled macroaggregated albumin (2.78–3.78 MBq) in 0.04 ml of 0.9% NaCl solution was injected into the tail vein. 2 minutes later an accumulative 120s image was acquired with the gamma camera. Following each imaging segment, the rats were permitted to recover and were returned to housing until the next imaging date. Scintigraphy images were analyzed using rectangular regions of interest (ROIs), which circumscribed the left and right lung fields of each animal separately. Total activity in each lung was recorded (counts/ROI/120s) and lung perfusion ratio was calculated as the ratio of the right (irradiated) to the left (non-irradiated) lung activity. The mean and standard errors of lung perfusion ratios were calculated and evaluated as a function of time.
Histology
Five animals each were sacrificed before and at 1, 3, and 7 days, and 2, 4, 6, 8, 10, 14, and 20 weeks after irradiation for immunohistochemistry and histopathology studies. Animals were euthanized with pentobarbital overdose. Both lungs were infused by tracheal instillation of a solution containing 10% neutral-buffered formalin, 2% glutaraldehyde, and 0.085 M sodium cacodylate buffer for 25 minutes for fixation prior to removal of the lung. After removal the four lobes of the irradiated right lung were separated and embedded in paraffin. The tissue was then cut into 5μm thick sections at a microtome, stored on slides and stained.
Immunohistochemistry
Lungs were assessed for the number and activity of macrophages with ED-1 staining, the level of expression of CA-9 (carbonic anhydrase-9) as a tissue hypoxia marker, the level of expression of 8-OHdG (8-hydroxy-2′-deoxyguanosine) as a marker for oxidative stress/ ROS production, and levels of TGF-β as a profibrogenic and VEGF as a proangiogenic cytokine.
Immunohistochemistry was performed as described by Hsu et al 18. Briefly, the tissue sections were deparaffinized and rehydrated with xylene and alcohol concentrations between 100% and 80%. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 minutes. The slides were then placed in a citrate buffer (Biogenex, San Ramon, CA) and heated in a microwave for 10 minutes for antigen retrieval. The tissue sections were rinsed with phosphate-buffered saline and incubated with primary antibodies to activated macrophage marker ED1 (MCA341, 1:100, Serotec, Oxford, UK), CA-9 (ab15086, 1:2000, Abcam Inc, Cambridge, MA), 8-OHdG (mouse monoclonal, 1:2000, JaICA, Shizuoka, Japan), VEGF (SC-152, 1:100, Santa Cruz Biotechnology Inc., Santa Cruz, CA), and active TGF-β1 (Sc-146, 1:200, Santa Cruz Biotechnology Inc., Santa Cruz, CA) overnight at 4°C. Slides were then washed three times in phosphate-buffered saline for 5 minutes followed by the incubation with the appropriate secondary antibody (1:200, Jackson ImmunoResearch, West Grove, PA) for 30 minutes at room temperature. Again slides were washed three times in phosphate-buffered saline for 5 minutes followed by incubation with ABC-Elite (Vector Laboratories, Burlingame, CA) for 30 minutes at room temperature and developed using DAB working solution (Laboratory Vision, Fremont, CA). Finally, the slides were counterstained with Harris hematoxylin (Fisher Scientific, Pittsburgh, PA) and mounted with coverslips.
Image Analysis
For image analysis tissue sections from the lower right lobe from each animal were examined. Image analysis was carried out as previously described 19. Briefly, slides were systematically scanned under lower magnification in a microscope to define the injury of that specific timepoint by evaluating hematoxylin and eosin stained slides along with immunohistochemistry stained consecutive slides by an experienced pathologist. Then, five digital images representing the injury of that timepoint were acquired from each slide using a 20x objective. Activated macrophage marker ED1 and 8-OHdG staining were quantified through image analysis with ImageJ (Version 1.32j; National Institute of Health, Bethesda, MD). The digital image was thresholded to have a clear visualization of the positive expression. Using an automatic analyse particle function the positive expression was quantified. Results were expressed as the number of positively stained macrophages or 8-OHdG positive nuclei per digital image (average of five digital images per animal, average of five animals per time-point).
CA-9, active TGF-β and VEGF were analysed in Adobe Photoshop (Version 6.0; Adobe Systems, San Jose, CA). The magic wand tool was used to select a dark brown positive stain on the digital image. The “Similar” and “Grow” functions were then used to select all positive expression. After quantifying the positive expression per image the total tissue area regardless of expression was quantified. The percentage of positively stained pixels as the ratio of positive staining over total tissue area per digital image acquired at low magnification was recorded. Results represent the average percentage of positive staining from five digital images per animal and five animals per time point.
Statistical Analysis
All data are presented as mean ± standard error of the mean for each parameter. Time points for each parameter were compared against the pre-irradiation controls by t tests. All p values reported were two-sided and statistical significance was defined as p <0.05. All statistical analyses were performed using the JMP software version 6.0 (SAS Institute, Inc., Cary, North Carolina).
Results
Radionuclide lung perfusion studies were carried out up to 10 weeks after irradiation and the perfusion ratio of the irradiated right versus non-irradiated left lung was compared. Right/left perfusion ratio allows data comparison between animals even if the amount of tracer reaching the lungs varied due to tail vein injection difficulties (about 5% of animals) and inter-individual differences.
The baseline ratio of the perfusion of the right lung to the left lung before irradiation was 1.3. A significant decrease in lung perfusion occurred within three days following irradiation (Fig. 1) (perfusion ratio at 3 days vs. pre-radiation; p=0.0322). There was a short-term recovery (reperfusion) after the initial decline ending at week 2. After the second week, there was a significant and steady fall in perfusion of the irradiated right lung until the completion of the 10 weeks observation period (perfusion ratio at 4 weeks vs. pre-radiation; p=0.0259). At 10 weeks post irradiation, the lung perfusion was significantly reduced in the irradiated lung; for example, attaining only ∼15% of the initial perfusion values that were assessed prior to irradiation. These findings correlated with the histopathologic evidence of damage of the irradiated lung.
Fig. 1.
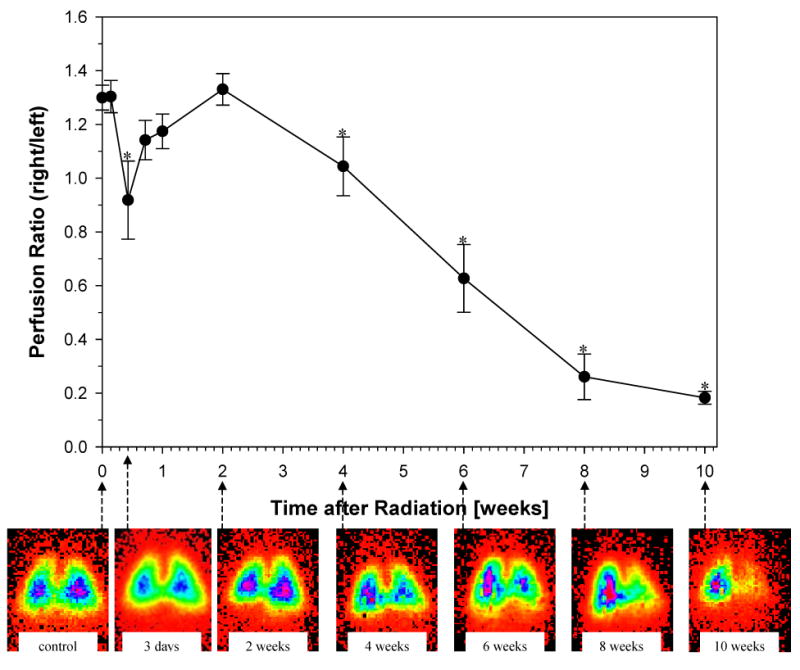
Time related changes in lung perfusion after 28 Gy single dose irradiation to the right hemithorax of rats. Mean perfusion ratio (see Methods) as a function of time (at pre-irradiation time point, T=0, for a period of 10 weeks post-irradiation). A significant decrease in lung perfusion was observed 3 days after irradiation (p=0.0322). After a short recovery period lung perfusion progressively declined from the 2 week post-irradiation time point, until the conclusion. Data are represented as mean (± 1 SEM) observed in 10 animals per time point. *p<0.05 compared with no radiation. Representative scintigraphic images (see Methods) in one animal at pre-irradiation (control) and throughout the 10 week time-course (3 days – 10 weeks) are shown for comparison. Color ramp is used to demonstrate radiolabled perfusion marker with activity levels from low to high indicated by orange < yellow < green < blue < majenta.
Five animals were harvested at each timepoint and the weight of the right lung was recorded immediately (Fig. 2). One day after irradiation an increase in the weight of the irradiated right lung was seen (p=0.0117), slightly decreasing again around 1 week following irradiation and steadily increasing afterwards peaking at week 6. Hereafter the weight of the irradiated right lung continued to decline reaching a lower weight 20 weeks after irradiation compared to the initial weight before irradiation (p=0.0384).
Fig. 2.
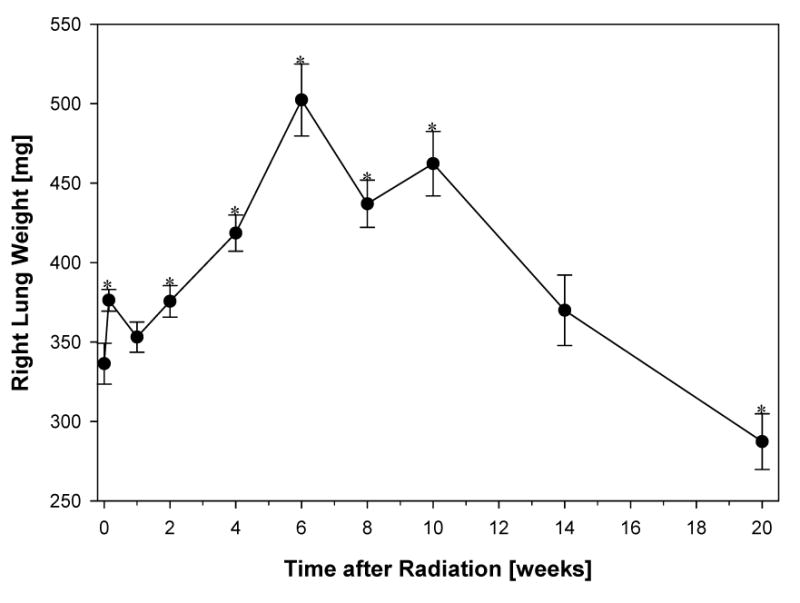
The wet weight of the right lung was recorded immediately after harvesting. At 6 weeks after irradiation the increase in lung weight coincides with edema and inflammation. This is followed by a steady decline in lung weight related to shrinkage and progressive fibrotic changes of the lung.
Data are represented as mean obtained from five animals per timepoint, error bars are ± 1 SEM. *p<0.05 compared with no radiation.
As expected, there was an increase in the number of total and activated macrophages detected in the right lung with time after irradiation (Fig. 3a and Fig. 4a). The number of macrophages increased significantly in the fourth week post radiation (p=0.0231) and continued to rise steadily until week 10. From this time point until the end of the study (week 20) the number of macrophages declined but still sustained on higher levels compared to the pre-radiation status.
Fig. 3.
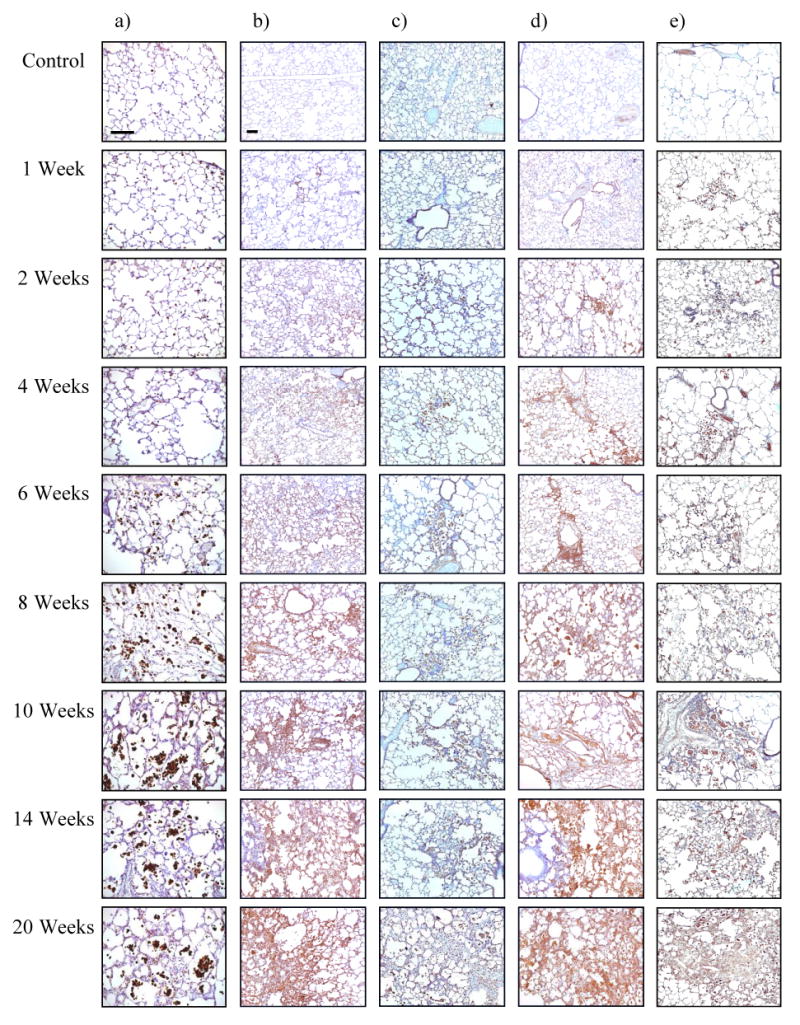
- activated macrophages (ED-1)
- hypoxia (CA-9)
- ROS (8-OHdG)
- VEGF
- TGF-β
Fig. 4.
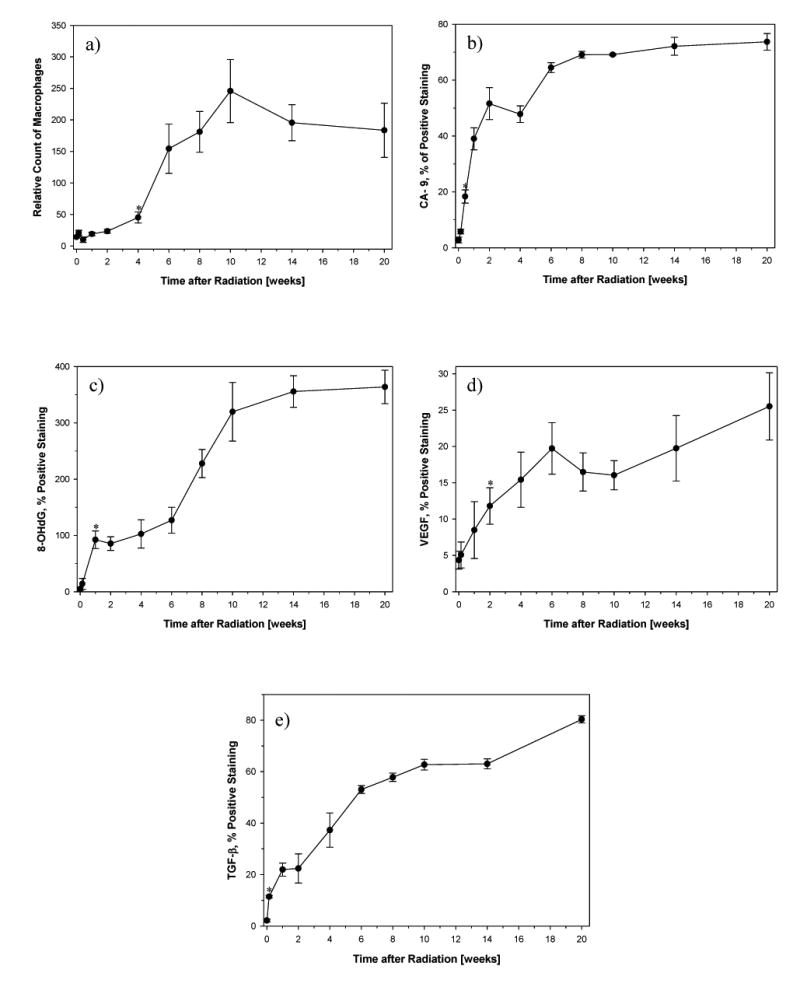
- Number of activated macrophages (ED-1): A significant increase in macrophages was seen 4 weeks after irradiation (p=0.0231) and peaked at 10 weeks (p=0.00979).
- Hypoxia (CA-9): A significant increase in CA-9 expression was already seen 3 days after irradiation (p=0.000327) and increased further with time.
- ROS (8-OHdG): A significant increase in 8-OHdG expression started 1 week after irradiation (p=0.0097) and increased further with time.
- VEGF: A significant increase in VEGF expression started 2 weeks after irradiation (p=0.0284).
- TGF-β: A significant increase in TGF-β expression was already seen 1 day after irradiation (p=0.00146) and continuously increased further with time.
Tissue hypoxia is associated with the increased expression of CA-9. A significant increase in CA-9 positive staining was observed at day 3 compared to the control (p=0.000327) (Fig. 3b and Fig. 4b) and corresponding to the drop in perfusion at day 3. Severe hypoxia (> 60% positive staining) developed between weeks 6–20 after irradiation. CA-9 positive staining was most intensely seen in macrophages but also in pulmonary epithelial cells.
Oxidative stress is associated with increased expression of 8-OHdG which was detected in the irradiated lung tissue as well (Fig. 3c and Fig. 4c). The amount of tissue positive for 8-OHdG rose significantly within the first week after irradiation (p=0.0097) and continued to increase during the entire 20 weeks of the study.
The proangiogenic cytokine VEGF was another marker used to examine the change in the irradiated lung (Fig. 3d and Fig. 4d). A significant rise in VEGF positive staining was seen at 2 weeks after radiation (p=0.0284) and continued to rise thereafter.
As expected, the expression of TGF-β began to increase shortly after irradiation (1 day post radiation vs control; p=0.00146) and continuously increased over time (Fig. 3e and Fig. 4e).
Discussion
In the present study we detected early vascular changes associated with hypoxia in rat lungs three days after irradiation. Our previous studies had shown hypoxia in rat lungs at 6 weeks after irradiation, before the onset of functional or histopathologic changes 12. Fourteen and twenty four weeks after irradiation more severe hypoxia was seen using pimonidazole and CA-9, respectively 12–14. Hypoxia was associated with macrophage accumulation and activation, oxidative stress and upregulation of TGF-β and VEGF. The results suggested hypoxia to be a significant driving force in maintaining the cycle of repair processes that lead to chronic injury. In the present study, we wanted to more closely examine earlier events within days to weeks and the timeline after irradiation. We used CA-9 in order to visualize hypoxia in the lung tissue at different time points. The CA-9 protein is strongly induced by hypoxia and its transcription is regulated by the von Hippel-Lindau tumor suppressor gene (VHL)/ HIF 1α system 20. In tumors, CA-9 was confirmed as a useful endogenous marker of hypoxia and showed an excellent co-localization with pimonidazole and a strong correlation with oxygen electrode measurements 21, 22. Our immunohistochemical studies using CA-9 showed a surge in expression at 3 days following 28 Gy single dose irradiation to the right lung. Tissue hypoxia steeply increased to more severe hypoxia (>60% positive staining) within 6 to 8 weeks after irradiation and further progressed throughout the observation period of 20 weeks.
We hypothesized that tissue hypoxia results from both an increase in oxygen consumption by activated macrophages and a decrease in oxygen delivery in the tissue resulting from the damage to the vasculature and reduced tissue perfusion. A reduction in lung perfusion following irradiation has already been described 23–25. Previous radionuclide lung perfusion studies were performed at 2, 4, 10, 16, and 34 weeks after 18-Gy single-dose irradiation to the right hemithorax in rats. 25. This prior study revealed a significant decrease in relative blood flow in the irradiated right lung 4 weeks after irradiation. In the study by Kwock et al 24 a single dose of 30 Gy delivered to the right hemithorax of rats resulted in a 70% decrease in pulmonary perfusion in the irradiated lung by 2–3 weeks after irradiation. In accordance to these previous investigations, our present study showed a significant decline in lung perfusion at 4 weeks post radiation. Looking at earlier time points, we observed a significant decrease in lung perfusion three days following irradiation along with the occurrence of tissue hypoxia at the same time. While tissue hypoxia progressed, we found a brief period of reperfusion until the perfusion of the right lung declined steadily from two weeks post radiation, again contributing to the progression of hypoxia in the irradiated tissue. At three days after irradiation no histopathologic changes were seen with light microscopy. From electron microscopy studies it is described that within hours to days a destruction of the alveolo-capillary membrane occurs with enhanced vascular permeability, interstitial edema, and fibrin exudation in the alveolar space 3, 4. In our study these acute changes were expressed in an immediate rise in lung weight one day after irradiation. Edema and exudation can both contribute to the development of hypoxia and reduced perfusion. Hypoxia in addition might have further lowered perfusion by hypoxic vasoconstriction. Another possibly intervening factor could be Angiotensin II, a potent vasoconstrictor. Recent research has brought up the role of the renin-angiotensin system in radiation induced injury 26, 27. Radiation via ROS could induce Angiotensin II, which in turn produces superoxide, upregulates TGF-β and mediates the release of proinflammatory cytokines through activation of transcription factors like protein-1 and NFκB. Other factors to be considered in this context are endothelin and connective tissue growth factor. Also, an initial depletion of nitric oxide (NO˙) through reaction with superoxide to peroxynitrite could play a role in these early vascular events 28. In addition to the degradation of NO˙ by ROS, hypoxia decreases the formation of NO˙ 29. Reduced NO˙ levels are assumed to be an initiating event in the pathogenesis of microvascular injury. Figure 5 depicts the timeline and correlations of the observed events of this study and a simplified view of possibly other intervening factors. The reperfusion period between 3 days and 2 weeks post radiation correlated with a drop in lung weight. This could indicate that edema and exudation partially resolved allowing for better perfusion. With time endothelial cells separate from the basement membrane due to the vascular damage and many capillaries swell and obstruct 30 contributing again to the decrease in perfusion and increasing hypoxia and overall tissue damage. As the cycle of tissue damage and repair continues causing fibrosis and shrinking of the lung, we recorded a decline in weight of the irradiated lung after week 6.
Fig. 5.
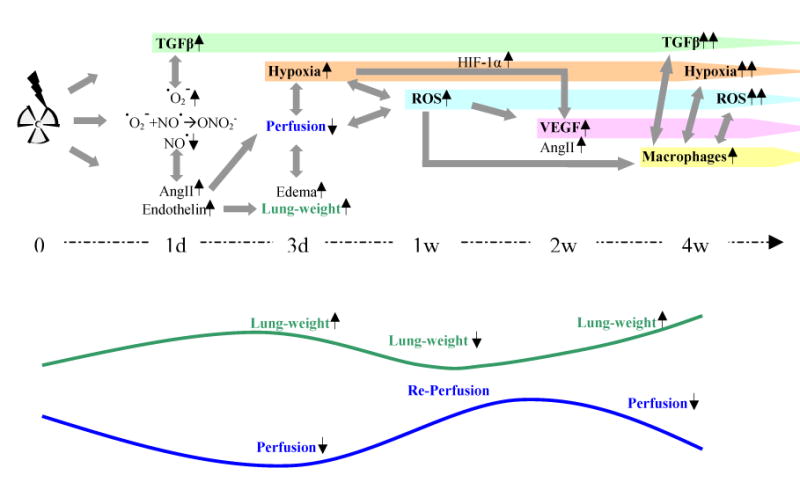
Proposed time related events: Based on the results of this study we hypothezise that early events in radation induced lung injury are based on early vascular changes. This is most likely mediated by several mechanisms: First, an imbalance of ROS and RNS leading to a depletion of nitric oxide (NO˙) due to reaction with superoxide (˙O2-) to peroxynitrite (ONO2-) 28 could cause vasoconstriction as seen in Fig. 1. Initial ˙O2- and ONO2- production activates TGF-β that continuously contributes to radiation induced damage. Second, an enhanced expression of other vasoconstrictive mediators such as Angiotensin II (Ang II) and Endothelin 26, 27 can also be implicated in this process. Endothelin may also lead to acute pulmonary edema. These early vascular changes contribute to the development of hypoxia, which further enhances oxidative stress, depletion of nitric oxide (NO˙) 29 and expression of HIF-1α and its downstream genes like VEGF and Antiotensin II. The combination of circumstances facilitates the recruitment of inflammatory cells like macrophages, which in turn enhance cytokine and ROS production and further promote hypoxia due to enhanced activation and proliferation maintaining the cycle of repair processes that lead to chronic injury.
The number of macrophages significantly increased 4 weeks after irradiation and escalated from there until 10 weeks after irradiation. Afterwards the number of macrophages fell until the end of the observation period of 20 weeks, but still remaining at elevated levels compared to unirradiated tissue. The observed increase in macrophages between week 4 and 10 post radiation falls within the inflammatory response phase. Looking at the CA-9 expression data, after the initial surge between 1 day and 2 weeks post radiation, there is a second steep rise starting at week 4. This second steep rise correlates with the macrophage accumulation and supports the hypothesis that tissue hypoxia also results from an increase in oxygen consumption by activated macrophages. It is known, that in turn hypoxia promotes macrophage migration into the disturbed microenvironment. Once recruited, macrophages become stimulated 31, 32. Our in vitro studies confirmed that hypoxia stimulates the oxidative burst in macrophages, increasing the extracellular superoxide concentration along with a pronounced upregulation of TGF-β (p<0.05) and VEGF (positive trend) 14.
To assess the oxidative damage we used 8-OHdG. 8-OHdG is a product of oxidative DNA damage and a well known marker for oxidative stress 33. Our study showed that oxidative stress occurs in previously healthy tissue within the first week following irradiation and continues for the entire 20 weeks of our study. This indicates chronic oxidative stress in the tissue is initially caused by irradiation and continues to persists after irradiation as a part of the repair processes. During the peak of macrophage accumulation in weeks 4 to 10 after irradiation we found the steepest rise in positive 8-OHdG staining, assuming that macrophages highly contribute to the production of reactive oxygen species.
As described above, hypoxia and oxidative stress induce marked changes in the secretory activity of macrophages and a wide variety of other cells involved in tissue repair. ROS have been implicated in cell signaling pathways, regulating transcription factors and changes in gene expression. ROS are also known to upregulate TGF-β and VEGF 14, 34–36. Both cytokines play essential roles in the process of radiation induced lung injury. Here, we have demonstrated that the level of VEGF, a main factor in angiogenesis and vascular permeability, is significantly elevated 2 weeks after irradiation when hypoxia and oxidative stress were already present. An explicit association with macrophage accumulation was not found.
TGF-β is a profibrogenic cytokine, which has a critical role in wound healing and appears to be one of the central factors in the development of radiation induced pulmonary injury 7, 19, 37–40. In accordance to other studies, we demonstrated an immediate increase in TGF-β after irradiation 41–43. Levels of TGF-β continued to rise steadily for the entire duration of the study underlining its central role in the process of radiation induced lung injury.
The present study confirms our hypothesis that tissue hypoxia is a key player in the onset and perpetuation of injury and early hypoxia is related to perfusion changes. It is generating a non-healing wound response through continuous generation of ROS and expression/activation of cytokines. We have further shown a correlation between macrophage accumulation and increase in hypoxia, oxidative stress and cytokines. This indicates that irradiation induced damage is not confined to events solely involving cells within the irradiated lung, but numerous recruited inflammatory cells may play a significant role. In addition, we were able to demonstrate a continuous pathogenetic process, assuming that the acute inflammatory and late fibrotic stages of radiation induced lung injury do not represent separate pathogenetic entities 44, 45.
Although our study is descriptive in character and the pathogenesis of radiation-induced lung injury is not a linear process but rather complex multifactorial, where one event causes many others, our findings are an important step in understanding the chronology of radiation induced lung injury and how different events correlate to each other. We had chosen high single-dose experiments to determine the temporal onset of hypoxia and oxidative stress. Studies with more clinically relevant fractionated radiation regiments are currently under investigation.
Acknowledgments
The facility for the lung perfusion studies is supported by the NIH ES 11961. We appreciate the advice of Dr. Daohai Yu, Department of Biostatistics and Information Systems, on the statistical analyses.
Footnotes
Conflict of interest statement
The authors declare no conflict of interest.
Supported in part by the NIH (National Institutes of Health), Grant RO1 # CA 098452, PI: Zeljko Vujaskovic, and in part by the DFG (German Research Society), Research Fellowship Grant # FL 551/1-1 for Katharina Fleckenstein
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abratt RP, Morgan GW. Lung toxicity following chest irradiation in patients with lung cancer. Lung Cancer. 2002;35:103–109. doi: 10.1016/s0169-5002(01)00334-8. [DOI] [PubMed] [Google Scholar]
- 2.Roach M, 3rd, Gandara DR, Yuo HS, et al. Radiation pneumonitis following combined modality therapy for lung cancer: analysis of prognostic factors. J Clin Oncol. 1995;13:2606–2612. doi: 10.1200/JCO.1995.13.10.2606. [DOI] [PubMed] [Google Scholar]
- 3.Moosavi H, McDonald S, Rubin P, et al. Early radiation dose-response in lung: an ultrastructural study. Int J Radiat Oncol Biol Phys. 1977;2:921–931. doi: 10.1016/0360-3016(77)90190-0. [DOI] [PubMed] [Google Scholar]
- 4.Travis EL. The sequence of histological changes in mouse lungs after single doses of x-rays. Int J Radiat Oncol Biol Phys. 1980;6:345–347. doi: 10.1016/0360-3016(80)90145-5. [DOI] [PubMed] [Google Scholar]
- 5.Rubin P, Finkelstein J, Shapiro D. Molecular biology mechanisms in the radiation induction of pulmonary injury syndromes: interrelationship between the alveolar macrophage and the septal fibroblast. Int J Radiat Oncol Biol Phys. 1992;24:93–101. doi: 10.1016/0360-3016(92)91027-k. [DOI] [PubMed] [Google Scholar]
- 6.Rubin P, Johnston CJ, Williams JP, et al. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol Biol Phys. 1995;33:99–109. doi: 10.1016/0360-3016(95)00095-G. [DOI] [PubMed] [Google Scholar]
- 7.Martin M, Lefaix J, Delanian S. TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys. 2000;47:277–290. doi: 10.1016/s0360-3016(00)00435-1. [DOI] [PubMed] [Google Scholar]
- 8.Denham JW, Hauer-Jensen M. The radiotherapeutic injury—a complex ‘wound’. Radiother Oncol. 2002;63:129–145. doi: 10.1016/s0167-8140(02)00060-9. [DOI] [PubMed] [Google Scholar]
- 9.O'Sullivan B, Levin W. Late radiation-related fibrosis: pathogenesis, manifestations, and current management. Semin Radiat Oncol. 2003;13:274–289. doi: 10.1016/S1053-4296(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 10.Stone HB, Coleman CN, Anscher MS, et al. Effects of radiation on normal tissue: consequences and mechanisms. Lancet Oncol. 2003;4:529–536. doi: 10.1016/s1470-2045(03)01191-4. [DOI] [PubMed] [Google Scholar]
- 11.Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 2006;6:702–713. doi: 10.1038/nrc1950. [DOI] [PubMed] [Google Scholar]
- 12.Vujaskovic Z, Anscher MS, Feng QF, et al. Radiation-induced hypoxia may perpetuate late normal tissue injury. Int J Radiat Oncol Biol Phys. 2001;50:851–855. doi: 10.1016/s0360-3016(01)01593-0. [DOI] [PubMed] [Google Scholar]
- 13.Vujaskovic Z, Marks LB, Anscher MS. The physical parameters and molecular events associated with radiation-induced lung toxicity. Semin Radiat Oncol. 2000;10:296–307. doi: 10.1053/srao.2000.9424. [DOI] [PubMed] [Google Scholar]
- 14.Jackson IL, Chen L, Batinic-Haberle I, et al. Superoxide Dismutase Mimetic Reduces Hypoxia-Induced O˙2-, TGF-β, and VEGF Production by Macrophages. Radiation Research. 2006 doi: 10.1080/10715760600913150. [DOI] [PubMed] [Google Scholar]
- 15.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 16.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 17.Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003;22:5734–5754. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 18.Hsu SM, Raine L, Fanger H. The use of antiavidin antibody and avidin-biotin-peroxidase complex in immunoperoxidase technics. Am J Clin Pathol. 1981;75:816–821. doi: 10.1093/ajcp/75.6.816. [DOI] [PubMed] [Google Scholar]
- 19.Anscher MS, Thrasher B, Rabbani Z, et al. Antitransforming growth factor-beta antibody 1D11 ameliorates normal tissue damage caused by high-dose radiation. Int J Radiat Oncol Biol Phys. 2006;65:876–881. doi: 10.1016/j.ijrobp.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 20.Wykoff CC, Beasley NJ, Watson PH, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–7083. [PubMed] [Google Scholar]
- 21.Olive PL, Aquino-Parsons C, MacPhail SH, et al. Carbonic anhydrase 9 as an endogenous marker for hypoxic cells in cervical cancer. Cancer Res. 2001;61:8924–8929. [PubMed] [Google Scholar]
- 22.Loncaster JA, Harris AL, Davidson SE, et al. Carbonic anhydrase (CA IX) expression, a potential new intrinsic marker of hypoxia: correlations with tumor oxygen measurements and prognosis in locally advanced carcinoma of the cervix. Cancer Res. 2001;61:6394–6399. [PubMed] [Google Scholar]
- 23.Marks LB, Munley MT, Spencer DP, et al. Quantification of radiation-induced regional lung injury with perfusion imaging. Int J Radiat Oncol Biol Phys. 1997;38:399–409. doi: 10.1016/s0360-3016(97)00013-8. [DOI] [PubMed] [Google Scholar]
- 24.Kwock L, Davenport WC, Clark RL, et al. The effects of ionizing radiation on the pulmonary vasculature of intact rats and isolated pulmonary endothelium. Radiat Res. 1987;111:276–291. [PubMed] [Google Scholar]
- 25.Vujaskovic Z, Down JD, van t' Veld AA, et al. Radiological and functional assessment of radiation-induced lung injury in the rat. Exp Lung Res. 1998;24:137–148. doi: 10.3109/01902149809099578. [DOI] [PubMed] [Google Scholar]
- 26.Duprez DA. Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: a clinical review. J Hypertens. 2006;24:983–991. doi: 10.1097/01.hjh.0000226182.60321.69. [DOI] [PubMed] [Google Scholar]
- 27.Robbins ME, Diz DI. Pathogenic role of the renin-angiotensin system in modulating radiation-induced late effects. Int J Radiat Oncol Biol Phys. 2006;64:6–12. doi: 10.1016/j.ijrobp.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 28.Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whorton AR, Simonds DB, Piantadosi CA. Regulation of nitric oxide synthesis by oxygen in vascular endothelial cells. Am J Physiol. 1997;272:L1161–1166. doi: 10.1152/ajplung.1997.272.6.L1161. [DOI] [PubMed] [Google Scholar]
- 30.Maisin JR. The influence of radiation on blood vessels and circulation. Chapter 3. Ultrastructure of the vessel wall. Curr Top Radiat Res Q. 1974;10:29–57. [PubMed] [Google Scholar]
- 31.Lewis JS, Lee JA, Underwood JC, et al. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999;66:889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- 32.Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. 2005;175:6257–6263. doi: 10.4049/jimmunol.175.10.6257. [DOI] [PubMed] [Google Scholar]
- 33.Fischer-Nielsen A, Jeding IB, Loft S. Radiation-induced formation of 8-hydroxy-2'-deoxyguanosine and its prevention by scavengers. Carcinogenesis. 1994;15:1609–1612. doi: 10.1093/carcin/15.8.1609. [DOI] [PubMed] [Google Scholar]
- 34.Patel B, Khaliq A, Jarvis-Evans J, et al. Oxygen regulation of TGF-beta 1 mRNA in human hepatoma (Hep G2) cells. Biochem Mol Biol Int. 1994;34:639–644. [PubMed] [Google Scholar]
- 35.Shweiki D, Neeman M, Itin A, et al. Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: implications for tumor angiogenesis. Proc Natl Acad Sci U S A. 1995;92:768–772. doi: 10.1073/pnas.92.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong M, Elson G, Legarda D, et al. Production of vascular endothelial growth factor by murine macrophages: regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am J Pathol. 1998;153:587–598. doi: 10.1016/S0002-9440(10)65601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rube CE, Uthe D, Schmid KW, et al. Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol Biol Phys. 2000;47:1033–1042. doi: 10.1016/s0360-3016(00)00482-x. [DOI] [PubMed] [Google Scholar]
- 38.Anscher MS, Vujaskovic Z. Mechanisms and potential targets for prevention and treatment of normal tissue injury after radiation therapy. Semin Oncol. 2005;32:S86–91. doi: 10.1053/j.seminoncol.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 39.Rabbani ZN, Anscher MS, Zhang X, et al. Soluble TGFbeta type II receptor gene therapy ameliorates acute radiation-induced pulmonary injury in rats. Int J Radiat Oncol Biol Phys. 2003;57:563–572. doi: 10.1016/s0360-3016(03)00639-4. [DOI] [PubMed] [Google Scholar]
- 40.Vujaskovic Z, Groen HJ. TGF-beta, radiation-induced pulmonary injury and lung cancer. Int J Radiat Biol. 2000;76:511–516. doi: 10.1080/095530000138510. [DOI] [PubMed] [Google Scholar]
- 41.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 42.Ewan KB, Henshall-Powell RL, Ravani SA, et al. Transforming growth factor-beta1 mediates cellular response to DNA damage in situ. Cancer Res. 2002;62:5627–5631. [PubMed] [Google Scholar]
- 43.Yi ES, Bedoya A, Lee H, et al. Radiation-induced lung injury in vivo: expression of transforming growth factor-beta precedes fibrosis. Inflammation. 1996;20:339–352. doi: 10.1007/BF01486737. [DOI] [PubMed] [Google Scholar]
- 44.Dorr W, Bertmann S, Herrmann T. Radiation induced lung reactions in breast cancer therapy. Modulating factors and consequential effects. Strahlenther Onkol. 2005;181:567–573. doi: 10.1007/s00066-005-1457-9. [DOI] [PubMed] [Google Scholar]
- 45.Williams J, Chen Y, Rubin P, et al. The biological basis of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13:182–188. doi: 10.1016/S1053-4296(03)00045-6. [DOI] [PubMed] [Google Scholar]