

Abstract
Investigations of the pathways involved in the metabolism of endocannabinoids have grown exponentially in recent years following the discovery of cannabinoid receptors (CB) and their endogenous ligands, such as anandamide (AEA) and 2-arachidonoylglycerol (2-AG). The in vivo biosynthesis of AEA has been shown to occur through several pathways mediated by N-acylphosphatidylethanolamide-phospholipase D (NAPE-PLD), a secretory PLA2 and PLC. 2-AG, a second endocannabinoid is generated through the action of selective enzymes such as phosphatidic acid phsophohydrolase, diacylglycerol lipase (DAGL), phosphoinositide-specific PLC (PI-PLC) and lyso-PLC. A putative membrane transporter or facilitated diffusion is involved in the cellular uptake or release of endocannabinoids. AEA is metabolized by fatty acid amidohydrolase (FAAH) and 2-AG is metabolized by both FAAH and monoacylglycerol lipase (MAGL). The author presents an integrative overview of current research on the enzymes involved in the metabolism of endocannabinoids and discusses possible therapeutic interventions for various diseases, including addiction.
Keywords: FAAH, Endocannabinoids, CNS, NAPE-PLD, MAGL, CB1 receptors, alcohol-drinking behavior, therapy
INTRODUCTION
The herb Cannabis sativa, commonly referred as the marijuana plant, produces a complex mixture of pharmacological substances known as the cannabinoids. The major active component of marijuana, Δ9-tetrahydrocannabinol (Δ9THC), binds to members of the cannabinoid family of G-protein-coupled receptors (GPCRs). To date, two cannabinoid receptors have been cloned (CB1 and CB2) and the two major endogenous cannabinoids were characterized as arachidonyl ethanolamine (anandamide, AEA) and 2-arachidonylglycerol (2-AG). Additional endocannabinoids that exhibit cannabimimetic activity have also been discovered [51, 83, 95].
Unlike classical neurotransmitters and neuropeptides, endocannabinoids are not stored in intracellular compartments; rather they are produced on demand by receptor-stimulated cleavage of lipid precursors by various enzymes [4–6, 20, 36, 84] and released from neurons immediately afterwards [4–6, 36, 44, 84]. Following their release, the signaling function of the endocannabinoids is terminated through the action of a specific reuptake protein or transporter [6, 52, 94] and subsequent hydrolysis of the molecules by either fatty acid amide hydrolase (FAAH) [26] or monoacylglycerol lipase (MAGL)[37] or by other uncharacterized enzymatic activities known to hydrolyze endocannabinoids in vivo. The various enzymes involved in endocannabinoid metabolism offer intriguing opportunities for targeted drug development. This review will provide an overview of our current understanding of the critical enzymes involved in endocannabinoid biosynthesis and degradation and discuss possible therapeutic interventions for various diseases, including addiction.
ENZYMES INVOLVED IN ANANDAMIDE BIOSYNTHESIS AND DEGRADATION
AEA has been identified and quantitated throughout the human brain and in peripheral tissues[12, 40]. Signal transduction and ligand binding studies have suggested that it can act at both the CB1 (Ki 61 nM) and the CB2 (Ki 1930 nM) receptors, although it may be more efficacious at CB1 [42]. AEA behaves as an affinity-driven CB1 receptor agonist. Thus its efficacy at CB1 receptors, although higher than that of Δ9- THC, is often found to be lower than those of other cannabinoid agonists [e.g., (+)-WIN55212-2 or CP-55940].
AEA was shown to be widely distributed in the brain and peripheral tissues [40, 41]. AEA levels vary by a factor of 4 to 6 between different regions of the rat brain, with the highest levels in the striatum and brainstem and the lowest levels in the cerebellum and cortex [12, 129]. AEA was found in regions of both the rat and human brains that contain high densities of CB1 receptors (e.g., the hippocampus, cerebellum, and striatum) and also in a region that is sparse in CB1 receptors, the thalamus [40]. In the rat, the concentration of AEA in the thalamus was approximately twice that measured in the cerebellum. It is clear from these data that for AEA, the relative regional abundance in the brain does not correlate with the distribution of CB1 receptors. AEA levels in the brain are equivalent to those of other neurotransmitters such as dopamine and serotonin, but at least 10-fold lower than the levels reported for GABA and glutamate. AEA has also been found in peripheral tissues such as the human and rat spleens, which express high levels of CB2 receptors—a finding that lends support to the idea that AEA is an agonist at both CB1 and CB2 receptors [40]. Small amounts of AEA were also found in the human heart and thalamus and also in rat skin, whereas only trace quantities were detected in human serum, plasma, and cerebrospinal fluid [40]. It should be mentioned here that the variation of the endocannabinoid levels under physiological or pathological conditions is most important for understanding the endocannabinoids role in normal neurotransmission and in variety of disease conditions rather than their absolute levels in various brain regions.
Enzymes responsible for the biosynthesis and degradation of AEA were characterized in various tissues. The postulated biosynthetic pathway for AEA involves the energy-independent condensation of ethanolamine and free arachidonic acid [29, 123]. The biochemical characteristics and inhibitor kinetics observed for the enzyme catalyzing this condensation reaction were found to be essentially identical to those described for the enzyme fatty acid amide hydrolase (FAAH; see below) [26, 29, 123]. However, several lines of evidence derived from both physical and biological data argue that the production of AEA in vivo does not occur through the energy-independent condensation of free arachidonic acid and ethanolamine [36, 59, 67]. Instead, the AEA precursor is an N-arachidonylphosphatidylethanolamine (N-ArPE), which is believed to originate from the transfer of arachidonic acid from the sn-1 position of 1,2-sn-di-arachidonylphosphatidylcholine to phosphatidylethanolamine catalyzed by a calcium-dependent N-acyltransacylase (NAT) (Fig. 1). N-ArPE is then cleaved by an N-acylphosphatidylethanolamine (NAPE)-specific phospholipase D (PLD) (NAPE-PLD) [36, 89, 100], which releases AEA and phosphatidic acid. It is not clear whether the N-acyltransferase or the NAPE-specific PLD controls the rate-limiting step of AEA synthesis [31, 49, 110]. The NAT enzyme has been identified biochemically in both brain and testis microsomal preparations [20, 114, 115]. However, the detailed purification, characterization and cloning of a specific NAT remains to be done. Another pathway which involves the conversion of NAPE into 2-lysol-NAPEs via the action of secretory PLA2 has also been reported. 2-Lysol-NAPEs are then converted into N-acyl-ethanolamides, including AEA, via a selective lyso phospholipase D (lyso-PLD)[117]. The NAPE-PLD (see details below) has only recently been purified, cloned, and characterized [90]. It has been reported recently that oleoylethanolamide (OEA) generation was followed by enhanced accumulation of OEA-generating NAPEs through Ca2+-independent NAT in the proximal small intestine[43].
Figure 1. The potential enzymes involved in anandamide biosynthesis.

Stimulation of adenylate cyclase and cAMP-dependent protein kinase potentiate the N-acyltransferase (Ca2+-dependent transacylase) NAT. A fatty arachidonic acid chain is transferred by NAT from the sn-1 position of phospholipids (phosphoglycerides, PG) to the primary amine of phosphatidylethanolamine (PE) in a Ca2+-dependent manner, forming an N-arachidonyl phosphatidylethanolamine (N-ArPE). This N-ArPE intermediate is then hydrolyzed by a phospholipase D (PLD)-like enzyme to yield anandamide (AEA). In addition, possible alternative pathways for AEA formation from N-ArPE are shown. Once synthesized, AEA can be transported to the outside of the cell through a process that has not yet been well characterized. AMT, anandamide membrane transporter.
A recent study proposed the existence of a parallel pathway for AEA synthesis in the mouse brain and RAW264.7 macrophages [71]. In this pathway, AEA is generated from NAPE in a two-step process involving the PLC-catalyzed cleavage of NAPE to yield pAEA, which is subsequently dephosphorylated by a protein tyrosine phosphatase(s) (PTPN22) [71] (Fig. 1).
NAPE-PLD
NAPE-PLD was cloned from mouse, rat, and human, and the three enzymes were found to be highly conserved with more than 89% identity at the amino acid level [90]. NAPE-PLD belongs to the zinc metallohydrolase family of the β-hydrolase fold and is structurally unrelated to other known PLD enzymes [90]. NAPE-PLD’s lack of transphosphatidylation activity indicates that the enzyme is not only structurally but also functionally distinct from PLD members of the HKD/phosphatidyltransferase gene family [70]. NAPE-PLD uses not only N-arachidonyl-PE (a precursor of AEA) as a substrate but also other NAPEs with different N-acyl groups [90]. NAPE-PLD is present in almost all organs of the mouse, with higher specific activities in the brain, kidney, and testis [90]. In bovine organs, the brain exhibits the highest NAPE-PLD specific activity, followed by kidney, spleen, lung, heart, and liver. In all animal species, the brain consistently displays high NAPE-PLD activity. Whether there are isozymes of NAPE-PLD is currently not known. An age-dependent change in NAPE-PLD activity was reported recently in the rat brain [86]. The regulatory mechanisms for the expression of NAPE-PLD mRNA and protein are not known. The generation of specific inhibitors of NAPE-PLD and gene-disrupted animals will be of great use for future studies. Indeed, in a recent study, AEA biosynthesis was unaffected in NAPE-PLD knockout mice, suggesting that other enzymes [68] or isozymes of NAPE-PLD also participate in the synthesis of AEA. These NAPE-PLD(−/−) mice possess lower brain levels of saturated NAEs but essentially unchanged levels of polyunsaturated NAEs, including AEA, suggesting the existence of additional enzymatic pathways for the biosynthesis of NAEs in vivo. The enzyme α/β-hydrolase 4 (Abh4), which selectively hydrolyzes NAPEs and lysoNAPEs bearing both saturated and polyunsaturated N-acyl chains has been described in NAPE-PLD(−/−) mice recently[102]. This report support the existence of an NAPE-PLD-independent route for NAE biosynthesis and suggest that Abh4 may have a role in this metabolic pathway by acting as a (lyso)NAPE-selective lipase[102].
As a putative neuromodulator, AEA that is released into the synaptic cleft is expected to be rapidly inactivated. In general, two mechanisms are known to remove endocannabinoids from the synaptic cleft to ensure rapid signal inactivation: re-uptake (see detailed discussion below) or enzymatic degradation by FAAH (see below).
Soon after the discovery of AEA, an enzyme that catalyzes the hydrolysis of AEA to arachidonic acid and ethanolamine was detected in rat brain tissue [29] (Fig. 2) and is variously known as anandamide amidase, anandamide hydrolase, anandamide amidohydrolase, or fatty acid amide hydrolase (FAAH) (see below). In addition to its hydrolysis by FAAH, AEA is metabolized by COX-2, lipoxygenase (LOX) and cytochrome P450. COX-2 has been shown to metabolize AEA in to PGE2-ethanolamide (PGE2-EA)[98]. 12- and 15-LOX, non-heme iron-containing enzymes convert AEA into 12- and 15-hydroxy-AEA (12- and 15-HAEA) in vitro, respectively [64, 124]. Cytochrome P450 also metabolizes AEA into several polar lipids [19]. cDNA cloning and functional expression of the enzyme termed “N-acylethanolamine-hydrolyzing acid amidase (NAAA)” from human, rat, and mouse has been reported recently and had no homology to FAAH but belonged to the choloylglycine hydrolase family [119]. This NAE-hydrolyzing enzyme (NAAA) was revealed to be a glycoprotein localizing mainly in lysosomes[119]. Recently, in the absence of FAAH, exogenously injected AEA was shown to be converted into o-phosphorylcholine (o-PC)-AEA in the brain and spinal cord. The choline-specific phosphodiesterase (NPP6) was found to convert PC-NAE into NAE [87] (Fig. 2). Further research is required to elucidate the exact mechanism and enzymes involved in this pathway of AEA metabolism.
Figure 2. Scheme illustrating the potential proteins and enzymes involved in anandamide uptake and degradation.

Anandamide can be internalized by neurons through an as yet unidentified transport mechanism, “the endocannabinoid transporter (AMT)”. Once inside neurons, AEA is rapidly cleaved by the hydrolytic enzyme fatty acid amide hydrolase (FAAH) to give arachidonic acid (AA) and ethanolamine (EA). Alternatively, different lipoxygenases (LOXs) and cyclooxygenase-2 (COX-2) can metabolize AEA, generating hydroxyl derivatives of AEA (HAEAs) and prostaglandins-ethanolamides (PG-EAs), respectively. On the other hand, in FAAH (−/−) mice, AEA is converted into o-phosphorylcholine (o-PC)-AEA by an unidentified biosynthetic pathway and o-PC-AEA can in turn be catabolized to AA by a choline-specific phosphodiesterase (NPP6). AMT, anandamide membrane transporter.
Enzymes Involved in 2-Arachidonylglycerol Biosynthesis and Degradation
2-AG has been characterized as a unique monoacylglycerol species isolated from the canine gut [82] and rat brain [116] and as an endogenous cannabinoid receptor ligand. According to its chemical structure, this new putative endocannabinoid is an arachidonyl ester rather than an amide [7, 8, 55]; it was found to bind to both CB1 (Ki 2.4 μM) and CB2 receptors. The CB1 receptor binding activity of 2-AG was 24-times less potent than that of AEA. 2-AG caused the typical effects of Δ9-THC, such as antinociception, immobility, immunomodulation, and inhibition of electrically evoked contractions of the mouse vas deferens [82, 111, 113]. However, further studies are necessary to determine the relative importance of 2-AG in the human body and brain. Information regarding tissue levels of 2-AG is still limited. Brain tissue concentrations of 2-AG are approximately 200-fold higher than those of AEA [12]. The spatial distributions of the two endocannabinoids are similar in different regions of the brain. The highest concentrations were found in the brainstem, medulla, limbic forebrain, striatum, and hippocampus and the lowest in the cortex, diencephalons, mesencephalon, hypothalamus, and cerebellum (for review see [110]). However, no correlation was found between 2-AG concentrations and CB1 receptor distribution. 2-AG was also detected in the peripheral nervous system, such as in the sciatic nerve, lumbar spinal cord, and lumbar dorsal root ganglion. 2-AG was also detected in the rat and bovine retinas (for review see [110]).
Generation of 2-AG was first described in ionomycin-stimulated N18TG2 cells [16], in electrically stimulated rat hippocampal slices, and in ionomycin-stimulated neurons [108]. Rapid generation of 2-AG was also observed in rat brain homogenate during incubation with Ca2+[62] and in thrombin- or A23187-stimulated human umbilical vein endothelial cells [112]. Intraperitoneal injection of picrotoxin stimulated 2-AG levels in the rat brain [116]. Generation of 2-AG was also observed in cerebellar granular neurons in culture after chronic alcohol exposure [5]. Substantial amounts of this stimulus-induced 2-AG were released from the cells [5, 15, 112].
2-AG biosynthesis occurs by two possible routes in neurons, which are illustrated in Fig. 3. Phospholipase C (PLC)-mediated hydrolysis of membrane phospholipids may produce diacylglycerol (DAG), which may be subsequently converted to 2-AG by diacylglycerol lipase (DAGL) activity [96, 113]. Alternatively, the formation of DAG involves the hydrolysis of phosphatidic acid through Mg2+ and Ca2+-dependent PA phsophohydrolase activity [14, 21]. There is a vast literature available on the biochemical properties and molecular structure of mammalian phospholipase C [23]. On the other hand, the molecular cloning of DAGLα was reported recently [13]. Both DAGL α and β are associated with the cell membrane and are stimulated by Ca2+ and blocked by DAGL inhibitors (p-hydroxy-mercuri-benzoate, HgCl2 and RHC-80267). DAGL appears to be targeted to postsynaptic spines; it is highly enriched at the base of the spine neck and in the adjacent somatodendritic membrane but is excluded from the main body of the spines and the excitatory synapses in cerebellar Purkinje cells. In hippocampal pyramidal cells, DAGL is distributed at the spine head or neck or at both structures[60, 130]. The different fine localizations in different neuron types suggest that the specificity and efficiency of endocannabinoid-mediated retrograde suppression of neurotransmission depend not only on CB1 expression levels in presynaptic elements but also on the distance between the postsynaptic site of 2-AG production and the presynaptic CB1 receptor. Alternatively, phospholipase A1 (PLA1) may generate a lysophospholipid, which may be hydrolyzed to 2-AG by lyso-PLC activity [113]. The enzyme activities (PLA1 and lyso-PLC) involved in this second pathway are well characterized [120–122]. LysoPI-specific PLC is distinct from various other types of PLCs that act on other inositol phospholipids, and is located in the synaptosomes [120–122]. Under certain conditions, 2-AG can also be synthesized through the conversion of 2-arachidonyl lysophosphatidic acid (LPA) (Fig. 3) by a phosphatase to yield 2-AG [88]. In certain conditions, 2-AG can also be synthesized through hydrolysis of PA and not through the activation of PLC in intact N18TG2 [14] and cultured rat microglial [21] cells. Various structurally distinct inhibitors of PLC and DGL activities prevent 2-AG formation in cultures of cortical neurons, suggesting that the PLC/DGL pathway may play a primary role in this process [108].
Figure 3. The enzymes involved in the biosynthetic pathways of 2-arachidonylglycerol.
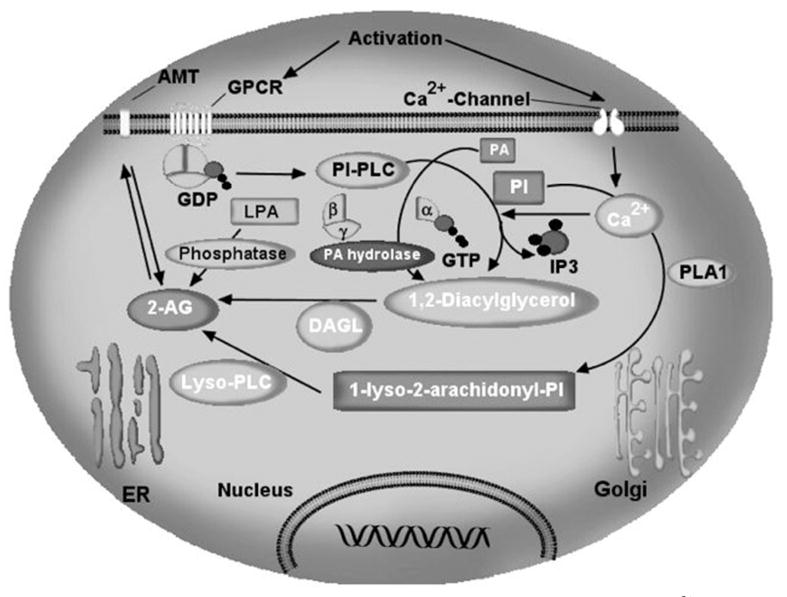
Intracellular Ca2+ initiates 2-AG biosynthesis by inducing the formation of diacylglycerol (DAG) in the membrane by stimulating the phosphatidyl-inositol-phospholipase C (PI-PLC) pathway. Alternatively, the formation of DAG involves the hydrolysis of phosphatidic acid through Mg2+ and Ca2+-dependent PA phsophohydrolase activity. 2-AG is the product of DAG-lipase (DAGL) acting on DAG. The second pathway involves hydrolysis of PI by phospholipase A1 (PLA1) and hydrolysis of the resultant lyso-PI by a specific lyso-PLC. In certain conditions 2-AG can also be synthesized through the conversion of 2-arachidonyl lysophosphatidic acid (LPA) by a phosphatase to yield 2-AG. Once synthesized, 2-AG can be transported to the outside of the cell through a process that has not yet been characterized. AMT, anandamide membrane transporter.
2-AG is inactivated by reuptake via a membrane transport molecule, the ‘AEA membrane transporter’ (AMT) (see below for details)[6, 9, 10, 44, 52, 53, 76], and subsequent intracellular enzymatic degradation [28, 30, 36] by monoacylglycerol lipase (MAGL) (see below for details) in the same manner as other monoacylglycerols [63]. Similarly, 2-AG is metabolized by porcine MAGL from brain cytosol and particulate fractions [47]. FAAH (see below) has also been shown to metabolize 2-AG (Fig. 4) [34, 46]. 2-AG is metabolized to 2-arachidonyl LPA through the action of monoacyl glycerol kinase(s). 2-Arachidonyl LPA is then converted into 1-steroyl-2-arachidonyl PA [103]. 1-Steroyl-2-arachidonyl PA is further utilized in the “PI cycle” or is used in the de novo synthesis of phosphatidylcholine (PC) and phosphatidylethanolamine (PE). Furthermore, 2AG is metabolized by enzymatic oxygenation of 2-AG by cyclooxygenase-2 (COX-2) into prostaglandin H2 (PGH2) glycerol esters. The biological activity and the role of oxygenated 2-AG have yet to be determined. Inhibition of COX-2 prolongs DSI, suggesting that COX-2 limits endocannabinoid action in retrograde signaling and synaptic plasticity [61, 99, 106].
Figure 4. Schematic summary of the proteins and enzymes involved in 2-AG uptake and degradation.
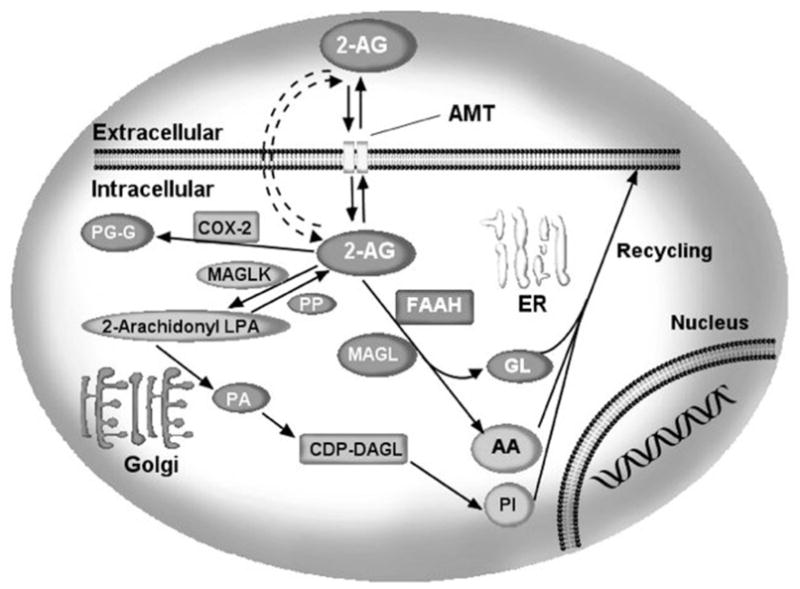
2-Arachidonylglycerol can be internalized by neurons through an as yet unidentified transport mechanism, “the endocannabinoid transporter (AMT)”. Once inside neurons, 2-AG can be hydrolyzed by the enzymes fatty acid amide hydrolase (FAAH) or monoacylglycerol lipase (MAGL). Alternatively, 2-AG can be metabolized to 2-arachidonyl LPA (2-AG-LPA) through the action of monoacyl glycerol kinase(s) (MAGLK). 2-AG-LPA is then converted into 1-steroyl-2-arachidonyl PA. 1-Steroyl-2-arachidonyl PA is further utilized in the de novo synthesis of PC and PE. Furthermore, 2AG is metabolized by enzymatic oxygenation of 2-AG by COX-2 into PGH2 glycerol esters (PG-G). AMT, anandamide membrane transporter.
Uptake of Endocannabinoids
AEA appears to be inactivated by a two-step process involving the transport of this lipid into cells [10] (Fig. 2) followed by intracellular hydrolysis by the integral membrane enzyme FAAH. However, with respect to the inactivation of AEA, only FAAH has been molecularly characterized and structurally studied. Indeed, the actual mechanism of AEA uptake or, more generally, the movement of this fatty acid amide not only through facilitated diffusion by a protein transporter but also through passive diffusion across the plasma membrane remains an enigmatic and controversial subject. Some authors have suggested that AEA uptake occurs by a facilitated diffusion process mediated by a protein transporter [6, 9, 10, 52], whereas others have described it as a passive diffusion process driven by FAAH [28, 30]. For a current discussion of these two points of view, see the recent papers [45] and [54].
AEA uptake by cells occurs via diffusion through the cell membrane, facilitated by a saturable, temperature-dependent and selective transport system [36]. The transporter, the ‘anandamide membrane transporter’ (AMT), has been identified in most cells analyzed so far [53] (Figs. 1,2 and 4), and AMT inhibitors capable of enhancing AEA actions in vitro and in vivo have been developed [10, 33]. Also, structure-activity relationship studies have been carried out on the AMT with a large variety of AEA analogues [32, 53, 56, 85, 92, 94]. It was established that at least one to four cis double bonds in the fatty acyl chain are necessary for binding to the AMT or to be transported into cells [85]. On the other hand, it was also observed that the ethanolamine ‘head’ of AEA could be substituted with bulkier groups, especially aromatic structures, to yield compounds still capable of binding to the AMT [56, 85]. Finally, it was reported that the AMT is present in endothelial cells [72], is activated by nitric oxide (NO) [73–76], and is inhibited by the plant cannabinoids Δ9THC and cannabidiol at micromolar concentrations [97]. The role of intracellular catabolism of AEA in driving, in part, the AMT was also pointed out [30, 97].
The uptake of 2-AG by cells was first observed in rat basophilic RBL-2H3 and mouse neuroblastoma N18TG2 cells, and was shown to be inhibited by unsaturated monoacylglycerols such as 2-oleoyl- and 2-linoleoyl-glycerols [11]. In studies carried out in RBL-2H3 and J774 cells [34, 35, 97] it was reported that 2-AG did not effectively inhibit the uptake of [14C] AEA when the two substances were present at the same concentration, thus suggesting that the AMT does not recognize 2-AG as a substrate. However, very recently, a study carried out in human astrocytoma cells [9] confirmed that 2-AG is taken up by cells, and showed for the first time that this process can be inhibited by the previously developed AMT inhibitor, AM404 [10].
Despite the accumulation of new data from various studies, little is known regarding the identity of the AEA and 2-AG transport molecule. Cloning and in vitro expression of the transporter gene will be required for a clear understanding of the endocannabinoid signaling system, including the biosynthetic, uptake, and degradation pathways.
FAAH
The actual enzymes involved in fatty acid amide (FAA) metabolism remained unknown until the late 1990s, when a rat oleamide hydrolase activity was affinity purified and its cDNA cloned [26]. Oleamide amidase could be connected to AEA hydrolysis because the same enzyme catalyzed AEA and oleamide hydrolysis before the cloning of FAAH [80]. FAAH is a membrane-bound enzyme, which belongs to the family of amidase proteins. They are characterized by a highly conserved region that is rich in serine, glycine and alanine residues. This “amidase signature” region (AS) is common to more than 80 amidases and it corresponds to amino acids 215–257 in mammalian FAAH. Recombinantly expressed FAAH was shown to hydrolyze oleamide, AEA, and several other endogenous FAAs. 2-Arachidonylglycerol (2-AG) also appears to be a substrate of FAAH in vitro [47], and thus FAAH is generally required for inactivation of endocannabinoids in the brain. FAAH was first isolated from rat liver through its ability to catalyze the hydrolysis of the sleep-inducing lipid, oleamide [26, 27].
The mouse and human FAAH genes, each composed of 15 exons, are localized on chromosomes 1 and 4, respectively [126]. Recently, the first X-ray crystal structure of FAAH was reported [18]. This 2.8 Å structure of FAAH in a complex with the irreversible inhibitor methoxy arachidonyl fluorophosphonate revealed several unusual features of the enzyme. First, the core catalytic machinery of FAAH is composed of a serine–serine–lysine catalytic triad (S241–S217–K142), in contrast to the serine–histidine–aspartate triad typical of most serine hydrolases. These results are consistent with previous enzymological studies indicating that S241 and K142 play key catalytic roles as the nucleophile and acid/base, respectively [93]. The structure of FAAH also revealed that this enzyme possesses a remarkable collection of channels that appear to grant it simultaneous access to both the membrane and cytoplasmic compartments of the cell, possibly to facilitate substrate binding, product release, and catalytic turnover. These unusual mechanistic and structural features of FAAH have inspired new strategies for the design of specific inhibitors (e.g., URB 597) that display high selectivity for this enzyme relative to the hundreds of serine hydrolases present in the human proteome [81].
Despite biochemical and cell biological studies supporting a role for AEA as an endogenous CB1 agonist, the behavioral effects elicited by AEA are very weak and transient compared with those produced by Δ9-THC [1]. The limited pharmacological activity of AEA may be due to its rapid catabolism in vivo, as this lipid is hydrolyzed to arachidonic acid within minutes of exogenous administration [128]. Nonetheless, the relative contribution made by FAAH to the hydrolysis of anandamide in vivo remained largely unclear until it was cloned and a mouse model was generated in which this enzyme was genetically deleted (FAAH-knockout) [25]. FAAH-KO mice were shown to be born at the expected Mendelian frequency and were viable, fertile and normal in their general cage behavior. Tissues from FAAH- KO mice were found to display a 50–100-fold reduction in hydrolysis rates for AEA and related FAAs. In contrast to FAAH wild-type mice, in which administered AEA failed to produce significant behavioral effects, FAAH-KO mice exhibited robust responses to AEA, becoming hypomotile, analgesic, cataleptic, and hypothermic. Furthermore, all of the behavioral effects of AEA in FAAH-KO mice were blocked by pre-treatment with the CB1 receptor antagonist SR141716A (rimonabant), indicating that AEA acts as a potent and selective CB1 agonist in these animals. Consistent with this notion, AEA in brain homogenates from FAAH-KO mice showed approximately 15-fold higher apparent binding affinity for the CB1 receptor [69].
These FAAH-KO mice have been shown to have elevated (10–15-fold) levels of AEA and other N-acylethanol-amines (NAE) in several brain regions, including the hippocampus, cortex, and cerebellum [22, 25]. Interestingly, these increased levels of AEA and NAEs in the brain correlated with a CB1-dependent reduction in pain sensation in FAAH-KO mice [25]. These observations taken together suggest that FAAH is a key enzyme involved in the catabolism of AEA and other NAEs in vivo and suggest that pain pathways are under the influence of a FAAH-regulated endocannabinoid tone. However, it should be noted that FAAH-KO mice exhibited normal motility, body weight, and body temperature [25], suggesting that several other neurobehaviors affected by exogenously applied CB1 agonists were not controlled by AEA tone in these animals.
Localization and Distribution of FAAH in the Brain
The majority of FAAH in the mouse brain is associated with neurons; this is particularly interesting and probably most directly relevant to endocannabinoid signaling. FAAH appears not to be confined to neuronal somata but also extends into the areas surrounding the neural tissue. This is most clearly seen in the hippocampal formation where, although the highest concentration of FAAH is clearly located in the somata of hippocampal pyramidal cells and granule cells of the dentate gyrus, there is also a widespread presence of FAAH in the adjacent layers that contain their axons and dendrites [38]. The most striking and intensely stained FAAH-immunoreactive neurons were found in the mesencephalic trigeminal nucleus (Me5) [38]. In addition to neuronal FAAH expression, oligodendrocytes and ventricular ependymal cells of the mouse brain [38] also express FAAH. The functional significance of FAAH expression in these non-neuronal cells is not yet known. More typically, FAAH and CB1 are anatomically associated, with FAAH-immunoreactive neuronal somata surrounded by CB1-immunoreactive fibers, in the rat cerebellar cortex, hippocampus, and neocortex [39]. FAAH-expressing neurons were also found in brain regions (thalamus, mid-brain, and hind brain) known to express few or no CB1 receptors. However, there clearly are some regions of the brain (e.g., thalamic nuclei, mesencephalic trigeminal nuclei, cerebellar nuclei) where FAAH-expressing neurons are widespread but lack associated CB1 expression. In regions of the brain such as these, the role of FAAH is likely to be unrelated to CB1-dependent endocannabinoid signaling mechanisms [38]. This may be due to the presence of other potential targets for AEA in the brain, different from CB1. This could explain the lack of complementarity’s between CB1 and FAAH distribution in some brain areas. For example, in these brain areas AEA could act as endogenous ligand of TRPV1 receptors instead [78].
FAAH is likely to influence retrograde synaptic signaling molecules in several brain regions, including the cerebellar cortex where endocannabinoids mediate retrograde suppression of both excitatory and inhibitory synapses onto Purkinje cells [65, 66, 77]. In the cerebellar cortex, FAAH is located in Purkinje cell somata, while CB1 is located on the presynaptic terminals of granule cells (glutamatergic parallel fibers), basket cells (GABAergic), and probably also climbing fibers (glutamatergic). Depolarization of Purkinje cells causes transient endocannabinoid-mediated inhibition of excitatory inputs from parallel fibers and climbing fibers, with maximal inhibition occurring after approximately 5 s, followed by a gradual restoration of basal excitatory input within 50 s [65]. Because FAAH is located in the somatodendritic compartment of Purkinje cells, the duration of the inhibitory action of Purkinje cell-derived endocannabinoids is likely to be largely influenced by the uptake and FAAH-mediated inactivation of endocannabinoids in those cells [38]. FAAH is also present in fibers of the olfactory nerves that project into the olfactory bulb glomeruli where they form synapses with the glomeruli dendrites of mitral cells. This is of particular interest because FAAH is targeted to the axonal compartment of these neurons whereas in all other regions of the mouse and rat brains it is targeted to the somatodendritic compartment. It also provides a unique example of a synapse where FAAH is located both presynaptically (olfactory-receptor neuron terminals) and postsynaptically (mitral cells) [38]. The functional significance of this complex pattern of expression has yet to be investigated. Recently using FAAH inhibitors, it was shown that FAAH inhibition does not affect depolarization-induced suppression of inhibition (DSI) [79, 118].
The distribution of FAAH in the brain, although strikingly complementary with CB1 receptors in many regions of the forebrain and in the cerebellar cortex, is not always associated with CB1 receptor-expressing neurons. For example, there is a population of striatal GABAergic medium-spiny projection neurons that target the CB1 receptor to their axonal terminals in the globus pallidus (GP), entopeduncular nucleus (EP), and substantia nigra pars reticulata (SNR). The density of CB1 receptors in these output nuclei is particularly high, while FAAH-expressing neurons are sparse or absent [38].
Recent proteomic data suggest the existence of a second mammalian AS enzyme with FAAH activity [127]. The FAAH-2 gene exists in primates and in distantly related vertebrates but not in mice and rats. This FAAH-2 enzyme exhibits an overlapping but distinct tissue distribution, substrate selectivity, and inhibitor sensitivity compared to the original FAAH enzyme (FAAH-1; discussed above). Both FAAH-1 and FAAH-2 share 20% amino acid sequence identity. Similar to FAAH-1, FAAH-2 possess an N-terminal transmembrane domain and an AS sequence containing the serine-serine-lysine catalytic triad, along with other amino acid residues required for enzyme activity [127]. FAAH-2 showed greater activity with monounsaturated (C18: 1 NAE) substrates than with polyunsaturated (C20: 4 NAE) acyl chains. Moreover, FAAH-2 is situated within the membrane whereas FAAH-1 has a predominantly cytoplasmic orientation on the membrane [127]. These observations indicate that FAAH-2 may be important for the regulation of monounsaturated lipid amides in the CNS and peripheral tissues; however, further investigation is needed.
A naturally occurring single nucleotide polymorphism in the human FAAH gene, 385A, was found to be strongly associated with an endocannabinoid risk factor in overweight/obesity [105], street drug use and problem drug/alcohol use. This association seems especially relevant to illegal drug use, because neither alcohol nor nicotine abuse alone showed any significant relationship to the 385C→A missense mutation. The association of a naturally occurring human FAAH mutation with problem drug use provides further support that the endogenous cannabinoid system plays an important role in the neural circuits that underlie drug abuse and dependence [104], including alcoholism [8]. Inhibition of FAAH by URB-597 at any dose tested did not increase the reinforcing efficacy of heroin, suggesting that FAAH is not involved in heroin addiction [107]. Mice deficient in FAAH have reduced levels of the peptide product of the cocaine- and amphetamine-related transcript (CART) in nerve fibers and terminals in several brain regions implicated in appetite control [91].
There is strong evidence that some of the pharmacological action of alcohol is mediated by the endocannabinoid signaling system [2, 3, 24]. FAAH activity was not affected by chronic alcohol treatment in cerebellar granular neurons [6]. However, mice exposed to chronic alcohol showed decreased FAAH activity in the cortex [125]. Overwhelming evidence suggests that the endocannabinoid signaling system regulates alcohol drinking behavior in various rodent models (see recent reviews [2, 3, 24]). However, a direct role for FAAH in the regulation of alcohol drinking behavior in mice was shown recently [8]. Increased alcohol consumption and preference and decreased alcohol sensitivity were observed in female FAAH knockout mice but not in male FAAH knockout mice [8]. Recently increased consumption and preference for alcohol, decreased sensitivity to alcohol-induced sedation, and faster recovery from alcohol-induced motor incoordination were observed in both male and female FAAH knockout mice and also in wild-type mice received specific inhibitor of FAAH activity—URB597 [17]. These results suggest that impaired FAAH function may present a phenotype of high voluntary alcohol consumption, and identify FAAH as both a regulator of endocannabinoid function and a possible therapeutic target for alcohol-related disorders. Decreased expression and activity of FAAH was found in the prefrontal cortex (PFC) of alcohol-preferring rats with a compensatory down-regulation of CB1 signaling [50]. Furthermore, intra-PFC injections of the FAAH inhibitor URB-597 increased alcohol self-administration in Wistar rats [50]. These data taken together indicate that the enzymes which regulate the endocannabinoid signaling system could be important for alcohol’s reinforcing effects. These findings are significant for the development of potential therapeutic drugs targeting FAAH for the treatment of alcoholism.
MAG Lipase
MAGL has been shown to regulate endocannabinoid levels in the CNS [37]. Recently, MAGL was cloned from mouse adipose tissue [57] and the rat brain [37]. The human MAGL cDNA was also cloned and shows 84% identity with the mouse MAGL [58] and 85% identity with the rat MAGL. The mouse MAGL gene is located on chromosome 6, in a region with known homology to human chromosome 3q21 [57]. The mouse and human MAGL genes consist of seven exons ranging in size from 90 bp to over 2580 bp and the intron sizes are not conserved between the two species [57]. The mouse MAGL contains 302 amino acids with a molecular weight of 33 kD; its mRNA is a single transcript of 4 kb [58] and is expressed in various regions of the mouse brain. The catalytical triad was identified as Ser-122, Asp-239, and His-269 [57] and the residues are conserved between mouse and human MAGL. MAGL is found in all rat organs [57]. It has a heterogeneous distribution in the rat brain, with the highest levels of expression being in regions where the CB1 receptor is abundant, such as the hippocampus, cortex, and cerebellum [37]. MAGL was localized to axon terminals of granule cells and CA3 pyramidal cells of the hippocampus [48]. Interestingly, MAGL is expressed in presynaptic terminals [37, 48], suggesting it has a role in terminating retrograde signaling at presynaptic neurons [109]. The generation and availability of MAGL knockout mice will be of great use in understanding the role of MAGL in regulating endocannabinoid levels in the CNS. Ethanol intake for 15–30 days caused a marked impairment of heparin-releasable MAGL activity in rat heart tissue [101]. It has yet to be determined whether MAGL has any role in regulating endocannabinoid levels in drug addiction, including alcoholism.
CONCLUSION
The enzymes involved in endocannabinoid metabolism are emerging as very important regulators of endocannabinoid function at peripheral and neuronal levels. In the case of AEA and 2-AG, the role of FAAH and MAGL in controlling their cellular activity seems more critical than that of NAT, NAPE-PLD or AMT. Considering various biological and physiological functions of endocannabinoids in the animal tissues, the enzymes involved in the biosynthesis and metabolism of endocannabinoids have been attractive targets. Better understandings of these enzymes in pathophysiological conditions will largely contribute to develop selective inhibitors, which may be useful for the development of novel therapeutic drugs.
Acknowledgments
The author acknowledges support in part by grants from the National Institutes of Health (NIH AA11031) and the New York State Psychiatric Institute.
References
- 1.Adams IB, Compton DR, Martin BR. J Pharmacol Exp Ther. 1998;284:1209. [PubMed] [Google Scholar]
- 2.Basavarajappa BS. J Clin Res. 2005;11:16. [Google Scholar]
- 3.Basavarajappa BS. In: New Research on Alcoholism. Baye DR, editor. Nova Science Publishers, Inc; New York: 2006. in press. [Google Scholar]
- 4.Basavarajappa BS, Hungund BL. J Neurochem. 1999;72:522. doi: 10.1046/j.1471-4159.1999.0720522.x. [DOI] [PubMed] [Google Scholar]
- 5.Basavarajappa BS, Saito M, Cooper TB, Hungund BL. Biochemica Biophysica Acta. 2000;1535:78. doi: 10.1016/s0925-4439(00)00085-5. [DOI] [PubMed] [Google Scholar]
- 6.Basavarajappa BS, Saito M, Cooper TB, Hungund BL. Eur J Pharmacol. 2003;466:73. doi: 10.1016/s0014-2999(03)01557-7. [DOI] [PubMed] [Google Scholar]
- 7.Basavarajappa BS, Yalamanchili R, Cooper TB, Hungund BL. In: Handbook of Neurochemistry and Molecular Neurobiology. Hamon M, Sylvester VE, editors. Springer; NY: 2007. in press. [Google Scholar]
- 8.Basavarajappa BS, Yalamanchili R, Cravatt BF, Cooper TB, Hungund BL. Neuropharmacology. 2006;50:834. doi: 10.1016/j.neuropharm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Beltramo M, Piomelli D. NeuroReport. 2000;11:1231. doi: 10.1097/00001756-200004270-00018. [DOI] [PubMed] [Google Scholar]
- 10.Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Science. 1997;277:1094. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- 11.Ben-Shabat S, Fride E, Sheskin T, Tamiri T, Rhee MH, Vogel Z, Bisogno T, De Petrocellis L, Di Marzo V, Mechoulam R. Eur J Pharmacol. 1998;353:23. doi: 10.1016/s0014-2999(98)00392-6. [DOI] [PubMed] [Google Scholar]
- 12.Bisogno T, Berrendero F, Ambrosino G, Cebeira M, Ramos JA, Fernandez-Ruiz JJ, Di Marzo V. Biochem Biophys Res Commun. 1999;256:377. doi: 10.1006/bbrc.1999.0254. [DOI] [PubMed] [Google Scholar]
- 13.Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. J Cell Biol. 2003;163:463. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bisogno T, Melck D, De Petrocellis L, Di Marzo V. J Neurochem. 1999;72:2113. doi: 10.1046/j.1471-4159.1999.0722113.x. [DOI] [PubMed] [Google Scholar]
- 15.Bisogno T, Sepe N, De Petrocellis L, Di Marzo V. Adv Exp Med Biol. 1997;433:201. doi: 10.1007/978-1-4899-1810-9_42. [DOI] [PubMed] [Google Scholar]
- 16.Bisogno T, Sepe N, Melck D, Maurelli S, De Petrocellis L, Di Marzo V. Biochem J. 1997;322:671. doi: 10.1042/bj3220671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blednov YA, Cravatt BF, Boehm SL, II, Walker D, Harris RA. Neuropsychopharmacology. 2006 doi: 10.1038/sj.npp.1301274. In Press. [DOI] [PubMed] [Google Scholar]
- 18.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Science. 2002;298:1793. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 19.Burstein SH, Rossetti RG, Yagen B, Zurier RB. Prostaglandins Other Lipid Mediat. 2000;61:29. doi: 10.1016/s0090-6980(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 20.Cadas H, di Tomaso E, Piomelli D. J Neurosci. 1997;17:1226. doi: 10.1523/JNEUROSCI.17-04-01226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K, Pfister SL, Campbell WB, Hillard CJ. Mol Pharmacol. 2004;65:999. doi: 10.1124/mol.65.4.999. [DOI] [PubMed] [Google Scholar]
- 22.Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. J Neurosci. 2003;23:3916. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cocco L, Faenza I, Fiume R, Maria Billi A, Gilmour RS, Manzoli FA. Biochim Biophys Acta. 2006;1761:509. doi: 10.1016/j.bbalip.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Colombo G, Serra S, Vacca G, Carai MA, Gessa GL. Pharmacol Biochem Behav. 2005;81:369. doi: 10.1016/j.pbb.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 25.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Proc Natl Acad Sci USA. 2001;98:9371. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 27.Cravatt BF, Prospero-Garcia O, Siuzdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Science. 1995;268:1506. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 28.Day TA, Rakhshan F, Deutsch DG, Barker EL. Mol Pharmacol. 2001;59:1369. doi: 10.1124/mol.59.6.1369. [DOI] [PubMed] [Google Scholar]
- 29.Deutsch DG, Chin SA. Biochem Pharmacol. 1993;46:791. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- 30.Deutsch DG, Glaser ST, Howell JM, Kunz JS, Puffenbarger RA, Hillard CJ, Abumrad N. J Biol Chem. 2001;276:6967. doi: 10.1074/jbc.M003161200. [DOI] [PubMed] [Google Scholar]
- 31.Di Marzo V. Biochim Biophys Acta. 1998;1392:153. doi: 10.1016/s0005-2760(98)00042-3. [DOI] [PubMed] [Google Scholar]
- 32.Di Marzo V, Bisogno T, De Petrocellis L. Curr Pharm Des. 2000;6:1361. doi: 10.2174/1381612003399365. [DOI] [PubMed] [Google Scholar]
- 33.Di Marzo V, Bisogno T, Melck D, Ross R, Brockie H, Stevenson L, Pertwee R, De Petrocellis L. FEBS Lett. 1998;436:449. doi: 10.1016/s0014-5793(98)01175-2. [DOI] [PubMed] [Google Scholar]
- 34.Di Marzo V, Bisogno T, Sugiura T, Melck D, De Petrocellis L. Biochem J. 1998;331:15. doi: 10.1042/bj3310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di Marzo V, De Petrocellis L, Sepe N, Buono A. Biochem J. 1996;316:977. doi: 10.1042/bj3160977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Nature. 1994;372:686. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 37.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Proc Natl Acad Sci USA. 2002;99:10819. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Egertova M, Cravatt BF, Elphick MR. Neuroscience. 2003;119:481. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 39.Egertova M, Giang DK, Cravatt BF, Elphick MR. Proc R Soc Lond B Biol Sci. 1998;265:2081. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felder C, Nielsen A, Briley E, Palkovits M, Priller J, Axelrod J, Nguyen D, Richardson J, RM R, Koppel G, Paul S, Becker G. FEBS Lett. 1996;393:231. doi: 10.1016/0014-5793(96)00891-5. [DOI] [PubMed] [Google Scholar]
- 41.Felder CC, Briley EM, Axelrod J, Simpson JT, Mackie K, Devane WA. Proc Natl Acad Sci USA. 1993;90:7656. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL. Mol Pharmacol. 1995;48:443. [PubMed] [Google Scholar]
- 43.Fu J, Astarita G, Gaetani S, Kim J, Cravatt BF, Mackie K, Piomelli D. J Biol Chem. 2007;282:1518. doi: 10.1074/jbc.M607809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Nat Neurosci. 1999;2:358. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- 45.Glaser ST, Abumrad NA, Fatade F, Kaczocha M, Studholme KM, Deutsch DG. Proc Natl Acad Sci USA. 2003;100:4269. doi: 10.1073/pnas.0730816100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goparaju SK, Kurahashi Y, Suzuki H, Ueda N, Yamamoto S. Biochim Biophys Acta. 1999;1441:77. doi: 10.1016/s1388-1981(99)00143-2. [DOI] [PubMed] [Google Scholar]
- 47.Goparaju SK, Ueda N, Yamaguchi H, Yamamoto S. FEBS Lett. 1998;422:69. doi: 10.1016/s0014-5793(97)01603-7. [DOI] [PubMed] [Google Scholar]
- 48.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Eur J Neurosci. 2004;20:441. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 49.Hansen HH, Hansen SH, Schousboe A, Hansen HS. J Neurochem. 2000;75:861. doi: 10.1046/j.1471-4159.2000.0750861.x. [DOI] [PubMed] [Google Scholar]
- 50.Hansson AC, Bermudez-Silva FJ, Malinen H, Hyytia P, Sanchez-Vera I, Rimondini R, Rodriguez de Fonseca F, Kunos G, Sommer WH, Heilig M. Neuropsychopharmacol. 2007;32:117. doi: 10.1038/sj.npp.1301034. [DOI] [PubMed] [Google Scholar]
- 51.Hanus L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE, Kustanovich I, Mechoulam R. Proc Natl Acad Sci USA. 2001;98:3662. doi: 10.1073/pnas.061029898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB. J Neurochem. 1997;69:631. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- 53.Hillard CJ, Jarrahian A. Chem Phys Lipids. 2000;108:123. doi: 10.1016/s0009-3084(00)00191-2. [DOI] [PubMed] [Google Scholar]
- 54.Hillard CJ, Jarrahian A. Br J Pharmacol. 2003;140:802. doi: 10.1038/sj.bjp.0705468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. Pharmacol Rev. 2002;54:161. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 56.Jarrahian A, Manna S, Edgemond W, Campbell W, Hillard C. J Neurochem. 2000;74:2597. doi: 10.1046/j.1471-4159.2000.0742597.x. [DOI] [PubMed] [Google Scholar]
- 57.Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. J Biol Chem. 1997;272:27218. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 58.Karlsson M, Reue K, Xia YR, Lusis AJ, Langin D, Tornqvist H, Holm C. Gene. 2001;272:11. doi: 10.1016/s0378-1119(01)00559-5. [DOI] [PubMed] [Google Scholar]
- 59.Katayama K, Ueda N, Katoh I, Yamamoto S. Biochim Biophys Acta. 1999;1440:205. doi: 10.1016/s1388-1981(99)00124-9. [DOI] [PubMed] [Google Scholar]
- 60.Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. J Neurosci. 2006;26:5628. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim J, Alger BE. Nat Neurosci. 2004;7:697. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- 62.Kondo S, Kondo H, Nakane S, Kodaka T, Tokumura A, K W, Sugiura T. FEBS Lett. 1998;429:152. doi: 10.1016/s0014-5793(98)00581-x. [DOI] [PubMed] [Google Scholar]
- 63.Konrad RJ, Major CD, Wolf BA. Biochemistry. 1994;33:13284. doi: 10.1021/bi00249a015. [DOI] [PubMed] [Google Scholar]
- 64.Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, Jakobsson PJ, Marnett LJ. J Biol Chem. 2002;277:44877. doi: 10.1074/jbc.M206788200. [DOI] [PubMed] [Google Scholar]
- 65.Kreitzer AC, Regehr WG. J Neurosci. 2001;21:RC174. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kreitzer AC, Regehr WG. Neuron. 2001;29:71. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 67.Kurahashi Y, Ueda N, Suzuki H, Suzuki M, Yamamoto S. Biochem Biophys Res Commun. 1997;237:512. doi: 10.1006/bbrc.1997.7180. [DOI] [PubMed] [Google Scholar]
- 68.Leung D, Saghatelian A, Simon GM, Cravatt BF. Biochemistry. 2006;45:4720. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lichtman AH, Hawkins EG, Griffin G, Cravatt BF. J Pharmacol Exp Ther. 2002;302:73. doi: 10.1124/jpet.302.1.73. [DOI] [PubMed] [Google Scholar]
- 70.Liscovitch M, Czarny M, Fiucci G, Tang X. Biochem J. 2000;345(Pt 3):401. [PMC free article] [PubMed] [Google Scholar]
- 71.Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, Kunos G. Proc Natl Acad Sci USA. 2006;103:13345. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maccarrone M, Attina M, Bari M, Cartoni A, Ledent C, Finazzi-Agro A. J Neurochem. 2001;78:339. doi: 10.1046/j.1471-4159.2001.00413.x. [DOI] [PubMed] [Google Scholar]
- 73.Maccarrone M, Bari M, Lorenzon T, Bisogno T, Di Marzo V, Finazzi-Agro A. J Biol Chem. 2000;275:13484. doi: 10.1074/jbc.275.18.13484. [DOI] [PubMed] [Google Scholar]
- 74.Maccarrone M, Bari M, Menichelli A, Del Principe D, Agro AF. FEBS Lett. 1999;447:277. doi: 10.1016/s0014-5793(99)00308-7. [DOI] [PubMed] [Google Scholar]
- 75.Maccarrone M, Fiorucci L, Erba F, Bari M, Finazzi-Agro A, Ascoli F. FEBS Lett. 2000;468:176. doi: 10.1016/s0014-5793(00)01223-0. [DOI] [PubMed] [Google Scholar]
- 76.Maccarrone M, van der Stelt M, Rossi A, Veldink GA, Vliegenthart JF, Agro AF. J Biol Chem. 1998;273:32332. doi: 10.1074/jbc.273.48.32332. [DOI] [PubMed] [Google Scholar]
- 77.Maejima T, Ohno-Shosaku T, Kano M. Neurosci Res. 2001;40:205. doi: 10.1016/s0168-0102(01)00241-3. [DOI] [PubMed] [Google Scholar]
- 78.Maione S, Bisogno T, de Novellis V, Palazzo E, Cristino L, Valenti M, Petrosino S, Guglielmotti V, Rossi F, Di Marzo V. J Pharmacol Exp Ther. 2006;316:969. doi: 10.1124/jpet.105.093286. [DOI] [PubMed] [Google Scholar]
- 79.Makara JK, Mor M, Fegley D, Szabo SI, Kathuria S, Astarita G, Duranti A, Tontini A, Tarzia G, Rivara S, Freund TF, Piomelli D. Nat Neurosci. 2005;8:1139. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- 80.Maurelli S, Bisogno T, De Petrocellis L, Di Luccia A, Marino G, Di Marzo V. FEBS Lett. 1995;377:82. doi: 10.1016/0014-5793(95)01311-3. [DOI] [PubMed] [Google Scholar]
- 81.McKinney MK, Cravatt BF. Annu Rev Biochem. 2005;74:411. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 82.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. Biochem Pharmacol. 1995;50:83. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 83.Mechoulam R, Fride E. In cannabinoid receptors. Academic Press; 1995. [Google Scholar]
- 84.Mechoulam R, Fride E, Di Marzo V. Eur J Pharmacol. 1998;359:1. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- 85.Melck D, Bisogno T, De Petrocellis L, Chuang H, Julius D, Bifulco M, Di Marzo V. Biochem Biophys Res Commun. 1999;262:275. doi: 10.1006/bbrc.1999.1105. [DOI] [PubMed] [Google Scholar]
- 86.Moesgaard B, Petersen G, Jaroszewski JW, Hansen HS. J Lipid Res. 2000;41:985. [PubMed] [Google Scholar]
- 87.Mulder AM, Cravatt BF. Biochemistry. 2006;45:11267. doi: 10.1021/bi061122s. [DOI] [PubMed] [Google Scholar]
- 88.Nakane S, Oka S, Arai S, Waku K, Ishima Y, Tokumura A, Sugiura T. Arch Biochem Biophys. 2002;402:51. doi: 10.1016/S0003-9861(02)00038-3. [DOI] [PubMed] [Google Scholar]
- 89.Natarajan V, Reddy PV, Schmid PC, Schmid HH. Biochim Biophys Acta. 1981;664:445. doi: 10.1016/0005-2760(81)90067-9. [DOI] [PubMed] [Google Scholar]
- 90.Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. J Biol Chem. 2004;279:5298. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- 91.Osei-Hyiaman D, Depetrillo M, Harvey-White J, Bannon AW, Cravatt BF, Kuhar MJ, Mackie K, Palkovits M, Kunos G. Neuroendocrinology. 2005;81:273. doi: 10.1159/000087925. [DOI] [PubMed] [Google Scholar]
- 92.Palmer SL, Khanolkar AD, Makriyannis A. Curr Pharm Des. 2000;6:1381. doi: 10.2174/1381612003399419. [DOI] [PubMed] [Google Scholar]
- 93.Patricelli MP, Lovato MA, Cravatt BF. Biochemistry. 1999;38:9804. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- 94.Piomelli D, Beltramo M, Glasnapp S, Lin SY, Goutopoulos A, Xie XQ, Makriyannis A. Proc Natl Acad Sci USA. 1999;96:5802. doi: 10.1073/pnas.96.10.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, Nomikos GG, Carter P, Bymaster FP, Leese AB, Felder CC. J Pharmacol Exp Ther. 2002;301:1020. doi: 10.1124/jpet.301.3.1020. [DOI] [PubMed] [Google Scholar]
- 96.Prescott SM, Majerus PW. J Biol Chem. 1983;258:764. [PubMed] [Google Scholar]
- 97.Rakhshan F, Day TA, Blakely RD, Barker EL. J Pharmacol Exp Ther. 2000;292:960. [PubMed] [Google Scholar]
- 98.Ross RA, Craib SJ, Stevenson LA, Pertwee RG, Henderson A, Toole J, Ellington HC. J Pharmacol Exp Ther. 2002;301:900. doi: 10.1124/jpet.301.3.900. [DOI] [PubMed] [Google Scholar]
- 99.Sang N, Zhang J, Chen C. J Physiol. 2006;572:735. doi: 10.1113/jphysiol.2006.105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmid PC, Reddy PV, Natarajan V, Schmid HH. J Biol Chem. 1983;258:9302. [PubMed] [Google Scholar]
- 101.Sevilla E, Valette A, Gastaldi M, Boyer J, Verine A. Biochem Pharmacol. 1991;41:2005. doi: 10.1016/0006-2952(91)90142-r. [DOI] [PubMed] [Google Scholar]
- 102.Simon GM, Cravatt BF. J Biol Chem. 2006;281:26465. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- 103.Simpson CM, Itabe H, Reynolds CN, King WC, Glomset JA. J Biol Chem. 1991;266:15902. [PubMed] [Google Scholar]
- 104.Sipe JC, Chiang K, Gerber AL, Beutler E, Cravatt BF. Proc Natl Acad Sci USA. 2002;99:8394. doi: 10.1073/pnas.082235799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sipe JC, Waalen J, Gerber A, Beutler E. Int J Obes (Lond) 2005;29:755. doi: 10.1038/sj.ijo.0802954. [DOI] [PubMed] [Google Scholar]
- 106.Slanina KA, Roberto M, Schweitzer P. Neuropharmacology. 2005;49:660. doi: 10.1016/j.neuropharm.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 107.Solinas M, Panlilio LV, Tanda G, Makriyannis A, Matthews SA, Goldberg SR. Neuropsychopharmacology. 2005;30:2046. doi: 10.1038/sj.npp.1300754. [DOI] [PubMed] [Google Scholar]
- 108.Stella N, Schweitzer P, Piomelli D. Nature. 1997;388:773. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 109.Straiker A, Mackie K. J Physiol. 2005;569:501. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sugiura T, Kobayashi Y, Oka S, Waku K. Prostaglandins Leukot Essent Fatty Acids. 2002;66:173. doi: 10.1054/plef.2001.0356. [DOI] [PubMed] [Google Scholar]
- 111.Sugiura T, Kodaka T, Kondo S, Tonegawa T, Nakane S, Kishimoto S, Yamashita A, K W. Biochem Biophys Res Commun. 1996;229:58. doi: 10.1006/bbrc.1996.1757. [DOI] [PubMed] [Google Scholar]
- 112.Sugiura T, Kodaka T, Nakane S, Kishimoto S, Kondo S, Waku K. Biochem Biophys Res Commun. 1998;243:838. doi: 10.1006/bbrc.1998.8187. [DOI] [PubMed] [Google Scholar]
- 113.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. Biochem Biophys Res Commun. 1995;215:89. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 114.Sugiura T, Kondo S, Sukagawa A, Tonegawa T, Nakane S, Yamashita A, Waku K. Biochem Biophys Res Commun. 1996;218:113. doi: 10.1006/bbrc.1996.0020. [DOI] [PubMed] [Google Scholar]
- 115.Sugiura T, Kondo S, Sukagawa A, Tonegawa T, Nakane S, Yamashita A, Waku K. Lipid Mediat Cell Signal. 1996;14:51. doi: 10.1016/0929-7855(96)00508-1. [DOI] [PubMed] [Google Scholar]
- 116.Sugiura T, Yoshinaga N, Kondo S, Waku K, Ishima Y. Biochem Biophys Res Commun. 2000;271:654. doi: 10.1006/bbrc.2000.2686. [DOI] [PubMed] [Google Scholar]
- 117.Sun YX, Tsuboi K, Okamoto Y, Tonai T, Murakami M, Kudo I, Ueda N. Biochem J. 2004;380:749. doi: 10.1042/BJ20040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Szabo B, Urbanski MJ, Bisogno T, Di Marzo V, Mendiguren A, Baer WU, Freiman I. J Physiol. 2006;577:263. doi: 10.1113/jphysiol.2006.119362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N. J Biol Chem. 2005;280:11082. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- 120.Tsutsumi T, Kobayashi T, Ueda H, Yamauchi E, Watanabe S, Okuyama H. Neurochem Res. 1994;19:399. doi: 10.1007/BF00967316. [DOI] [PubMed] [Google Scholar]
- 121.Ueda H, Kobayashi T, Kishimoto M, Tsutsumi T, Okuyama H. J Neurochem. 1993;61:1874. doi: 10.1111/j.1471-4159.1993.tb09829.x. [DOI] [PubMed] [Google Scholar]
- 122.Ueda H, Kobayashi T, Kishimoto M, Tsutsumi T, Watanabe S, Okuyama H. Biochem Biophys Res Commun. 1993;195:1272. doi: 10.1006/bbrc.1993.2181. [DOI] [PubMed] [Google Scholar]
- 123.Ueda N, Kurahashi Y, Yamamoto S, Tokunaga T. J Biol Chem. 1995;270:23823. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]
- 124.Ueda N, Yamamoto K, Yamamoto S, Tokunaga T, Shirakawa E, Shinkai H, Ogawa M, Sato T, Kudo I, Inoue K, et al. Biochim Biophys Acta. 1995;1254:127. doi: 10.1016/0005-2760(94)00170-4. [DOI] [PubMed] [Google Scholar]
- 125.Vinod KY, Yalamanchili R, Xie S, Cooper TB, Hungund BL. Neurochem Int. 2006;49:619. doi: 10.1016/j.neuint.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 126.Wan M, Cravatt BF, Ring HZ, Zhang X, Francke U. Genomics. 1998;54:408. doi: 10.1006/geno.1998.5597. [DOI] [PubMed] [Google Scholar]
- 127.Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. J Biol Chem. 2006;281:36569. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- 128.Willoughby KA, Moore SF, Martin BR, Ellis EF. J Pharmacol Exp Ther. 1997;282:243. [PubMed] [Google Scholar]
- 129.Yang HY, Karoum F, Felder C, Badger H, Wang TC, Markey SP. J Neurochem. 1999;72:1959. doi: 10.1046/j.1471-4159.1999.0721959.x. [DOI] [PubMed] [Google Scholar]
- 130.Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, Watanabe M. J Neurosci. 2006;26:4740. doi: 10.1523/JNEUROSCI.0054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]