

Abstract
The α-galactosylceramide (α-GalCer) known as KRN7000 remains the best studied ligand of the lipid-binding MHC class I-like protein CD1d. The KRN7000:CD1d complex is highly recognized by invariant natural killer T (iNKT) cells, an evolutionarily conserved subset of T lymphocytes that express an unusual semi-invariant T cell antigen receptor, and mediate a variety of proinflammatory and immunoregulatory functions. To facilitate the study of glycolipid antigen presentation to iNKT cells by CD1d, we undertook the production of mouse monoclonal antibodies (mAbs) specific for complexes of KRN7000 bound to mouse CD1d (mCD1d) proteins. Three such monoclonal antibodies were isolated that bound only to mCD1d proteins that were loaded with KRN7000 or closely-related forms of α-GalCer. These mAbs showed no reactivity with mCD1d proteins that were not loaded with α-GalCer, nor did they bind to complexes formed by loading mCD1d with the self-glycolipid and putative iNKT cell ligand isoglobotrihexosylceramide. These complex-specific monoclonal antibodies allow the direct detection and monitoring of complexes formed by the binding of KRN7000 and other α-GalCer analogues to mCD1d. The availability of these mAbs should facilitate a wide range of studies on the biology and potential clinical applications of CD1d-restricted iNKT cells.
1. Introduction
Type 1 or invariant natural killer T cells (iNKT cells) comprise a unique and evolutionarily conserved subset of T lymphocytes with a variety of regulatory and effector functions (Yu and Porcelli, 2005). A hallmark of these cells is their expression of unusual T cell receptors (TCR) composed of an invariant TCR α chain rearrangement, encoded by mouse Vα14 or human Vα24 joined to Jα18 with little or no N-region diversity, and somewhat restricted TCR β chains encoded by rearrangements of only one or a few Vβ gene segments (mouse Vβ8.2, 7 or 2, and human Vβ11). These cells also differ from conventional T cells in that they recognize glycolipid antigens presented by the nonclassical β2-microglobulin (β2m)-associated MHC class I-like protein CD1d. Recognition of CD1d-presented glycolipids by iNKT cells elicits the rapid secretion of both T helper type 1 (TH1)- and T helper type 2 (TH2)-associated cytokines (e.g., IFN-γ and IL-4, respectively), without the need for priming. This innate-like behavior enables iNKT cells to influence the outcome of developing or ongoing immune reactions, making them important effectors and regulators of both innate and adaptive immunity.
Studies of the potential functions of iNKT cells have been greatly facilitated by the discovery that they can be activated by KRN7000, which is a synthetic α-galactosylceramide (α-GalCer) based on the structure of a natural product derived from the sponge Agelas mauritianus (Kawano et al., 1997). This compound binds strongly to CD1d to form a complex that is specifically recognized by the semi-invariant TCRs of iNKT cells, eliciting a broad range of effector activities including the production of both TH1- and TH2-type cytokines and the maturation of dendritic cells. Therapy with KRN7000 has shown beneficial effects in several mouse models of autoimmune disease, infection, and cancer (Yu and Porcelli, 2005).
Recently, the development of structurally related analogues of KRN7000 has raised the possibility of designing improved therapeutics that activate a more limited range of the multiple effector and regulatory properties of iNKT cells. For example, several variants of KRN7000, such as those designated OCH, PBS-25 and α-GalCer C20:2, can preferentially activate the TH2-type cytokine production of iNKT cells while stimulating much reduced systemic IFN-γ (Miyamoto et al., 2001; Goff et al., 2004; Yu et al., 2005). This TH2-biased response may represent a viable mode of NKT cell activation for therapy in certain autoimmune diseases.
The mechanism leading to the induction of TH2-biased responses by certain α-GalCer analogues is a controversial topic. It has been shown that one of the relevant compounds, OCH, forms a glycolipid:CD1d complex that binds the iNKT cell TCR with lower avidity than KRN7000:CD1d complexes (Stanic et al., 2003; Forestier et al., 2007). This is not true, however, of other TH2-biasing analogues, such as α-GalCer C20:2, which forms complexes with CD1d that bind to the iNKT TCR with an avidity similar to or greater than that of KRN7000:CD1d complexes. In these studies, it was also shown that C20:2 exhibited more permissive loading requirements onto CD1d than KRN7000 (Yu et al., 2005). For example, whereas KRN7000 required internalization to an endocytic compartment for efficient presentation, the C20:2 analogue had no such requirement and appeared to rapidly and efficiently associate with cell surface CD1d molecules. These results suggested that the altered cytokine response seen with C20:2 stimulation could be due to presentation by different types of antigen-presenting cells, or by distinct subpopulations of CD1d molecules in different subcellular compartments of the cell.
In most studies to date, iNKT cell hybridomas have been used as a sensitive readout for formation of α-GalCer:CD1d complexes. This technique is indirect and has many limitations that prevent its use for directly imaging the complexes of interest or assessing their formation and distribution in real time. To explore the mechanistic and kinetic details of ligand association with CD1d molecules, a more direct method of detecting α-GalCer:CD1d complexes would be of great value. The straightforward technique of using fluorescent, radio- or biotin-labeled α-GalCer molecules as probes for complex formation is hampered by the problem that these probes can associate with the cell surface independently of CD1d, probably through nonspecific interactions with membranes and hydrophobic protein domains (Wallner et al., 2004; data not shown).
Thus, to address and overcome these experimental conditions, we have undertaken the production of monoclonal antibodies (mAbs) that react specifically with the α-GalCer:CD1d complex. This strategy is similar to the generation of mAbs against peptide:MHC complexes that have been used as probes to detect the presence of these complexes in various in vitro and ex vivo settings. Here we describe the successful generation of three mAbs that react specifically with murine CD1d molecules only when they have bound KRN7000 or other closely related glycolipids.
2. Materials and Methods
2.1 Reagents
Mice with genetic deletions of both CD1.1 and CD1.2 in the BALB/c background (BALB/c.CD1d−/−) were obtained from the Jackson Laboratory (Bar Harbor, ME) (Smiley et al., 1997). Wild type BALB/c mice were from Taconic Farms (Germantown, NY). Mice were housed in a specific pathogen-free facility, and were handled according to institutional guidelines. Female mice were used between 8 and 15 weeks of age.
RMA-S cells transfected with mouse CD1d (mCD1d) were provided by S. Behar (Brigham and Women’s Hospital, Harvard Medical School) (Behar et al., 1999). mCD1d-transfected A20 cells provided by M. Kronenberg (La Jolla Institute for Allergy and Immunology, La Jolla, CA) (Prigozy et al., 2001) were further subcloned by limiting dilution. One high-expressing subclone, designated “K8,” was used in this study. The Vβ8.2+ iNKT hybridoma DN3A4-1.2 was provided by M. Kronenberg (Brossay et al., 1998). HeLa cells transfected with human CD1d (hCD1d) and cloned by limiting dilution (“subclone 6”) have been described (Spada et al., 1998). The non-secreting myeloma NSO/1 was obtained from M. Scharff (Albert Einstein College of Medicine) (Galfre and Milstein, 1981). The C57BL/6.p53−/−-derived dendritic cell line JAWS II was purchased from the ATCC (Manassas, VA) (MacKay and Moore, 1997). Rat anti-mCD1d mAb-secreting hybridomas 1B1 and 19G11.2 (both IgG2b/κ) were provided by M. Kronenberg and A. Bendelac (University of Chicago, IL), respectively (Brossay et al., 1997; Roark et al., 1998).
Most of the above lines were maintained in RPMI-1640 medium supplemented with 10 mM HEPES, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 55 μM 2-mercaptoethanol, 20 μg/ml gentamicin (Gibco, Grand Island, NY) and 10% heat-inactivated (55°C, 30 min) fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA). HeLa, NSO/1 and hybridoma cells were grown in similarly supplemented DMEM. JAWS II dendritic cells were maintained in modified MEMα medium with ribonucleosides and deoxyribonucleosides (Gibco) and supplemented as above, but with 20% FBS (instead of 10%), 1 mM sodium pyruvate and 5 ng/ml mouse GM-CSF (Peprotech, Rocky Hill, NJ).
Glycolipids KRN7000 [(2S, 3S, 4R)-1-O-(α-D-galactopyranosyl)-N-hexacosanoyl-2-amino-1,3,4-octadecanetriol], OCH [(2S, 3S, 4R)-1-O-(α-D-galactopyranosyl)-N-tetracosanoyl-2-amino-1,3,4-nonanetriol], and α-GalCer C20:2 [(2S, 3S, 4R)-1-O-(α-D-galactopyranosyl)-N-11,14-cis-eicosadienoyl-2-amino-1,3,4-octadecanetriol] were synthesized as described (Yu et al., 2005; Ndonye et al., 2005). Isoglobotrihexosylceramide (iGb3) was obtained from Alexis Biochemicals (San Diego, CA). These lipids were initially solubilized in chloroform:methanol (2:1) for aliquoting. Solvent was removed from the aliquots by drying in vacuo, and the glycolipids were then resuspended to 100 μM in dimethylsulfoxide (DMSO) for in vitro use.
Soluble recombinant mCD1d, co-expressed with mouse β2m, was prepared through a baculovirus-driven Drosophila melanogaster expression system provided by M. Kronenberg (Matsuda et al., 2000). Soluble single chain hCD1d:β2m and hCD1b:β2m fusion proteins were produced using a Chinese hamster ovary cell system as described (Im et al., 2004). These CD1 proteins had a biotin ligase site and a hexahistidine (His6) tag appended to their C-termini. For some experiments, mCD1d:IgG1 dimers were used with similar results (BD Pharmingen, San Jose, CA). CD1d proteins were loaded overnight (ON) with a 4- to 10-fold molar excess of glycolipid, diluted from DMSO stocks, in PBS + 0.05% Triton X-100 at room temperature (RT). The addition of Triton X-100 was required to obtain full loading of most lipids, including KRN7000 (unpublished observations).
2.2 Immunization protocol and fusion
For preparation of the primary immunogen, the purified protein derivative of Mycobacterium tuberculosis strain H37Ra (PPD; Statens Serum Institut, Copanhagen, Denmark) was covalently coupled on a 1:1 mass basis with complexes of KRN7000:mCD1d in PBS in the presence of 0.1% glutaraldehyde (grade I, Sigma) for 2 h at RT, with continuous stirring. Fixative was quenched with one volume of 0.2 M L-lysine, pH 7.4, and the product was then dialyzed against PBS through a 3 kDa MW cutoff membrane (Pierce, Rockford, IL). The composition of the final product (KRN7000:mCD1d:PPD) was studied by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) through a 4–20% Tris/Glycine gradient PAGEr Gold precast gel (Cambrex, East Rutherford, NJ). The gel was stained using GelCode Blue (Pierce).
Four BALB/c mice and 3 BALB/c.CD1d−/− mice were each vaccinated i.p. with 0.2 ml of live Mycobacterium bovis Bacille Calmette-Guerin (BCG vaccine SSI, Statens Serum Institut), reconstituted as per the manufacturer’s instructions. This dose was equivalent to twice the adult i.d. human dose, and contained between 0.4 and 1.6 × 106 colony forming units of attenuated M. bovis BCG, Danish strain 1331.
At day 23 post-BCG vaccination, the mice were injected i.p. with 5 μg KRN7000:mCD1d:PPD, in 200 μl of a 1:1 emulsion of PBS with Imject Alum (Pierce). This was followed by another injection at day 42, and test bleeds were taken at day 48. The mice were then boosted i.p. at day 61 with 6 – 7 × 106 KRN7000-loaded A20.mCD1d cells (loaded with 250 nM KRN7000 in FBS-free medium ON, irradiated at 5000 rad, and fixed for 30 s with 0.05% glutaraldehyde in PBS), given in 200 μl PBS.
After three months of rest, responding BALB/c and BALB/c.CD1d−/− mice were given a final i.v. boost of 10 μg KRN7000:mCD1d (not aldehyde-fixed, and without PPD) in 400 μl PBS. This time point corresponded to day 155 of the protocol. These mice were sacrificed and their spleens prepared for fusion 3 days later. Myeloma/splenocyte fusions were conducted with minor deviations from the classic procedures (Galfre and Milstein, 1981). Spleens were aseptically dispersed in PBS, and the recovered cells were then washed in PBS, followed by erythrocyte lysis using a red blood cell lysing buffer (Sigma, St. Louis, MO), used at 2 ml per spleen for 3 min at RT. This reaction was quenched by the addition of FBS- and HEPES-free supplemented DMEM.
The cells were then washed and resuspended in FBS/HEPES-free DMEM. One-fourth of the preparation was then mixed with the myeloma partner NSO/1 at a cell ratio of 4:1 in FBS/HEPES-free DMEM, and then centrifuged at 430g for 5 min. The tube containing the cell pellet was then placed in a 40°C water bath. Into this was added 1 ml of 37°C-heated 50% (w/v) polyethylene glycol (MW 1450 Da, in PBS, from Sigma) over 1 min with stirring. The suspension was then stirred for an additional minute. This was followed by the addition of 1 ml heated FBS/HEPES-free DMEM added over 1 min, then 7 ml FBS/HEPES-free DMEM added over 1 min. The cells were then centrifuged as above and resuspended in complete DMEM.
The fusomas were then plated at 200,000 cells in 100 μl/well together with MRC-5 fibroblast feeder cells (from the ATCC, irradiated 2500 rad, 2500 cells/well) in 96-well flat-bottom tissue culture plates. Medium containing 2X HAT (1X = 100 μM hypoxanthine, 0.4 μM aminopterin and 16 μM thymidine, from Sigma) was then added at 100 μl/well at day 2. Half of the culture medium was replaced with fresh DMEM/1X HAT every 2 days. Screening of culture supernatants (described below) was begun as each well reached half-confluence, beginning at ~2 weeks post fusion. Hybridomas of interest were subcloned twice by limiting dilution.
2.3 Production, purification and labeling of mAbs
For milligram-scale mAb production, hybridomas were gradually adapted to culture in RPMI supplemented as above, but with 8% heat-inactivated low IgG FBS (Gibco). 500 ml of medium in two-liter roller bottles were inoculated with 108 cells, and cultures were supplemented with OptiMAb supplement (Gibco) following the manufacturer’s instructions. mAbs were purified from clarified and filtered supernatants by standard protein G column chromatography (Amersham Biosciences/GE Healthcare, Piscataway, NJ).
Purified mAbs (~2.5 mg/ml in PBS) were biotinylated ON at 4°C using NHS-LC-biotin (Pierce; dissolved to 10 mM with DMSO just prior to use) with a biotin:mAb molar ratio of 30:1. Unbound biotin was not removed from the prep, as this would not interfere with downstream applications. mAbs were likewise labeled with the succinimidyl ester of the fluorescent dye AlexaFluor 647 (Molecular Probes, Eugene, OR; diluted 2.4 mM in 1 M NaHCO3 immediately before use) with a dye:mAb molar ratio of 14.4:1 (1:10 vol/vol), run for 1 hour at RT and then ON at 4°C. Excess dye was removed by dialysis against PBS through a 10 kDa MW cutoff membrane (Pierce).
For nonbinding negative control antibodies, chromatographically purified normal mouse and rat IgG was purchased from Jackson ImmunoResearch (West Grove, PA).
2.4 FACS analysis using α-GalCer:mCD1d complex-specific Ab
Antibody activity was assayed by FACS on CD1d+ cell lines (usually RMA-S.mCD1d or JAWS II). Cells were first loaded with KRN7000, iGb3, or the equivalent volume of DMSO vehicle (0.1% vol/vol final). Loading was done ON with 100 nM lipid in complete medium, unless otherwise stated. Cells were then washed, blocked with 10% heat-inactivated normal goat serum (NGS) with 0.05% NaN3 in PBS, and then incubated with hybridoma supernatants (in medium), purified mAb, or sera (diluted in 10% bovine serum) for 30 min at RT. After washing by suspending in cold PBS and pelleting the cells by centrifugation (430g, 5 min at 4°C), Ab binding was revealed with goat F(ab’)2 anti-mouse Ig(H+L)-phycoerythryn (PE) conjugate (Southern Biotech, Birmingham, AL), diluted 1/200 in 1% NGS/0.05% NaN3/PBS for 30 min at RT. For rat mAbs, goat F(ab’)2 anti-rat IgG(H+L)-PE (Caltag, South San Francisco, CA) was used instead. Cells were then washed, and flow cytometry data were acquired on a FACSCalibur (Becton Dickenson, Mountain View, CA), and data were analyzed using WINMDI 2.8 software (The Scripps Research Institute, La Jolla, CA).
2.5 ELISA for α-GalCer:mCD1d complex-specific Ab
ELISA plates (Corning Costar 3369) were coated with KRN7000:mCD1d or iGb3:mCD1d complexes (typically 0.2 – 0.5 μg/ml in PBS, 50 μl/well) ON at RT. After blocking with 0.1% bovine serum albumin in PBS (1 h at RT or ON at 4°C), plates were washed thrice with PBS + 0.05% Tween 20. Ab was then added as culture supernatant or diluted in PBS + 1% bovine serum for 2 – 3 h at RT. After washing, alkaline phosphatase (AP)-conjugated goat anti-mouse IgG (Southern Biotech; 1/2000 in 1% NGS/PBS + 0.05% Tween 20) was added for 45 min at RT. After washing five times, bound enzyme was revealed by reading absorbance at 405 nm following the addition of 1 mg/ml p-nitrophenylphosphate (Sigma) in 63 mM Na/CO3 buffer, pH 9.6, plus 2 mM MgCl2.
Alternatively, to detect biotinylated Ab, AP-streptavidin (from Zymed, South San Francisco, CA; diluted 1/2000 in 1% NGS/PBS + 0.05% Tween 20) was used. To detect rat mAb, AP-conjugated goat anti-rat IgG(H+L) (Southern Biotech, similarly diluted) was used.
mAb isotypes were determined by standard ELISA, using the corresponding purified capture and AP-conjugated detection isotype-specific reagents (goat polyclonal Ab) and purified mouse standard mAbs from Southern Biotech.
2.6 iNKT cell hybridoma in vitro activation assay
RMA-S.mCD1d or JAWS II dendritic cells were loaded with 100 nM KRN7000, iGb3, or the equivalent amount of DMSO vehicle ON. After washing three times in PBS, cells were resuspended in medium and plated in 96-well tissue culture-treated plates at 50,000 cells per well in 50 μl medium. To this was added 50 μl medium containing 30 μg/ml of various mAbs or IgG controls. Plates were then returned to 37°C for a 30 min pre-incubation. After this, the iNKT cell hybridoma DN3A4-1.2 was added at 50,000 cells per well in 50 μl medium. The final mAb concentration was then 10 μg/ml. After 12 – 15 hours culture supernatants were harvested and assayed for IL-2 by ELISA, using purified capture and biotinylated detection rat anti-mouse IL-2 mAbs from BD Pharmingen. AP-streptavidin and p-nitrophenylphosphate substrate were used as above. Recombinant mouse IL-2 (R&D Systems, Minneapolis, MN) was used as standard. This system was typically sensitive to ~20 pg/ml.
3. Results and Discussion
3.1 Immunization strategy
There are only a handful of peptide:MHC molecule complex-specific mAbs reported in the literature, suggesting that the derivation of such mAbs is not trivial. The principal concern in carrying out immunizations to produce these mAbs is to generate a sufficiently large repertoire of responding B cells that produce specific Ab against the peptide:MHC complex, while minimizing responses against the MHC protein itself. Even in the few successful attempts to generate mAbs specific for peptide:MHC complexes, this problem has been difficult to overcome. For instance, in the derivation of the ovalbumin peptide SIINFEKL:H-2Kb complex-specific mAb 25-D1.16, the investigators chose to immunize an allogeneic host (BALB/c, H-2Kd) (Porgador et al., 1997). This was thought to be necessary to elicit CD4 T cell help for the responding B cells. Although this approach was ultimately successful in isolating the complex-specific mAb-secreting hybridoma, there was a strong anti-H-2Kb response in the serum, and anti-H-2Kb Ab-secreting hybridomas were obtained at much higher frequency in their primary screen compared to complex-specific hybridomas.
Based on structural comparisons of CD1d and MHC molecules and their complexes with bound ligands, we speculated that achieving a B cell response primarily against the complex-specific epitopes might be even more difficult in the case of CD1d than for MHC molecules. For example, an available crystal structure of a short-chain α-GalCer:mCD1d complex shows that much of the α-GalCer structure is buried within the lipid-binding groove of the protein, with only the terminal sugar and a few adjacent atoms “visible” from the outside (Zajonc et al., 2005).
This suggested to us that immunization with α-GalCer:mCD1d complexes in a host animal not tolerant to the mCD1d protein (e.g., a CD1d−/− mouse, or a wild type rat), the secondary structure of the mCD1d polypeptide would be more immunodominant than the altered glycolipid/protein interface—probably much more so than the MHC protein sequence being immunodominant over the peptide/protein interface, since in the latter case side chains of several of the peptide’s amino acids are oriented away from the MHC surface, and possibly “visible” from the outside (Zhang et al., 1992).
These theoretical considerations caused us to search for immunization protocols that would strongly discourage an Ab response against the protein sequence itself, while still providing strong immunization against the complex-specific epitopes. One such strategy was used in earlier published studies to produce both the influenza virus hemagglutinin peptide FESTGNLI:H-2Kk complex-specific Fab 13.4.1 and the human myelin basic protein peptide ENPVVHFFKNIVTPR:HLA-DR2 (DRB1*1501) complex-specific Fab MK16 (Andersen et al., 1996; Krogsgaard et al., 2000). In these two reports, the immunized mouse hosts expressed the MHC protein of interest as a self-antigen. This was sufficient to prevent an Ab response against the MHC protein itself. To generate sufficient CD4 T cell help, Mycobacterium tuberculosis PPD was coupled to the peptide:MHC complexes used as the immunogen, and an M. bovis BCG-vaccinated host was used. This use of PPD as an atypical “carrier” has been reported to facilitate Ab responses against other difficult antigens, and has the added advantage of not eliciting anti-carrier Abs (Lachmann et al., 1986).
Following the above example, KRN7000:mCD1d complex was cross-linked with PPD using glutaraldehyde. The major species in the resultant product was identified by denaturing SDS-PAGE as one diffuse band migrating at 43 – 45 kDa (Fig. 1). This differed from the KRN7000:mCD1d complex itself before cross-linking, which migrated as two bands at 34 and ~8 kDa, corresponding to the CD1d polypeptide and β2m, respectively. There were no demonstrable differences between the gel migration of the fixed KRN7000:mCD1d:PPD complex as compared to glutaraldehyde-fixed KRN7000:mCD1d—indicating that the components of PPD that were coupled to KRN7000:mCD1d were either not large enough, or not uniform enough, to cause an identifiable shift (data not shown).
Figure 1.
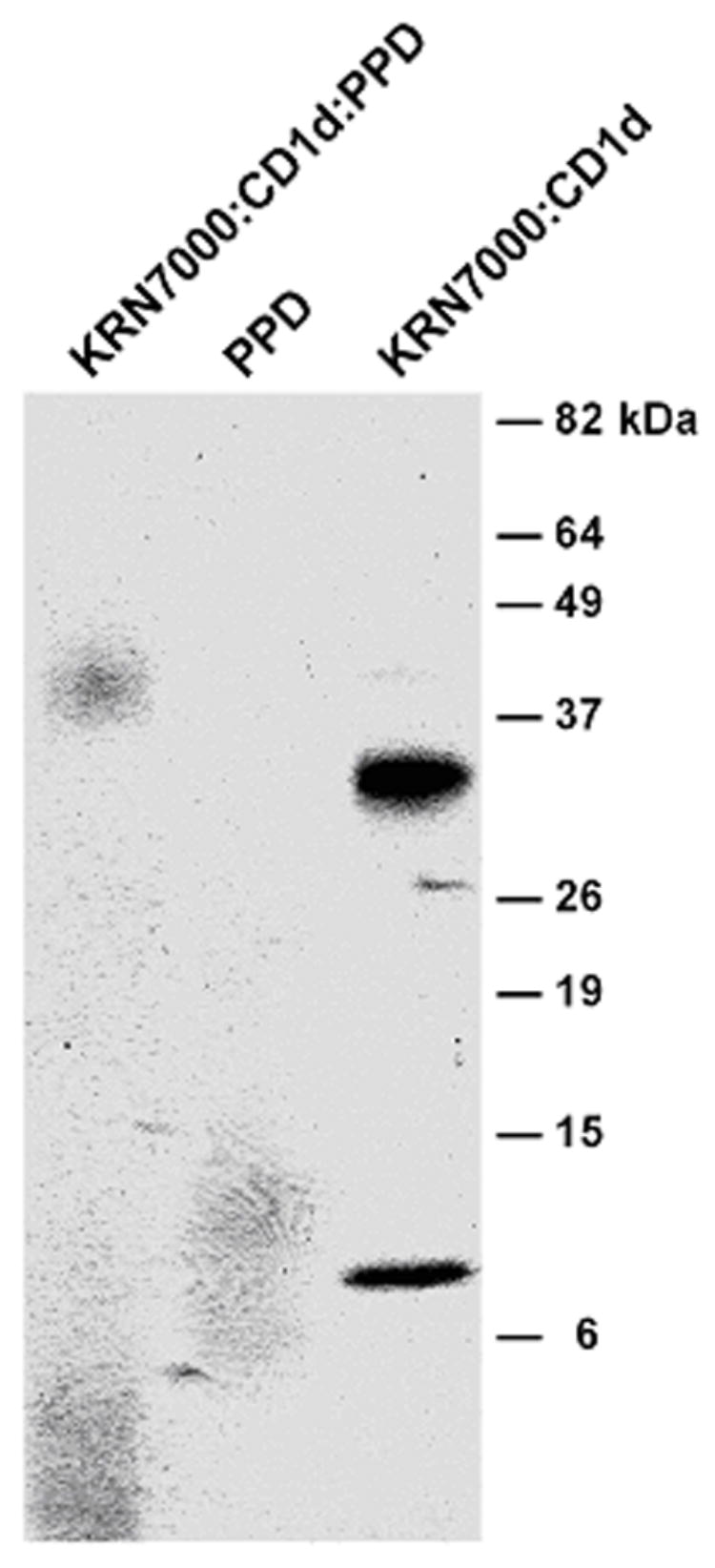
Nonreducing SDS-PAGE profile of the KRN7000:mCD1d:PPD complex used for the immunization protocol. Lane 1 shows 1 μg of the glutaraldehyde cross-linked immunogen. The constituents used to make this preparation are run separately at lane 2 (PPD, 4 μg) and lane 3 (KRN7000-loaded mCD1d, not aldehyde-fixed, 1 μg). Positions of MW markers are shown on the right.
We then proceeded to inject the KRN7000:mCD1d:PPD complex into BCG-vaccinated BALB/c and BALB/c.CD1d−/− hosts. After two immunizations, two of the three CD1d−/− mice gave strong Ab responses against CD1d, irrespective of whether this was loaded with KRN7000 or not (Fig. 2). This confirmed our hypothesis that in animals lacking immunological tolerance to CD1d, epitopes associated with the CD1d polypeptide alone were markedly immunodominant over epitopes associated with the specific complex of KRN7000 bound to CD1d. On the other hand, two of the four immunized wild type BALB/c mice gave demonstrable responses against KRN7000-loaded mCD1d, while showing no response against iGb3- or vehicle-loaded CD1d by ELISA or FACS analyses (Fig. 2). This indicated that putative α-GalCer:mCD1d-specific Ab-secreting B cells were present in these hosts. Note that we chose to assay binding to iGb3-loaded mCD1d as a representative of endogenous-ligand loaded mCD1d, as opposed to “unloaded” recombinant protein. This was done because we found that several anti-mCD1d mAbs and antisera, while not distinguishing lipid versus vehicle-loaded mCD1d+ cells by FACS, clearly show preferential binding to loaded recombinant mCD1d protein over the unloaded form by ELISA (data not shown).
Figure 2.
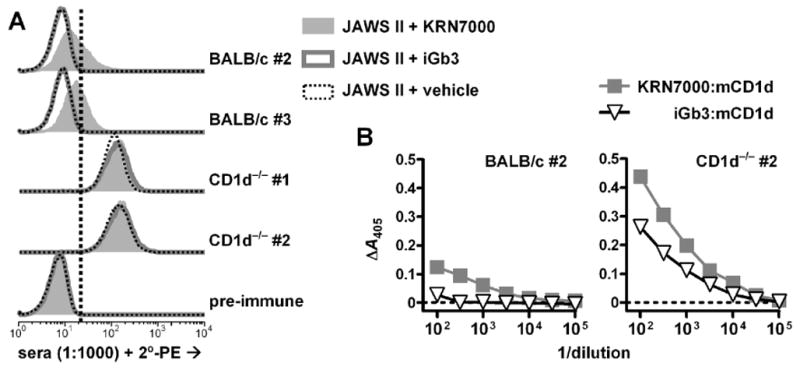
Serum reactivities in mice one week after the second injection of KRN7000:mCD1d:PPD complexes. (A) FACS-based detection of total antibody reactive to JAWS II dendritic cells loaded with KRN7000, iGb3, or DMSO vehicle. Sera were tested at 1:1000 dilution. Results from responding mice (2 of 4 BALB/c, 2 of 3 BALB/c.CD1d−/−) and a pre-immune mouse are shown. The vertical dotted line indicates the upper limit of fluorescence for negative control staining (i.e., minimum level that exceeded the fluorescence of >99% of cells stained with pre-immune serum). Staining with pre-immune serum was similar to staining with the secondary agent alone (data not shown). (B) ELISA-based detection of IgG reactive to platebound KRN7000- or iGb3-loaded mouse CD1d protein, from two samples as indicated. Sera diluted as indicated; y-axes represent specific absorbance values after subtraction of nonspecific binding of serum antibody to buffer-coated wells. Sera from non-responding mice did not show any detectable mCD1d-specific binding above background.
To generate hybridomas producing complex-specific mAbs, we initially boosted the mice with KRN7000-loaded A20.mCD1d cells (BALB/c-derived) to enrich for B cells that would respond to cell-associated mCD1d, thus increasing the probability of generating mAbs useful for FACS. A fusion attempted with one of these mice (BALB/c mouse #2) three days later proved unproductive.
A second attempt was then undertaken in which, after resting the mice for three months, a final boost with KRN7000:mCD1d complex (without PPD) was given i.v., and fusions were conducted three days later. The terminal sera maintained the original reactivity observed after the 2nd immunization: by FACS, the bleed from CD1d−/− mouse #1 demonstrated binding to cell surface mCD1d, regardless of association with KRN7000. Serum from the responding BALB/c mouse #3, on the other hand, showed staining of KRN7000-loaded mCD1d+ cells, but not those loaded with vehicle (data not shown). This reactivity was present at a titer of ~1:10,000 by ELISA. There was, however, a low level of ELISA reactivity against iGb3:mCD1d. Because of the absence of such reactivity in FACS, this may have represented a response against the biotin ligase site and His6 tag present in the recombinant protein, but not in cell surface CD1d.
Supernatants from the resultant fusomas were screened for reactivity to KRN7000- and iGb3-loaded recombinant mCD1d by ELISA, and secondarily assayed for reactivity to KRN7000-, iGb3- or vehicle-loaded mCD1d+ cells by FACS. Out of 216 wells seeded from splenocytes from the CD1d−/− mouse (fusion K), ten wells reproducibly produced Ab that bound both recombinant mCD1d regardless of bound ligand—though there was slightly higher signal when KRN7000 was used instead of iGb3 (Fig. 3). This property was similar to ELISA results observed with the rat anti-mCD1d mAbs 1B1 and 19G11. By FACS, mCD1d+ cells stained equally well whether or not they were pre-incubated with KRN7000.
Figure 3.
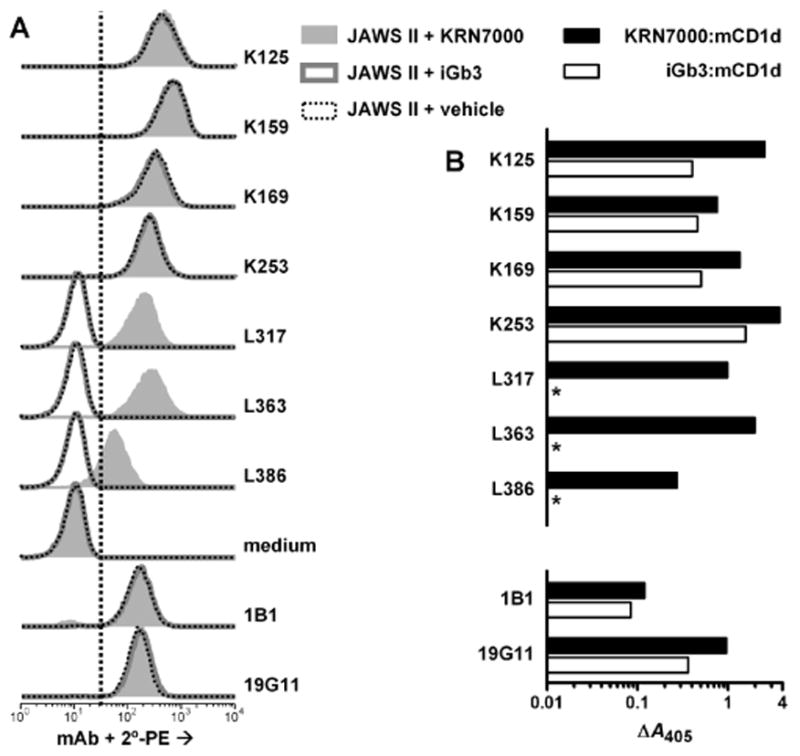
Reactivity of hybridomas from immunized BALB/c and CD1d−/− mice. Seven mAb culture supernatants were assayed by FACS on JAWS II cells (A) or by ELISA against plate bound mCD1d protein (B) as in Fig. 2. These supernatants were from confluent cultures of twice-subcloned hybridomas derived from either CD1d−/− mouse #1 (K125, K159, K169 and K253) or from BALB/c mouse #3 (L317, L363, L386). Rat mAbs 1B1 and 19G11, used at 1 μg/ml and detected with rat-specific reagents, were included for comparison. *, specific A405 < 0.010. For (A), vertical dotted line indicates upper limit for negative control staining, as described in Figure 2. Negative control staining was deterimined here by the sample incubated without primary mAb, followed by PE-conjugated secondary antibody (“medium”). Previous experiments showed that no signals above this background level were observed when a nonbinding control IgG1 mAb (P3X63Ag8), whole normal mouse serum or normal rat polyclonal IgG were used under these staining conditions, and similar results were obtained for staining of RMA-S.mCD1d cells (data not shown).
Of the 288 wells seeded from splenocytes from the immunized wild type BALB/c mouse (fusion L), supernatants from three wells reproducibly tested positive for reactivity against KRN7000:mCD1d by ELISA and FACS, without showing binding to iGb3:mCD1d or vehicle-loaded mCD1d+ cells (Fig. 3). Hybridomas from these three wells, designated L317, L363 and L386, as well as four of the wells containing conventional mCD1d-binding Ab from fusion K, were subcloned twice. Subcultures were adapted to growth in low IgG serum-containing medium and grown in roller bottle cultures, and mAbs were purified by protein G column chromatography. The isotype of mAb L363 was identified by ELISA to be IgG2a/κ, while the remaining six other subcloned hybridomas produced mAbs that had the IgG1/κ isotype.
3.2 Specificity of mAbs L317, L363 and L386 for KRN7000:mCD1d
An immediate concern was whether the putative KRN7000:mCD1d complex-specific mAbs isolated above were specific for KRN7000 itself, and not for the complex. Two lines of evidence showed this to be unlikely. First, while these mAbs bound KRN7000:mCD1d but not iGb3:mCD1d by ELISA, no binding was observed against complexes formed by loading recombinant human CD1d or CD1b with KRN7000. In addition, KRN7000-loaded hCD1d-transfected HeLa cells, which strongly activate mouse iNKT cell hybridomas, did not stain with mAb L363 above the background observed for iGb3- or vehicle-loaded cells (data not shown).
A second line of evidence was derived from inhibition ELISAs. Monoclonal antibodies L317, L363 and L386 were allowed to bind platebound KRN7000:mCD1d in the presence of increasing amounts of various inhibitors. Soluble KRN7000:mCD1d at ~5 μg/ml was able to inhibit 50% of specific binding, whereas free KRN7000, KRN7000:hCD1d, and iGb3:mCD1d were all unable to inhibit binding at any concentration (Fig. 4 and data not shown).
Figure 4.
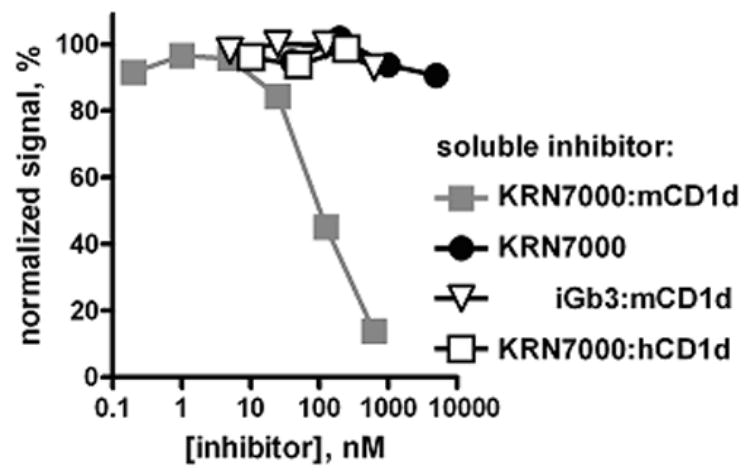
Specificity of mAb L317 for the KRN7000:mCD1d complex. Biotinylated L317 (1 nM) was allowed to bind to platebound KRN7000:mCD1d complexes in the presence of increasing concentrations of soluble KRN7000-loaded mCD1d, free KRN7000, iGb3-loaded mCD1d, or KRN7000-loaded human CD1d. Bound L317-biotin was detected with AP-streptavidin. Signal was normalized to mAb binding in the absence of inhibitor (100%), and binding to buffer-coated wells (0%). Similar results were obtained with mAbs L363 and L386.
3.3 Comparison of mAbs L317, L363 and L386 with other anti-mCD1d mAbs
We went on to further characterize these mAbs by comparison with other mAbs specific against mCD1d. Not including the mAbs detailed in this work, there are at least thirteen such mAbs in the literature (Bleicher et al., 1990; Brossay et al., 1997; Roark et al., 1998). We chose to compare the three complex-specific L-series mAbs and two of our K-series anti-mCD1d mAbs (K169 and K253) with the previously published and widely used rat anti-mCD1d mAbs 1B1 and 19G11. These two rat mAbs either compete weakly or not at all with each other for binding to mCD1d, suggesting that they bind to distinct epitopes on the protein. Furthermore, one study showed that 1B1 inhibited recognition of cell surface mCD1d (presumably loaded with endogenous ligands) by only six out of a panel of eleven iNKT cell hybridomas tested, whereas 19G11 inhibited recognition by all eleven (Roark et al., 1998). This suggests that 19G11 binding strongly blocks or otherwise abolishes an epitope on the CD1d protein required for TCR contact, compared to binding of 1B1, which is less disruptive of TCR:CD1d interactions.
We studied the binding of fixed amounts of biotin-labeled mAbs to platebound KRN7000- and iGb3-loaded mCD1d, in the presence of various amounts of different “cold” competitors. In one example (Fig. 5A), we found that binding of L363-biotin to KRN7000:mCD1d complex was strongly inhibited by 19G11, L317 and K253, less so by 1B1 and L386, and not at all by K169. This analysis was extended to the other mAbs, as summarized in Fig. 5B. One possible interpretation of our data is that the epitopes seen by L317 and L363 are either shared with or abolished by 19G11 and K253, and to a much lesser extent by 1B1. L386, on the other hand, may simply have a much lower avidity to KRN7000:mCD1d, since its binding to its cognate antigen is easily inhibited by nearly all mAbs tested. This competition analysis also provided additional evidence on the specificity of L317, L363 and L386, since each of these mAbs demonstrably inhibited binding of at least one of the anti-mCD1d mAbs to KRN7000:mCD1d, while showing no inhibition of the interaction of the anti-mCD1d mAbs to iGb3:mCD1d.
Figure 5.
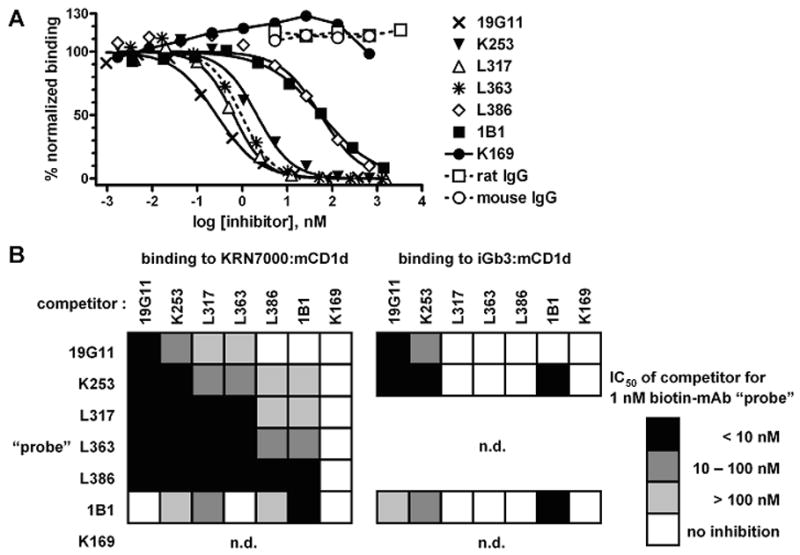
Inhibition of mAb binding to KRN7000:mCD1d by other mCD1d-reactive mAbs. (A) 1 nM mAb L363-biotin (“probe”) was allowed to bind to platebound KRN7000:mCD1d in the presence of increasing concentrations of various unlabeled mAbs. Purified total rat and mouse IgG were included as negative controls. Probe binding was revealed with AP-streptavidin, and signal was normalized as in Fig. 4. Where applicable, 4-parameter sigmoidal dose/response curves were drawn against the 0% and 100% asymptotes. The dotted curve represents inhibition of L363-biotin by “cold” L363. (B) checkerboard summary of inhibition of mAb-biotin probe binding to KRN7000:mCD1d or iGb3:mCD1d, by co-incubation with seven different mAbs. Inhibition was estimated by the concentration of mAb required for half-maximal inhibition (IC50). Darker-filled boxes indicate more potent inhibition. n.d., not done: K169-biotin was not included, since biotinylation of this mAb abolished specific binding (data not shown). Also, mAbs L317, L363 and L386 did not bind iGb3:mCD1d, so these were not included as probes in the analysis of binding to that complex.
We also studied the capability of these mAbs to block recognition of cell surface lipid:mCD1d complexes by one well-described iNKT cell hybridoma, DN3A4-1.2 (Fig. 6). mAb 19G11, at 10 μg/ml, inhibited recognition of mCD1d+ cells, whether they were loaded with KRN7000, iGb3, or vehicle. In these analyses, it is generally assumed that vehicle-pulsed cells have mCD1d loaded with a mixture of endogenous ligands—some probably antigenic to the iNKT TCR, and others that are not. 1B1, on the other hand, could inhibit recognition of iGb3- and vehicle-loaded cells, but not of KRN7000-loaded cells.
Figure 6.
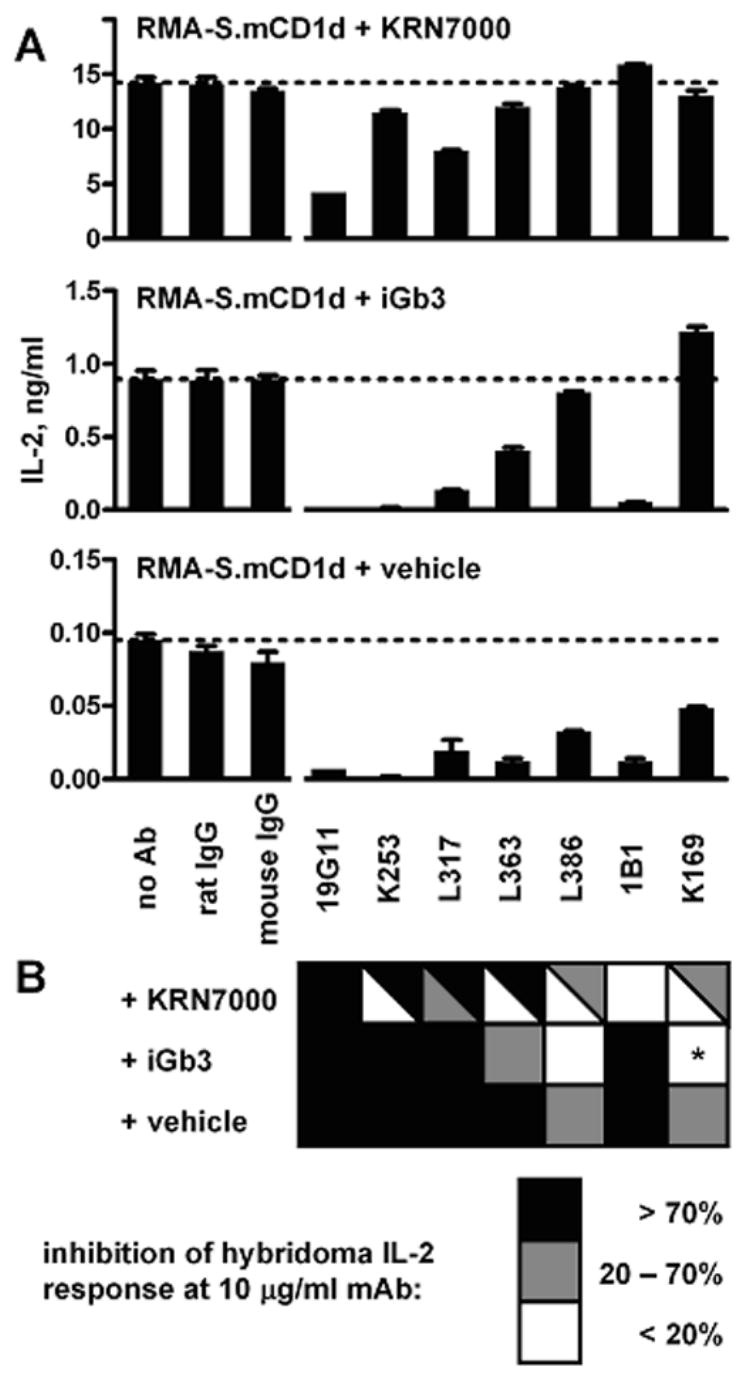
mAb inhibition of iNKT cell hybridoma recognition of cell surface mCD1d. IL-2 production by the iNKT cell hybridoma DN3A4-1.2 was measured after incubation with KRN7000-, iGb3-, or vehicle-loaded mCD1d+ cells, in the presence of various mAbs at 10 μg/ml. (A) one representative experiment, with RMA-S.mCD1d cells. Mean ± SE of IL-2 levels from duplicate wells shown. Horizontal dotted lines indicate the amount of IL-2 obtained in the absence of added Ab. (B) checkerboard summary of data from four experiments, using either RMA-S.mCD1d or JAWS II dendritic cells. Darker boxes indicate more inhibition by a particular mAb of the IL-2 response to mCD1d+ cells loaded with each lipid. Split boxes show the range of results obtained. *, mAb K169 consistently enhanced hybridoma responses to iGb3-loaded mCD1d+ cells.
The lack of binding by L317, L363 and L386 of mCD1d in the absence of KRN7000 has led us to predict that these mAbs would inhibit iNKT cell recognition of the KRN7000:mCD1d complex, but not recognition of mCD1d loaded with other ligands. This, however, was not the case. Inhibition by these mAbs of recognition of KRN7000-pulsed cells was variable, while there was some inhibition of iGb3- or vehicle-pulsed cells. One possible explanation for this unexpected result was that our complex-specific mAbs were actually specific for an “antigenic” conformation of mCD1d that corresponds to the conformation adopted when mCD1d molecules are loaded with any of a variety of lipid ligands. This conformation may have formed and accumulated at very high and detectable levels when mCD1d was loaded with KRN7000. In contrast, when iGb3 or other endogenous cellular lipids were the only ligands available for loading mCD1d, the putative antigenic conformation may have accumulated to much lower levels that were not detectable by FACS, but could be revealed by the more sensitive iNKT hybridoma response.
Another interesting observation from this analysis was the unusual behavior of the anti-mCD1d mAb K169, which was unique in that it did not inhibit the binding to mCD1d of any of the other mAbs. This indicated that the K169 mCD1d-binding site might be at some distance from the epitopes used by the others. It is likely that K169 was reactive with a less immunodominant region of mCD1d, such as that defined in an earlier study by the anti-mCD1d mAb 19F8, or by anti-mCD1d mAbs 16G9 and 4C4 (Roark et al., 1998). It was also remarkable that K169 actually increased iNKT cell hybridoma recognition of iGb3-loaded mCD1d+ cells, but did not have this impact on the antigenicity of mCD1d+ cells loaded with KRN7000 or with vehicle.
3.4 mAb recognition of mCD1d complexed with other α-GalCer analogues
We proceeded to determine whether our KRN7000:mCD1d complex-specific mAbs might also detect mCD1d loaded with other versions of α-GalCer. mCD1d protein was loaded with the α-GalCers KRN7000, C20:2, and OCH, as well as with iGb3 and DMSO vehicle in parallel. When coated onto ELISA plates, mAbs L317, L363 and L386 quantitatively and equally bound to both KRN7000- and C20:2-loaded mCD1d (Fig. 7 and data not shown). iGb3- and vehicle-loaded protein were not bound, as expected. OCH:mCD1d complexes, however, consistently elicited a signal lower than that observed with KRN7000:mCD1d. This may represent a lower avidity of the mAbs to this particular analogue:mCD1d complex. Alternatively, these data may actually reflect a loss of OCH from the lipid-binding groove of mCD1d in our experimental conditions, owing to a faster off-rate of this lipid from the protein (Stanic et al., 2003).
Figure 7.
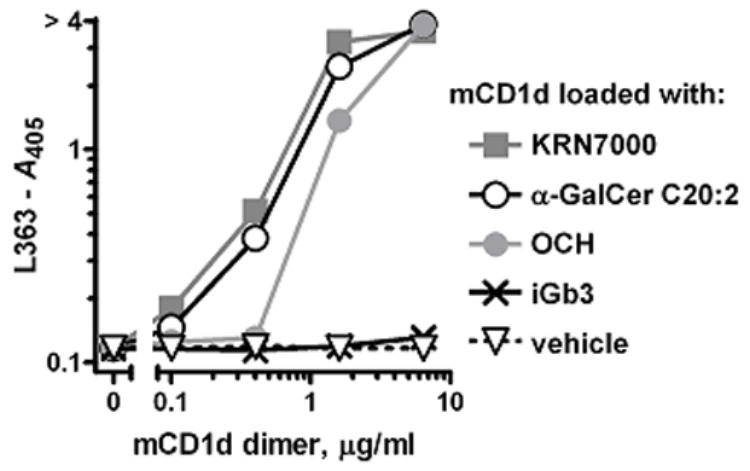
Binding of mAb L363 to mCD1d loaded with other α-GalCer lipids. Binding of mAb L363-biotin to ELISA plate wells coated with varying concentrations of mCD1d dimers was detected with AP-streptavidin. mCD1d protein was pre-loaded with KRN7000, α-GalCer C20:2, OCH, iGb3, or solvent vehicle in the presence of 0.05% Triton X-100.
The results above indicated that, at least for KRN7000 and C20:2, our mAbs may be used as a bona fide probe of formation of the α-GalCer:mCD1d complex. We then proceeded to test mAb L363 reactivity to mCD1d+ cells loaded with these different α-GalCer analogues. Our initial results showed that L363 stained C20:2-loaded cells more than cells loaded with an equimolar amount of KRN7000 (data not shown). This was in contrast with conventional iNKT cell hybridoma stimulation assays, which commonly show KRN7000 to be about one order of magnitude more potent than C20:2 (Yu et al., 2005). To evaluate this further, we used L363 to carefully examine the kinetics of antigen presentation for various α-GalCer analogues. JAWS II or RMA-S.mCD1d cells were loaded with 100 nM of α-GalCers KRN7000, C20:2, and OCH for various antigen pulse times (Fig. 8 and data not shown). L363 binding demonstrated that C20:2 association with cell surface CD1d was markedly faster than the loading of KRN7000, with maximal signal achieved at ~4 h, and signal decreasing after 10 h. OCH, on the other hand, had a much lower level of recognition by the mAb, but seemed to reach maximal signal at about the same time frame as α-GalCer C20:2. These data add to credence to the hypothesis that TH2-biasing variants of KRN7000 associate much more readily with CD1d—and indeed with a much faster velocity.
Figure 8.
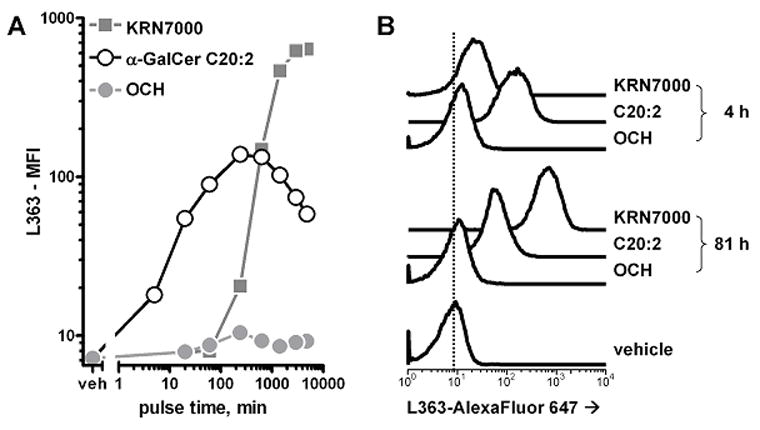
Binding of mAb L363 to cells loaded in vitro with various α-GalCers. JAWS II dendritic cells were loaded with 100 nM KRN7000, α-GalCer C20:2 or OCH for various times at 37°C, after which cells were collected and stained with AlexaFluor 647-labeled mAb L363. mAb staining was detected by FACS. (A) median fluorescence intensity graphed against lipid antigen pulse time. Note that both axes are displayed on logarithmic scales. (B) FACS histograms for 4- and 81-hour loaded cells. The vertical dotted line is for comparison of the histograms with the readout from vehicle-loaded cells. Similar results were obtained with RMA-S.mCD1d cells.
4. Concluding remarks
This work details the derivation and characterization of three mAbs against the most studied agonist of iNKT cells, the complex formed by α-GalCer and CD1d. These mAbs, together with two KRN7000:hCD1d complex-specific mAbs derived by phage display by V. Cerundulo’s group (Oxford University, U.K. Presented at the 4th International NKT Cell and CD1 Workshop, Italy, October 2006), represent the first ligand:antigen-presenting molecule complex-specific antibody reagents for study of the CD1 lipid antigen presentation system.
At least two of our mAbs, L317 and L363, were sensitive enough to be used as probes for in vitro loading of cell lines with α-GalCers KRN7000 and C20:2. Thus, we believe that the α-GalCer:mCD1d complex-specific mAbs described here should contribute significantly to studies of glycolipid antigen presentation by mCD1d using appropriate in vitro model systems. It will also be interest to determine whether these mAbs are reactive with mCD1d loaded with other related natural antigenic glycolipids, such as the Sphingomonas-derived α-glucoronosyl- and α-galactoronosylceramides, which could allow their use to be extended to models of CD1d presentation during bacterial infection (Mattner et al., 2005; Kinjo et al., 2005). Our studies have not yet addressed the use of these mAbs for the study of lipid antigen presentation by natural antigen presenting cells or using in vivo experimental systems. In the literature on complex-specific mAbs against conventional MHC antigen presenting molecules, so far only the I-Eα56–73:I-Ab complex-specific mAb Y-Ae has been successfully used to track in vivo formation of the ligand:protein complex, with physiologic doses of antigen (Murphy et al., 1989; Itano et al., 2003). This most likely reflects technical issues related to sensitivity and specificity of complex detection under normal physiologic conditions. Further studies will be needed to determine whether our novel complex-specific mAbs can be effectively applied to the study of glycolipid antigens by mCD1d expressed by relevant primary antigen-presenting cells in vitro and in vivo.
Acknowledgments
We thank S. Behar, M. Scharff, M. Kronenberg, and A. Bendelac for making available a number of cell lines used in this study. We are grateful to H. Hu for expert technical assistance in mAb purification. We especially appreciate technical advice and helpful discussions from M. Scharff and T. DiLorenzo (Albert Einstein College of Medicine). This work was supported by NIH grants RO1 AI45889 (to S.A.P.) and RO1 AI057519 (to A.R.H.). S.A.P. received partial support from an Irma T. Hirschl Career Scientist Award. G.S.B. is a former Lister Institute-Jenner Research Fellow and acknowledges support from the Medical Research Council (U.K.), the Wellcome Trust, and from the James Bardrick Research Chair. K.O.A.Y. was supported by the Medical Scientist Training Program of the Albert Einstein College of Medicine. Flow cytometry studies were supported by the FACS Core Facilities of the AECOM Center for AIDS Research (NIH/NIAID AI51519) and the AECOM Cancer Center (NIH/NCI P30 CA13330).
Abbreviations
- α-GalCer
α-galactosylceramide
- Ab
antibody
- AP
alkaline phosphatase
- β2m
β2-microglobulin
- DMSO
dimethylsulfoxide
- ELISA
enzyme-linked immunosorbent assay
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- hCD1d
human CD1d
- Ig
immunoglobulin
- iGb3
isoglobotrihexosylceramide
- iNKT cells
mouse Vα14 or human Vα24 invariant natural killer T cells
- mAb
monoclonal antibody
- mCD1d
mouse CD1d
- MW
molecular weight
- NGS
normal goat serum
- ON
overnight
- PBS
phosphate-buffered saline
- PE
phycoerythrin
- PPD
purified protein derivative of Mycobacterium tuberculosis strain H37Ra
- RT
room temperature
- TCR
T cell receptor
- TH1(2)
T helper type 1 (or 2)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen PS, Stryhn A, Hansen BE, Fugger L, Engberg J, Buus S. A recombinant antibody with the antigen-specific, major histocompatibility complex-restricted specificity of T cells. Proc Natl Acad Sci U S A. 1996;93:1820. doi: 10.1073/pnas.93.5.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar SM, Podrebarac TA, Roy CJ, Wang CR, Brenner MB. Diverse TCRs recognize murine CD1. J Immunol. 1999;162:161. [PubMed] [Google Scholar]
- Bleicher PA, Balk SP, Hagen SJ, Blumberg RS, Flotte TJ, Terhorst C. Expression of murine CD1 on gastrointestinal epithelium. Science. 1990;250:679. doi: 10.1126/science.1700477. [DOI] [PubMed] [Google Scholar]
- Brossay L, Jullien D, Cardell S, Sydora BC, Burdin N, Modlin RL, Kronenberg M. Mouse CD1 is mainly expressed on hemopoietic-derived cells. J Immunol. 1997;159:1216. [PubMed] [Google Scholar]
- Brossay L, Tangri S, Bix M, Cardell S, Locksley R, Kronenberg M. Mouse CD1-autoreactive T cells have diverse patterns of reactivity to CD1+ targets. J Immunol. 1998;160:3681. [PubMed] [Google Scholar]
- Forestier C, Takaki T, Molano A, Im JS, Baine I, Jerud ES, Illarionov P, Ndonye R, Howell AR, Santamaria P, Besra GS, DiLorenzo TP, Porcelli SA. Improved Outcomes in NOD Mice Treated with a Novel Th2 Cytokine-Biasing NKT Cell Activator. J Immunol. 2007;178:1415. doi: 10.4049/jimmunol.178.3.1415. [DOI] [PubMed] [Google Scholar]
- Galfre G, Milstein C. Preparation of monoclonal antibodies: strategies and procedures. Methods Enzymol. 1981;73:3. doi: 10.1016/0076-6879(81)73054-4. [DOI] [PubMed] [Google Scholar]
- Goff RD, Gao Y, Mattner J, Zhou D, Yin N, Cantu C, III, Teyton L, Bendelac A, Savage PB. Effects of Lipid Chain Lengths in α-Galactosylceramides on Cytokine Release by Natural Killer T Cells. J Am Chem Soc. 2004;126:13602. doi: 10.1021/ja045385q. [DOI] [PubMed] [Google Scholar]
- Im JS, Yu KO, Illarionov PA, LeClair KP, Storey JR, Kennedy MW, Besra GS, Porcelli SA. Direct Measurement of Antigen Binding Properties of CD1 Proteins Using Fluorescent Lipid Probes. J Biol Chem. 2004;279:299. doi: 10.1074/jbc.M308803200. [DOI] [PubMed] [Google Scholar]
- Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, Jenkins MK. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity. 2003;19:47. doi: 10.1016/s1074-7613(03)00175-4. [DOI] [PubMed] [Google Scholar]
- Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M. CD1d-restricted and TCR-mediated activation of Vα14 NKT cells by glycosylceramides. Science. 1997;278:1626. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- Krogsgaard M, Wucherpfennig KW, Cannella B, Hansen BE, Svejgaard A, Pyrdol J, Ditzel H, Raine C, Engberg J, Fugger L. Visualization of myelin basic protein (MBP) T cell epitopes in multiple sclerosis lesions using a monoclonal antibody specific for the human histocompatibility leukocyte antigen (HLA)-DR2-MBP 85–99 complex. J Exp Med. 2000;191:1395. doi: 10.1084/jem.191.8.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann PJ, Strangeways L, Vyakarnam A, Evan G. Raising antibodies by coupling peptides to PPD and immunizing BCG-sensitized animals. Ciba Found Symp. 1986;119:25. doi: 10.1002/9780470513286.ch3. [DOI] [PubMed] [Google Scholar]
- MacKay VL, Moore EE. Immortalized dendritic cells. 5,648,219 US patent. 1997
- Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, Koezuka Y, Kronenberg M. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med. 2000;192:741. doi: 10.1084/jem.192.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, III, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, Hoebe K, Schneewind O, Walker D, Beutler B, Teyton L, Savage PB, Bendelac A. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Miyake S, Yamamura T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature. 2001;413:531. doi: 10.1038/35097097. [DOI] [PubMed] [Google Scholar]
- Murphy DB, Lo D, Rath S, Brinster RL, Flavell RA, Slanetz A, Janeway CA., Jr A novel MHC class II epitope expressed in thymic medulla but not cortex. Nature. 1989;338:765. doi: 10.1038/338765a0. [DOI] [PubMed] [Google Scholar]
- Ndonye RM, Izmirian DP, Dunn MF, Yu KO, Porcelli SA, Khurana A, Kronenberg M, Richardson SK, Howell AR. Synthesis and evaluation of sphinganine analogues of KRN7000 and OCH. J Org Chem. 2005;70:10260. doi: 10.1021/jo051147h. [DOI] [PubMed] [Google Scholar]
- Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- Prigozy TI, Naidenko O, Qasba P, Elewaut D, Brossay L, Khurana A, Natori T, Koezuka Y, Kulkarni A, Kronenberg M. Glycolipid antigen processing for presentation by CD1d molecules. Science. 2001;291:664. doi: 10.1126/science.291.5504.664. [DOI] [PubMed] [Google Scholar]
- Roark JH, Park SH, Jayawardena J, Kavita U, Shannon M, Bendelac A. CD1.1 expression by mouse antigen-presenting cells and marginal zone B cells. J Immunol. 1998;160:3121. [PubMed] [Google Scholar]
- Smiley ST, Kaplan MH, Grusby MJ. Immunoglobulin E production in the absence of interleukin-4-secreting CD1-dependent cells. Science. 1997;275:977. doi: 10.1126/science.275.5302.977. [DOI] [PubMed] [Google Scholar]
- Spada FM, Koezuka Y, Porcelli SA. CD1d-restricted recognition of synthetic glycolipid antigens by human natural killer T cells. J Exp Med. 1998;188:1529. doi: 10.1084/jem.188.8.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanic AK, Shashidharamurthy R, Bezbradica JS, Matsuki N, Yoshimura Y, Miyake S, Choi EY, Schell TD, Van Kaer L, Tevethia SS, Roopenian DC, Yamamura T, Joyce S. Another view of T cell antigen recognition: cooperative engagement of glycolipid antigens by Vα14Jα18 natural T (iNKT) cell receptor. J Immunol. 2003;171:4539. doi: 10.4049/jimmunol.171.9.4539. [DOI] [PubMed] [Google Scholar]
- Wallner FK, Chen L, Moliner A, Jondal M, Elofsson M. Loading of the antigen-presenting protein CD1d with synthetic glycolipids. Chembiochem. 2004;5:437. doi: 10.1002/cbic.200300655. [DOI] [PubMed] [Google Scholar]
- Yu KO, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, Fujiwara N, Arias I, Miyake S, Yamamura T, Chang YT, Besra GS, Porcelli SA. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of α-galactosylceramides. Proc Natl Acad Sci U S A. 2005;102:3383. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu KO, Porcelli SA. The diverse functions of CD1d-restricted NKT cells and their potential for immunotherapy. Immunol Lett. 2005;100:42. doi: 10.1016/j.imlet.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Zajonc DM, Cantu C, Mattner J, Zhou D, Savage PB, Bendelac A, Wilson IA, Teyton L. Structure and function of a potent agonist for the semi-invariant natural killer T cell receptor. Nat Immunol. 2005;6:810. doi: 10.1038/ni1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Young AC, Imarai M, Nathenson SG, Sacchettini JC. Crystal structure of the major histocompatibility complex class I H-2Kb molecule containing a single viral peptide: implications for peptide binding and T-cell receptor recognition. Proc Natl Acad Sci U S A. 1992;89:8403. doi: 10.1073/pnas.89.17.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]