

Abstract
Oxygenated sterols such as 25-hydroxycholesterol kill Chinese hamster ovary cells because they inhibit the proteolytic processing of sterol regulatory element binding proteins (SREBPs), a pair of membrane-bound transcription factors that activate genes controlling cholesterol synthesis and uptake from lipoproteins. The unprocessed SREBPs remain membrane-bound, they cannot activate the cholesterol biosynthetic pathway, and the cells die of cholesterol deprivation. Several sterol-resistant hamster cell lines have been isolated previously by chemical mutagenesis and selection for resistance to killing by 25-hydroxycholesterol. We recently identified the defect in one such cell line (25-RA cells) as a point mutation in a newly discovered membrane protein of 1276 amino acids, designated SREBP cleavage-activating protein (SCAP). The mutation in the 25-RA cells resulted from a G-to-A transition in codon 443 of the SCAP gene, changing aspartic acid to asparagine. Wild-type SCAP, when overexpressed by transfection, stimulates the proteolytic processing of both SREBPs. The D443N substitution is an activating mutation that increases the activity of SCAP and renders it resistant to inhibition by 25-hydroxycholesterol. We here report the identical G-to-A transition in two additional lines of Chinese hamster ovary cells that were mutagenized and isolated by a similar protocol. The three mutations occurred independently as indicated by haplotype analysis of the mutant genes using two intragenic sequence polymorphisms. All three cell lines were mutagenized with alkylating agents (nitrosoethylurea or ethylmethane sulfonate) that favor G-to-A transitions. Nevertheless, the finding of the same nucleotide substitution at the same location in all three cell lines indicates that SCAP may be unique in its ability to stimulate SREBP cleavage, and residue 443 is a crucial determinant of the protein’s ability to be inhibited by 25-hydroxycholesterol.
Keywords: sterol regulatory element binding protein, cholesterol, low density receptors, somatic cell mutations
Animal cells in tissue culture protect themselves against the overaccumulation of cholesterol by suppressing transcription of genes encoding several enzymes of the cholesterol biosynthetic pathway, including 3-hydroxy-3-methylglutaryl CoA synthase (HMG CoA synthase), HMG CoA synthase, farnesyl diphosphate synthase, and squalene synthase, as well as receptors for low-density lipoprotein, which provide exogenous cholesterol (1, 2, 3, 4). These genes are actively transcribed when cells grow in a low-cholesterol environment, thereby allowing cells to satisfy their cholesterol demands from endogenous and exogenous sources. When sterols overaccumulate, transcription is repressed, thereby preventing overaccumulation of cholesterol.
Over the past 20 years several laboratories have isolated mutant Chinese hamster ovary (CHO) cells that fail to undergo sterol-mediated repression of transcription (5, 6, 7, 8, 9). The strategies for mutant isolation depended on the observation that hydroxylated sterols such as 25-hydroxycholesterol are potent suppressors of the sterol-regulated genes, but these sterols cannot replace cholesterol in cell membranes (10, 11). When wild-type cells are grown in the absence of cholesterol and in the presence of 25-hydroxycholesterol, their cholesterol synthesis is repressed and they die of cholesterol deficiency. In all cases studied to date, mutant cells that survive this selection have a global defect in which 25-hydroxycholesterol and other sterols (including low-density lipoprotein-derived cholesterol) no longer repress the genes encoding enzymes of the cholesterol biosynthetic pathway or the low-density lipoprotein receptor.
All of the known sterol-regulated genes are under control of a pair of transcription factors designated sterol regulatory element binding proteins (SREBPs)-1 and -2 whose activity is regulated by a unique proteolytic mechanism (12, 13, 14). The SREBPs are synthesized as tripartite proteins of approximately 1150 amino acids that are attached to membranes of the endoplasmic reticulum and nuclear envelope. The NH2-terminal segment of ≈500 amino acids projects into the cytosol, followed by a hairpin membrane anchor composed of two membrane-spanning regions separated by a short loop of ≈30 amino acids that projects into the endoplasmic reticulum lumen (15). This is followed by a COOH-terminal segment of ≈500 amino acids that projects into the cytosol. The NH2-terminal segment is a transcriptional activator of the basic-helix-loop-helix-leucine zipper family that binds to sterol regulatory elements in the promoters of the sterol-regulated genes (16, 17). The NH2-terminal segment cannot enter the nucleus unless it is released from its membrane anchor, and this is where the sterol-regulated proteolytic event is crucial.
When cells are grown in the absence of cholesterol, a protease cleaves each of the SREBPs at site 1, which is in the lumenal loop at or near a crucial arginine (18). This breaks the connection between the two transmembrane domains, and it allows a second protease to cleave the NH2-terminal fragment at site 2, which is within the first transmembrane domain (18). This cleavage releases the NH2-terminal segment, which travels to the nucleus and activates transcription. Sterols, including 25-hydroxycholesterol, abolish cleavage at site 1, thereby precluding cleavage at site 2. As a result, the NH2-terminal fragments cannot enter the nucleus, and transcription of the sterol-regulated genes declines.
Two classes of sterol-resistant CHO cells have been described, each of which has a defect in the regulation of SREBP processing. Class 1 mutants have undergone a genomic rearrangement that results in the production of a truncated form of SREBP-2 that includes all of the amino acids up to residue 460. The truncated protein contains the transcriptional activating domain and the basic-helix-loop-helix-leucine zipper domain, but it lacks the membrane attachment domain, so it enters the nucleus without a requirement for proteolysis. It is therefore active under all conditions, and it cannot be turned off by sterols (13, 14).
Class 2 sterol-resistant mutants produce normal full-length SREBP-1 and SREBP-2, but they cannot turn off the proteolytic processing of either protein in response to sterols (19). Recently, we traced the defect in one class 2 cell line (25-RA cells) (7) to an activating mutation in a gene that encodes a novel protein named SREBP cleavage-activating protein (SCAP) (20). We isolated the mutant SCAP cDNA through use of an expression cloning strategy in which pools of cDNAs from 25-RA cells were transfected into human embryonic kidney 293 cells in the presence of 25-hydroxycholesterol. We assayed for a cDNA that increased transcription of a reporter gene driven by a promoter that contained three copies of a sterol regulatory element. We identified a cDNA encoding a mutant form of SCAP with a G-to-A substitution that changes codon 443 from aspartic acid to asparagine. The mutation increases the ability of SCAP to stimulate the proteolytic cleavage of SREBPs at site 1, and it renders the reaction resistant to suppression by sterols (20).
SCAP is a membrane-bound protein of 1276 amino acids that is divided into two domains (20). The NH2-terminal domain of ≈800 amino acids consists of alternating hydrophobic and hydrophilic segments that encode up to 10 membrane-spanning segments. Some of these bear a sequence relationship to membrane-spanning regions in HMG CoA reductase, a membrane-bound enzyme whose degradation is accelerated by sterols (21, 22). The COOH-terminal domain of ≈475 amino acids is hydrophilic and contains four WD-repeats, which are found in a variety of regulatory proteins (23). Overexpression of wild-type SCAP in transfected cells increases the proteolysis of both SREBPs at site 1, and the reaction is rendered partially resistant to sterol suppression (20). The D443N mutant form of SCAP is up to 10-fold more active at low levels of expression, and it is highly resistant to suppression by sterols. We concluded that SCAP is a normal activator of SREBP cleavage at site 1 and that the D443N mutation increases its activity and decreases its sensitivity to inhibition by sterols, thus accounting for the sterol-resistance phenotype of the 25-RA cells.
In the current studies we have explored the status of SCAP in two other sterol-resistant class 2 CHO cell lines, designated SRD-4 (19) and SRD-8. Remarkably, we have found that both of these cell lines contain the same mutation as the 25-RA cells, namely a G-to-A transition in codon 443 of SCAP, changing aspartic acid to asparagine. We use haplotype analysis to show that the three G-to-A transitions arose independently. These data illustrate (i) the power of the selection scheme for 25-hydroxycholesterol resistance; (ii) the importance of the aspartic acid residue at the 443 position in determining the activity of SCAP; and (iii) the importance of SCAP as the only identified gene, other than SREBP-2, whose mutation can give rise to 25-hydroxycholesterol resistance in CHO cells.
MATERIALS AND METHODS
Cell Culture and Cell Fractionation.
All cells were grown in monolayer at 37°C in an atmosphere of 8–9% CO2. Stock cultures of CHO-K1 cells (American Type Culture Collection catalog no. CCL-61) and CHO-7 cells, a clone of CHO-K1 cells that were adapted to growth in lipoprotein-deficient serum (8), were grown in medium A (a 1:1 mixture of Ham’s F-12 medium and Dulbecco’s MEM containing 100 units/ml penicillin and 100 μg/ml streptomycin sulfate) supplemented with either 5% fetal calf serum (CHO-K1 cells) or 5% newborn calf lipoprotein-deficient serum (CHO-7 cells). 25-RA cells (7), SRD-4 cells (19, 24), and SRD-8 cells (see below) were grown in medium A supplemented with 5% newborn calf lipoprotein-deficient serum and 1 μg/ml 25-hydroxycholesterol.
For metabolic experiments, CHO-7, 25-RA, SRD-4, and SRD-8 cells were incubated in medium A containing 5% newborn calf lipoprotein-deficient serum, 50 μM compactin, and 50 μM sodium mevalonate under inducing (−25-hydroxycholesterol) or suppressing (+25-hydroxycholesterol) conditions and fractionated into a nuclear extract and 105 × g membrane pellet as described (18).
Immunoblot Analysis.
Immunoblot analysis was carried out on SDS/8% polyacrylamide gels as described (15) using monoclonal antibodies IgG-2A4 (17) and IgG-7D4 (14), which are directed against the NH2-terminal domains of SREBP-1 and SREBP-2, respectively. Bound antibodies were stained with the Enhanced Chemiluminescence Western Blotting Detection Kit (Amersham) as described (15, 20). Gels were calibrated with prestained molecular markers (New England Biolabs), and the filters were exposed to Reflection film (DuPont/NEN) at room temperature.
Isolation of 25-Hydroxycholesterol-Resistant SRD-8 Cells.
SRD-8 cells were isolated from pools of CHO-7 cells that had been mutagenized with nitrosoethylurea and selected in medium containing lipoprotein-deficient serum and 0.3 μg/ml 25-hydroxycholesterol as previously described (8). Surviving colonies were isolated with cloning cylinders, cloned by dilution plating, and then subjected to further selection with 1 μg/ml 25-hydroxycholesterol. One surviving clone, designated SRD-8, was expanded and maintained in medium A supplemented with 5% newborn calf lipoprotein-deficient serum and 1 μg/ml 25-hydroxycholesterol.
PCR, Single-Strand Conformational Polymorphism (SSCP) Analysis, and DNA Sequencing.
Standard molecular biology techniques were used (25). For haplotype analysis in Fig. 3, we isolated total RNA from subconfluent cultures using RNA STAT 60 solution (Tel-Test, Friendswood, TX) according to the manufacturer’s instructions. The RNA was used as a template for reverse transcription with a SCAP-specific primer (5′-TACAGTCAGGAGACAACAGT-3′) located in the 3′- untranslated region of the SCAP cDNA (20). Genomic DNA was isolated with a DNA extraction kit (Stratagene). Unless otherwise noted, PCR reactions were carried out in the presence of 50 mM Tris·HCl (pH 9.1), 16 mM (NH4)2SO4, 3.5 mM MgCl2, 0.15 mg/ml BSA, 125 μM dNTP (GIBCO/BRL), 100 nM primers, 0.1 unit/μl Klentaq polymerase (Ab Peptides, St. Louis), and 6 × 10−4 unit/μl cloned Pfu polymerase (Stratagene) in a Perkin–Elmer Model 9600 Thermocycler. PCR reactions for SSCP analysis were supplemented with 0.5 mCi (1 Ci = 37 GBq)/ml [32P]dCTP (Amersham). To verify sequence variations, PCR products were cloned into a plasmid, pNoTA/T7 (5 Prime → 3 Prime, Inc.) and sequenced on both strands with an Applied Biosystems model 377 DNA Sequencer.
Figure 3.
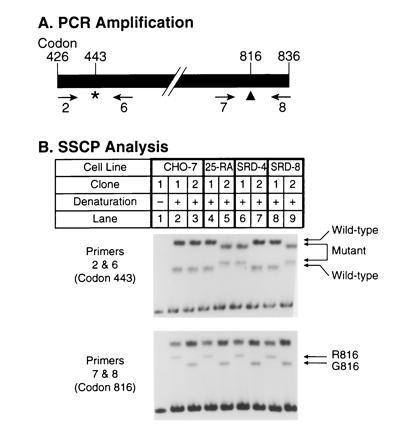
Haplotype analysis of codons 443 and 816 in the SCAP gene of 25-RA, SRD-4, and SRD-8 cells. (A) Scheme for PCR amplification of fragments from hamster SCAP cDNAs. Primers used for amplification in B are indicated below the map. The asterisk (∗) denotes the position of the G-to-A mutation in codon 443 of the SCAP gene. The arrowhead (▴) denotes the position of the G-to-A polymorphism at codon 816 that reflects an glycine/arginine (G/R) polymorphism in SCAP protein. (B) SSCP analysis. First-strand cDNA was prepared as described in Methods from RNA isolated from wild-type CHO-7 cells and mutant 25-RA, SRD-4, and SRD-8 cells. The cDNA was used as a template to amplify a 1.2-kb fragment by PCR using primers 2 and 8 (see A). Primer 2 was described in Fig. 2. Primer 8 (5′-AGCCTTTCCCAGTTCTCCTG-3′) corresponds to codons 830–836 of the SCAP cDNA (20). PCR was carried out at 95°C for 5 min, followed by 35 cycles at 94°C for 30 sec, 60°C for 30 sec, and 72°C for 90 sec in the presence of 60 mM Tris·HCl (pH 9), 15 mM (NH4)2SO4, and 1.5 mM MgCl2. The resulting fragment was cloned into the plasmid vector pNoTA/T7. Plasmid DNA from single clones containing the correct insert was re-amplified by PCR in two separate reactions using primers 2 and 6 (B Upper), or primers 7 and 8 (B Lower). After preliminary analysis, we chose two clones, arbitrarily indicated as clones 1 and 2, which represented the two alleles at this locus in each cell line. Primer 6 corresponds to codons 474–480 (5′-AACAGCTAGCTGTCTCTCATA-3′) and primer 7 corresponds to codons 776–782 (5′-TCATGGACATCGAGTGTCTG-3′) of the SCAP cDNA. PCR was carried out in the presence of [32P]dCTP at 95°C for 5 min, followed by 25 cycles at 94°C for 15 sec, 62°C for 15 sec, and 72°C for 15 sec. Aliquots of the reaction products were denatured for 5 min at 100°C in the presence of formamide and separated on an 8% polyacrylamide gel (lanes 2–9). Lane 1 shows a nondenatured control.
SSCP analysis was carried out essentially as described (26). Electrophoresis was performed in 2× TBE (25) at 300 V for ≈24 h at room temperature. The gels were then dried and exposed to Reflection film overnight at −70°C using an intensifying screen.
RESULTS
In the current experiments we studied three independently derived lines of class 2 sterol-resistant mutants (Fig. 1). The 25-RA cells were isolated in Hanover, NH, by Chang and Limanek (7) in 1980 after mutagenesis with ethylmethane sulfonate, and their defect in SCAP has been recently reported (20). The SRD-4 cells were isolated in our laboratory in Dallas after mutagenesis with nitrosoethylurea and were originally described by Metherall et al. (24) and subsequently studied by Cao et al. (19). The SRD-8 cells were also derived in Dallas after nitrosoethylurea mutagenesis, but the experiment was performed at a different time than the experiment that yielded the SRD-4 cells. The SRD-8 cells have not previously been described.
Figure 1.
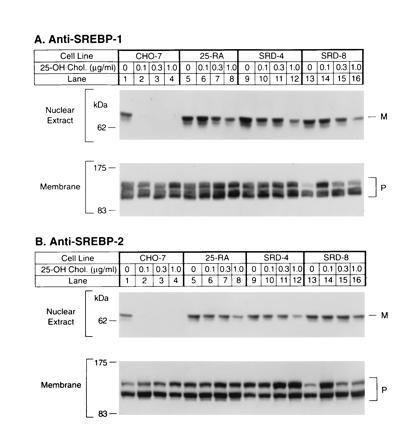
Immunoblot analysis of SREBP-1 (A) and SREBP-2 (B) in wild-type CHO-7 cells and mutant 25-hydroxycholesterol-resistant cells. On day 0, the indicated cell line was set up for experiments as described. On day 2, cells were switched to medium A containing 5% newborn calf lipoprotein-deficient serum, 50 μM sodium mevalonate, and 50 μM compactin supplemented with the indicated concentration of 25-hydroxycholesterol. After incubation for 16 h, the cells were harvested and fractionated as described, and aliquots of the nuclear extract (50 μg protein) and membrane fractions (100 μg protein) were subjected to SDS/PAGE. Immunoblot analysis was carried out with 30 μg/ml of IgG-2A4 (A) or 5 μg/ml IgG-7D4 (B) as described. Filters were exposed to film for 30 sec (A Upper) or 5 sec (A Lower and B). P and M denote the precursor and mature forms of SREBP-1 or SREBP-2, respectively.
For the experiment in Fig. 1 the three mutant cell lines, plus wild-type CHO-7 cells, were grown in sterol-deprivation medium containing lipoprotein-deficient serum, compactin to inhibit endogenous cholesterol synthesis, and a small amount of mevalonate to supply essential nonsterol products. Groups of dishes received 25-hydroxycholesterol at concentrations ranging from 0.1–1.0 μg/ml. After 16 h the cells were harvested, and nuclear extracts were subjected to electrophoresis and immunoblotted with an antibody against the NH2-terminal segment of SREBP-1 (Fig. 1A) or SREBP-2 (Fig. 1B). In the absence of 25-hydroxycholesterol, the CHO-7 nuclear extracts contained the mature NH2-terminal segments of both SREBPs, which migrate as anomalously large species with apparent molecular masses of about 68 kDa (lane 1). At the lowest concentration of 25-hydroxycholesterol (0.1 μg/ml), the NH2-terminal segments of both proteins disappeared from the nucleus (lane 2). We have shown previously that this results from an inhibition of proteolysis at site-1 (18). In sterol-deprivation medium, nuclei from the 25-RA, SRD-4, and SRD-8 cells each contained normal amounts of the NH2-terminal segments (Fig. 1, lanes 5, 9, and 13). The amounts of these segments were reduced only slightly by 25-hydroxycholesterol, even at 1.0 μg/ml (lanes 8, 12, and 16), which was 10 times higher than the concentration that caused a complete disappearance in the CHO-7 cells.
In 25-RA cells the G-to-A mutation in SCAP codon 443 destroys a site for the restriction endonuclease PvuI (20). To determine whether the SRD-4 and SRD-8 cells have a similar mutation, we amplified this region of genomic DNA with primers 2 and 4 (Fig. 2A) and digested the amplified products with PvuI (Fig. 2B). The amplified products from wild-type CHO cells (CHO-K1) were completely cut by PvuI, and no uncut DNA was seen (Fig. 2B, lane 2). In contrast, PvuI was able to cut only ≈50% of the amplified products from each of the three mutant cell lines (Fig. 2B, lanes 4, 6, and 8). Previous experiments with 25-RA cells have shown that this partial cleavage results from heterozygosity for the dominant G-to-A mutation in codon 443 (20).
Figure 2.
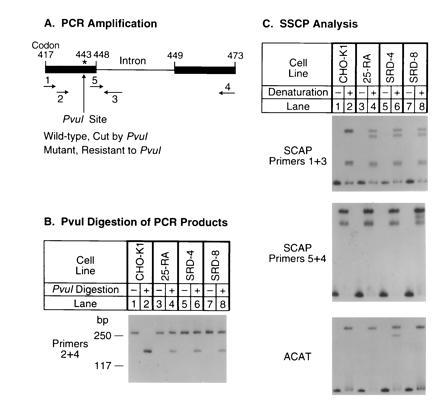
Identification of the same G-to-A mutation in SCAP genes from three sterol-resistant CHO cell lines. (A) Scheme for PCR amplification of fragments from the hamster SCAP gene. The asterisk (∗) denotes the position of the G-to-A mutation in codon 443 (D443N) of the SCAP gene. Primers used for amplification in B and C are indicated below the genomic map. (B) PvuI digestion of PCR products amplified from the SCAP gene. Genomic DNA from CHO-K1 (lanes 1 and 2), 25-RA (lanes 3 and 4), SRD-4 (lanes 5 and 6), and SRD-8 (lanes 7 and 8) cells was subjected to PCR with primers 2 and 4, which correspond to the codons for amino acids 426–432 and 467–473, respectively, in the hamster SCAP cDNA (20). Sequences of these two primers, respectively, are 5′-TGTCTGACTTCTTCCTCCAG-3′ and 5′-GCGTGCTGGCCTTCCCACAGG-3′. PCR was carried out in the presence of [32P]dCTP at 95°C for 5 min, followed by 30 cycles at 94°C for 30 sec, 62°C for 30 sec, and 72°C for 30 sec. Aliquots of the PCR products were incubated at 37°C for 24 h in the absence or presence of 230 units/ml PvuI as indicated. The treated DNA samples were separated on a 7% polyacrylamide gel containing 10% (vol/vol) glycerol. The resulting 271-bp PCR product included the 128-bp intron between codons 448 and 449 (see A). Only the larger cleavage product is shown. (C) SSCP analysis. (C Top) Genomic DNA from CHO-K1 (lanes 1 and 2), 25-RA (lanes 3 and 4), SRD-4 (lanes 5 and 6), and SRD-8 (lanes 7 and 8) cells was amplified as described above in B with primers 1 and 3 except that the annealing temperature was 61°C. Primer 1 corresponds to codons 417–423 (5′-TTCTGCCTCTTTGCTGTTGTG-3′), and primer 3 is derived from the intron located between codons 448 and 449 (5′-CTCCCATGTCTGAAGAGAGC-3′). The amplified 162-bp PCR product contained 96 bp of the SCAP cDNA sequence, followed by 66 bp of the intron sequence. Aliquots of the PCR products were denatured and applied to an 8% polyacrylamide gel (lanes 2, 4, 6, and 8) adjacent to aliquots of the nondenatured samples (lanes 1, 3, 5, and 7). (C Middle) SSCP analysis was carried out exactly as in the C Top, except that primers 5 and 4 were used for amplification. Primer 5 (5′-ATTCGGCGAATGGAGGTAAG-3′) corresponds to codons 444–448 of the SCAP cDNA plus nucleotides 5′-GTAAG-3′ from the intron between codons 448 and 449. (C Bottom) A pair of primers from the hamster ACAT (19) gene, one corresponding to an intron sequence 100-bp upstream of codon 256 (5′-GCTAACTTGAGTTAGAATGTG-3′) and the other corresponding to codons 268–274 (5′-TAGTACTCTAGGTACATTCTC-3′), were used to amplify a 178-bp fragment. Conditions for PCR and SSCP analysis were as in C Top.
The identity of the mutation in the three cell lines was also suggested by SSCP analysis (Fig. 2C). Genomic DNA was amplified with primers 1 and 3 (shown in Fig. 2A), and the products were denatured and subjected to nondenaturing gel electrophoresis. Wild-type DNA showed several bands (Fig. 2C Top). DNA from all three mutant cell lines showed this normal band pattern plus a novel band. The position of this novel band was the same in all three mutant DNA samples, further suggesting that the three mutant cell lines contained the same G-to-A substitution at codon 443.
To confirm this hypothesis, genomic DNA from the three mutant cell lines was amplified by PCR with primers 2 and 4 (see Fig. 2A) and cloned into a plasmid vector. Sequencing of several independent clones revealed that all three mutant cell lines carried the same A mutation at codon 443 on one allele while the nucleotide on the other allele was G (wild type).
Sequencing the genomic clones from SRD-8 cells revealed a new polymorphic marker, an A-to-G substitution at position 72 in the intron separating codons 448 and 449. In the SRD-8 cells, the G nucleotide in the intron was found on the same allele as the G-to-A substitution at codon 443. In contrast, in the two other mutant cell lines as well as in the wild-type CHO-7 cells, the A nucleotide in the intron was found on both alleles.
To rule out the possibility that the three mutant cell lines or DNA samples had been switched or cross-contaminated, we checked the DNA samples for two specific markers using the SSCP method (Fig. 2C Middle and Bottom). Taking advantage of the nucleotide change in the intron between codons 448 and 449 from SRD-8 cells, we amplified this region with primers 5 and 4 (see Fig. 2A). The data showed that the DNA sample from the SRD-8 cells had a different pattern on SSCP analysis as compared with the other two mutant cell lines, confirming that the SRD-8 DNA was unique (Fig. 2C Middle). To confirm the identity of the SRD-4 DNA, we took advantage of the fact that these cells harbor a point mutation in the ACAT gene (19). When this region of the ACAT gene was amplified and subjected to SSCP analysis, the SRD-4 cells had a unique band that was not present in any of the other cell lines, confirming the origin of this DNA (Fig. 2C Bottom).
As a final demonstration that the mutations in all three cell lines arose independently, we took advantage of an additional polymorphism in codon 816 of the SCAP gene, which we previously noted (20). CHO-7 cells are heterozygous for this polymorphism, which involves a G-to-A transition that changes codon 816 from glycine to arginine. This change does not affect the activity of SCAP. To determine whether the three codon 443 mutations arose on the same chromosome, we amplified a 1.2-kb segment of the SCAP mRNA that encompasses codons 443 and 816 using primers 2 and 8, respectively (Fig. 3A). These segments were cloned in a plasmid vector, and individual clones were analyzed by SSCP for the codon 443 and 816 sequences using primers 2 and 6 and primers 7 and 8, respectively. For each cell line, we obtained evidence of two different types of mRNA corresponding to each of the two alleles. Clones representing each allele were chosen for the demonstrative analysis that is shown in Fig. 3B. For each cell line, the two allele-specific clones are arbitrarily designated 1 and 2. As shown previously (20), the two alleles in CHO-7 cells differed at codon 816 (Fig. 3B Lower, lanes 2 and 3), but they both contained the same sequence at codon 443 (Fig. 3B Upper). In the 25-RA cells the mutant sequence at codon 443 occurred on the chromosome encoding G816 (lane 5). The same was true for the SRD-8 cells (lane 9). On the other hand, in the SRD-4 cells the mutation at codon 443 occurred on the chromosome encoding R816 (lane 6). DNA sequencing of several independent clones from each cell line confirmed the conclusions drawn from the SSCP analysis. These experiments demonstrate that the D443N mutation in SRD-4 cells occurred on a different chromosome than the one that gave rise to the D443N mutations in the 25-RA and SRD-8 cells.
Fig. 4 summarizes the haplotype analysis of the SCAP gene. The nucleotide differences at three sites in the SCAP gene allow each allele in the wild-type and mutant cells to be assigned to one of five haplotypes. Haplotypes 3, 4, and 5 contain the adenine in codon 443 and thus produce the superactive mutant version of SCAP. Each of these alleles differs from the other two at either intron nucleotide 72 or codon 816, thereby confirming that all three mutations at codon 443 arose independently on different chromosomal backgrounds.
Figure 4.
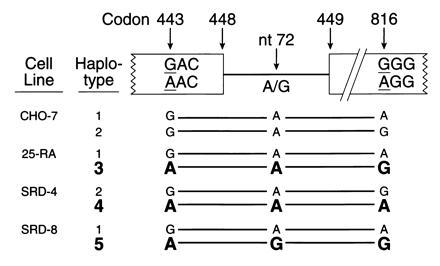
Summary of haplotype analysis of the SCAP gene from wild-type CHO cells and three mutant 25-hydroxycholesterol-resistant cell lines. DNA sequence analysis at the indicated sites was performed on cloned PCR products as described. Alleles encoding the D443N mutation are shown in boldface.
DISCUSSION
The current data document a single recurrent nucleotide substitution in three independently derived lines of mutant CHO cells with a class 2 sterol-resistant phenotype. All three cell lines exhibit a G-to-A transition in codon 443 of the SCAP gene, changing the codon from aspartic acid to asparagine. Through analysis of polymorphic variants at two other sites within the SCAP gene, we were able to demonstrate that all three D443N mutations arose independently on different chromosomal backgrounds. These findings rule out the possibility that the D443N mutation preexists in a small proportion of CHO cells, which grow preferentially during selection in 25-hydroxycholesterol. They also rule out the possibility of cross-contamination of the cell lines or the DNA samples.
What are the reasons for the recurrent G-to-A transitions at codon 443? One reason is the nature of the mutagens employed. Nitrosoethylurea and ethylmethane sulfonate are both monoalkylating agents that favor G-to-A transitions (27, 28). These agents ethylate guanine residues at the O6 position, creating a modified nucleotide that pairs preferentially with thymidine rather than cytosine (29). This eventually places an adenine in the coding strand. Although this propensity plays a role in the recurrent G-to-A transitions, it cannot account for the observation that the G-to-A transition occurs only at one site, namely, codon 443 of SCAP. This phenomenon is most likely attributable to the stringency of the selective growth conditions, which demand that cells continue to produce a nuclear form of SREBP in the presence of 25-hydroxycholesterol. Apparently, there are only a limited number of ways in which this can be accomplished.
So far, selection for 25-hydroxycholesterol resistance has produced only two classes of mutants. Three class 1 mutants have been studied, and all produce a truncated form of SREBP-2 (13, 14). In each case the SREBP-2 sequence terminates at residue 460, owing to a genomic rearrangement in the intron following this codon. Class 2 mutants produce a superactive, nonsuppressible form of SCAP that stimulates cleavage-activation of both SREBPs at site 1, and renders the cleavage reaction resistant to inhibition by sterols (20). In all three class 2 mutants identified to date, this phenotype results from a substitution of aspartic acid for asparagine at codon 443 of SCAP. Apparently, SCAP plays a unique role in regulating SREBP cleavage, and there are only a limited number of ways in which SCAP can be modified so as to render its action resistant to inhibition by sterols.
Aspartic acid 443 lies at the COOH-terminal end of one of the putative membrane-spanning segments of SCAP (20). This segment resembles membrane spanning segment 6 in mammalian HMG CoA reductase (30, 31). It is part of a sequence encompassing membrane-spanning segments 2–6 in HMG CoA reductase that is conserved in SCAP (20). In HMG CoA reductase these membrane-spanning segments form part of a sterol-sensing mechanism that accelerates degradation of the protein in response to sterols (21, 22). It is tempting to speculate that this segment of SCAP also functions as a sterol sensor and that the D443N substitution reduces its sensitivity. We do not believe that the putative sterol-sensing region of SCAP controls degradation of the protein since we found no difference in the steady-state levels of the protein encoded by the wild-type or mutant genes (20). Rather, we believe that sterols interact with SCAP so as to diminish its ability to stimulate the cleavage of SREBPs at site 1, and this function is impaired by the D443N mutation. We are currently designing experiments to determine whether this phenotype is attributable to the loss of a required aspartic acid or to the insertion of a disruptive asparagine.
Acknowledgments
We thank Jianxin Yang for invaluable help in isolation of SRD-8 cells; Lisa Beatty for excellent help with tissue culture; and Michelle Laremore and Jeff Cormier for oligonucleotide synthesis and DNA sequencing. This work was supported by research grants from the National Institutes of Health (HL20948) and the Perot Family Foundation.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: CHO, Chinese hamster ovary; HMG CoA, 3-hydroxy-3-methylglutaryl CoA; SREBP, sterol regulatory element binding protein; SCAP, SREBP cleavage-activating protein; SSCP, single-strand conformational polymorphism.
References
- 1.Brown M S, Goldstein J L. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein J L, Brown M S. Nature (London) 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 3.Ericsson J, Jackson S M, Lee B C, Edwards P A. Proc Natl Acad Sci USA. 1996;93:945–950. doi: 10.1073/pnas.93.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan G, Jiang G, Koch R L, Shechter I. J Biol Chem. 1995;270:21958–21965. doi: 10.1074/jbc.270.37.21958. [DOI] [PubMed] [Google Scholar]
- 5.Chen H W, Cavenee W K, Kandutsch A A. J Biol Chem. 1979;254:715–720. [PubMed] [Google Scholar]
- 6.Leonard S, Sinensky M. Biochim Biophys Acta. 1988;947:101–112. doi: 10.1016/0304-4157(88)90021-4. [DOI] [PubMed] [Google Scholar]
- 7.Chang T-Y, Limanek J S. J Biol Chem. 1980;255:7787–7795. [PubMed] [Google Scholar]
- 8.Metherall J E, Goldstein J L, Luskey K L, Brown M S. J Biol Chem. 1989;264:15634–15641. [PubMed] [Google Scholar]
- 9.Dawson P A, Metherall J E, Ridgway N D, Brown M S, Goldstein J L. J Biol Chem. 1991;266:9128–9134. [PubMed] [Google Scholar]
- 10.Kandutsch A A, Chen H W. J Biol Chem. 1974;249:6057–6061. [PubMed] [Google Scholar]
- 11.Brown M S, Goldstein J L. J Biol Chem. 1974;249:7306–7314. [PubMed] [Google Scholar]
- 12.Wang X, Sato R, Brown M S, Hua X, Goldstein J L. Cell. 1994;77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Sato R, Goldstein J L, Brown M S. Genes Dev. 1994;8:1910–1919. doi: 10.1101/gad.8.16.1910. [DOI] [PubMed] [Google Scholar]
- 14.Yang J, Brown M S, Ho Y K, Goldstein J L. J Biol Chem. 1995;270:12152–12161. doi: 10.1074/jbc.270.20.12152. [DOI] [PubMed] [Google Scholar]
- 15.Hua X, Sakai J, Ho Y K, Goldstein J L, Brown M S. J Biol Chem. 1995;270:29422–29427. doi: 10.1074/jbc.270.49.29422. [DOI] [PubMed] [Google Scholar]
- 16.Yokoyama C, Wang X, Briggs M R, Admon A, Wu J, Hua X, Goldstein J L, Brown M S. Cell. 1993;75:187–197. [PubMed] [Google Scholar]
- 17.Sato R, Yang J, Wang X, Evans M J, Ho Y K, Goldstein J L, Brown M S. J Biol Chem. 1994;269:17267–17273. [PubMed] [Google Scholar]
- 18.Sakai J, Duncan E A, Rawson R B, Hua X, Brown M S, Goldstein J L. Cell. 1996;85:1037–1046. doi: 10.1016/s0092-8674(00)81304-5. [DOI] [PubMed] [Google Scholar]
- 19.Cao G, Goldstein J L, Brown M S. J Biol Chem. 1996;271:14642–14648. doi: 10.1074/jbc.271.24.14642. [DOI] [PubMed] [Google Scholar]
- 20.Hua X, Nohturfft A, Goldstein J L, Brown M S. Cell. 1996;87:415–426. doi: 10.1016/s0092-8674(00)81362-8. [DOI] [PubMed] [Google Scholar]
- 21.Gil G, Faust J R, Chin D J, Goldstein J L, Brown M S. Cell. 1985;41:249–258. doi: 10.1016/0092-8674(85)90078-9. [DOI] [PubMed] [Google Scholar]
- 22.Kumagai H, Chun K T, Simoni R D. J Biol Chem. 1995;270:19107–19113. doi: 10.1074/jbc.270.32.19107. [DOI] [PubMed] [Google Scholar]
- 23.Neer E J, Schmidt C J, Nambudripad R, Smith T F. Nature (London) 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- 24.Metherall J E, Ridgway N D, Dawson P A, Goldstein J L, Brown M S. J Biol Chem. 1991;266:12734–12740. [PubMed] [Google Scholar]
- 25.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 26.Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Proc Natl Acad Sci USA. 1989;86:2766–2770. doi: 10.1073/pnas.86.8.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Zeeland A A, Jansen J G, de Groot A, van Hees S, van Teijlingen C M, op het Veld C W, Zdzienicka M Z, Lohman P H, Vrieling H. Toxicol Lett. 1995;77:49–54. doi: 10.1016/0378-4274(95)03271-1. [DOI] [PubMed] [Google Scholar]
- 28.Ingle C A, Drinkwater N R. Mutat Res. 1989;220:133–142. doi: 10.1016/0165-1110(89)90019-5. [DOI] [PubMed] [Google Scholar]
- 29.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 33–34. [Google Scholar]
- 30.Liscum L, Finer-Moore J, Stroud R M, Luskey K L, Brown M S, Goldstein J L. J Biol Chem. 1985;260:522–530. [PubMed] [Google Scholar]
- 31.Olender E H, Simoni R D. J Biol Chem. 1992;267:4223–4235. [PubMed] [Google Scholar]