

Summary
Mutations in the EGFR kinase are a cause of non-small cell lung cancer. To understand their mechanism of activation and effects on drug binding, we studied the kinetics of the L858R and G719S mutants and determined their crystal structures with inhibitors including gefitinib, AEE788 and a staurosporine. We find that the mutations activate the kinase by disrupting autoinhibitory interactions, and that they accelerate catalysis as much as 50-fold in vitro. Structures of inhibitors in complex with both wild-type and mutant kinases reveal similar binding modes for gefitinib and AEE788, but a marked rotation of the staurosporine in the G719S mutant. Strikingly, direct binding measurements show that gefitinib binds 20-fold more tightly to the L858R mutant than to the wild-type enzyme.
Significance
Mutations in the EGFR kinase domain occur in approximately 16% of NSCLCs, but at much higher frequencies in selected populations including non-smokers, women, and East Asian patients. The presence of these mutations correlates with response to small-molecule tyrosine kinase inhibitors targeting EGFR. Because the diverse mutations cluster around the catalytic cleft and because differences in inhibitor sensitivity of the mutants have been reported, it is important to understand the effect of the mutations on inhibitor binding at a structural level. The present work provides a structural foundation for understanding the differential inhibitor sensitivities of the L858R and G719S mutants, and will help guide rational application of currently available EGFR inhibitors and development of more potent and perhaps mutation-specific inhibitors.
Introduction
The epidermal growth factor receptor (EGFR, also called ErbB1, Her1) is a transmembrane receptor tyrosine kinase that transduces signals critical for cell proliferation, differentiation and motility. Upon growth factor binding, EGFR and other ErbB family members homo- or hetero- dimerize and activate their cytoplasmic tyrosine kinase domains to initiate intracellular signaling (Yarden and Sliwkowski, 2001; Schlessinger, 2004). Overexpression or mutational activation of the EGFR is implicated in the development and progression of numerous human malignancies, and a number of small-molecule tyrosine kinase inhibitors (TKIs) have been developed to target the ATP-binding cleft of the EGFR (Hynes and Lane, 2005). Several of these inhibitors are currently in clinical trials or have been approved for clinical use, including the 4-anilinoquinazolines gefitinib (Iressa) (Wakeling et al., 2002), erlotinib (Tarceva) (Pollack et al., 1999), and lapatinib (Rusnak et al., 2001), the pyrrolopyrimidine AEE788 (Traxler et al., 2004), and the irreversible inhibitor HKI-272 (Tsou et al., 2005).
Deletion mutations in the extracellular portion of the EGFR have long been known to cause constitutive, growth factor independent activation of the EGFR and are frequently found in glioblastoma (Ekstrand et al., 1992). More recently, somatic mutations in the EGFR kinase domain have been discovered in a subset of non-small cell lung cancers (NSCLC) (Paez et al., 2004; Lynch et al., 2004; Pao et al., 2004; Johnson and Janne, 2005; Gazdar et al., 2004; Chan et al., 2006; Shigematsu and Gazdar, 2006). The presence of these mutations in NSCLC correlates with responsiveness to TKIs including gefitinib and erlotinib. A number of distinct mutations have been identified including point mutations within the nucleotide-binding loop in exon 18, small deletions in exon 19 or insertions in exon 20, as well as point mutations in the activation loop in exon 21. Structurally, these mutations cluster around the active site cleft of the tyrosine kinase domain. The two most frequent mutations are the exon 19 deletion that removes residues 746–750 of the expressed protein and the exon 19 point substitution that replaces leucine 858 with arginine (L858R) (Chan et al., 2006; Shigematsu and Gazdar, 2006). The L858R substitution is the single most common mutation (approximately 40% of all mutations), and it lies in the activation loop (A-loop) of the kinase. Point mutations are also observed in glycine 719, albeit less frequently. Gly719 is found in the adjacent phosphate-binding “P-loop” of the kinase, and is substituted with serine, cysteine or alanine. The L858R and G719S point mutations, as well as the exon 19 deletions and exon 20 insertions, are transforming when introduced into a variety of cell lines including fibroblasts and lung epithelial cells (Lynch et al., 2004; Sordella et al., 2004; Arao et al., 2004; Greulich et al., 2005; Amann et al., 2005; Engelman et al., 2005).
Interestingly, the clinical correlation between the presence of specific mutations and therapeutic response to TKIs is mirrored in cell lines and EGFR-transfected cells. Cells bearing the mutant EGFR are in general more sensitive to TKIs than cells expressing the wild-type kinase. In particular, the L858R mutant is 10–100 fold more sensitive to erlotinib and gefitinib than the wild-type kinase (Pao et al., 2004; Greulich et al., 2005; Mukohara et al., 2005) and significantly more sensitive than the G719S mutant (Jiang et al., 2005). At the same time, the exceptions to this rule—for example, the exon 20 insertion mutants are highly resistant to both gefitinib and erlotinib (Greulich et al., 2005)—further underscore the dependence of inhibitor responses on specific mutations.
The structure of the wild-type EGFR kinase domain has been previously determined in both active and inactive conformations. The crystal structure was first reported alone and in complex with erlotinib (Stamos et al., 2002). Both the apo-EGFR and the EGFR/erlotinib structures reveal an active conformation of the kinase, although Tyr 869 in activation loop is not phosphorylated. This observation is consistent with the previous finding that the phosphorylation of the EGFR activation loop is not required for activity (Tice et al., 1999). The structure of wild-type EGFR kinase in complex with lapatinib (Wood et al., 2004) showed that the kinase can adopt an inactive conformation closely resembling that previously observed in Src-family (Williams et al., 1997; Xu et al., 1997; Sicheri et al., 1997) and cyclin-dependent kinases (De Bondt et al., 1993). In this inactive conformation, the regulatory “C-helix” is displaced from the active site. The bulky 4-anilino substituent in lapatinib cannot be accomodated in the active conformation of the kinase, and thus lapatinib appears to trap the kinase in the inactive conformation with the C-helix displaced (Wood et al., 2004). Very recently, a structural dissection of the mechanism of activation of the EGFR confirmed the Src/CDK-like state of the inactive kinase, and demonstrated a cyclin-like mechanism of activation involving asymmetric dimerization of the EGFR kinase domain (Zhang et al., 2006).
The discovery of activating mutation in the EGFR kinase domain and their differential sensitivity to inhibitors poses a number of structurally interesting and clinically relevant questions. How do the mutations activate the kinase? How do they affect inhibitor binding? And most importantly, to what extent will optimal therapeutic strategies require inhibitors tailored to specific mutations? While the sensitivity of EGFR-mutant tumors and cell lines may derive from an “oncogene addiction” effect (Gazdar et al., 2004), it may also stem, at least in part, from structural differences in the mutant kinases that confer intrinsic susceptibility to particular inhibitors (Lynch et al., 2004; Greulich et al., 2005).
In order to begin to address these questions, we have determined the structure of the wild-type EGFR kinase and the L858R and G719S mutants in complex with inhibitors including gefitinib, AEE788, the staurosporine compound AFN941, and the ATP-analog AMP-PNP. Additionally, we have characterized the kinetics of the wild-type and mutant kinases in vitro. We find that the L858R mutant is 50-fold more active than the wild-type kinase, and the G719S mutant approximately 10-fold more active than wild-type. Examination of the mutant structures reveals an overall conformation very similar to that of the wild-type kinase in the active state. Comparisons with the inactive wild-type EGFR structure indicate that the L858R and G719S mutations activate the kinase by disrupting interactions that stabilize the inactive conformation. Analysis of the inhibitor complexes shows that the binding mode of AMP-PNP, AEE788 and gefitinib is very similar among the wild-type, L858R and G719S kinases. In contrast, we observe a marked rotation of the staurosporine analog AFN941 in the binding cleft of the G719S mutant as compared with its orientation in the wild-type protein. Direct measurement of the binding of gefitinib and AEE788 to the wild-type and mutant kinases reveals that both compounds bind with greater affinity to the L858R mutant than to the wild-type kinase or G719S mutant. Notably, gefitinib binds 20 times more tightly to the L858R mutant.
Results
Structure and activity of the L858R and G719S mutants
The structures of the L858R and G719S mutants of the EGFR tyrosine kinase domain were determined in complex with the non-hydrolyzable ATP analog AMP-PNP, or with inhibitors including gefitinib, AEE788, or AFN941. In order to facilitate direct comparison, the wild-type EGFR kinase was also analyzed. A total of 12 structures were determined at resolutions ranging from 2.4 to 3.4 Å as summarized in Table 1. The wild-type and mutant proteins crystallized with the same crystal packing, but crystals of the L858R mutant were only obtained using polyethylene glycol 400 as a precipitant while the wild-type and G719S proteins produced better crystals in potassium sodium tartrate. When possible, we compare structures obtained in the same buffer condition (see Methods).
Table 1.
Crystallographic data and refinement statistics§
| PDB ID | mutation | inhibitor | Resolution Range (Å) | Overall Rsym (outmost) | Completeness, % (outmost) | Rcryst/Rfree (%) | RMSD bond length/angle |
|---|---|---|---|---|---|---|---|
| 2itx* | Wild-Type | AMP-PNP | 25-3.0 | 0.089 (0.395) | 99.9 (100.0) | 18.8/26.1 | 0.016/1.75 |
| 2j6m* | Wild-Type | AEE788 | 25-3.1 | 0.073 (0.385) | 96.7 (83.8) | 19.9/24.9 | 0.010/1.57 |
| 2itw* | Wild-Type | AFN941 | 25-2.9 | 0.066 (0.395) | 100.0 (100.0) | 18.7/25.6 | 0.017/1.85 |
| 2ity | Wild-Type | gefitinib | 25-3.4 | 0.123 (0.398) | 99.7 (100.0) | 21.1/26.0 | 0.022/1.81 |
| 2itn | G719S | AMP-PNP | 25–2.4 | 0.065 (0.398) | 99.9 (100) | 19.7/26.7 | 0.017/1.71 |
| 2ito | G719S | gefitinib | 25-3.2 | 0.086 (0.387) | 99.9 (100) | 18.9/26.6 | 0.015/1.69 |
| 2itp* | G719S | AEE788 | 25-2.7 | 0.058 (0.367) | 99.9 (100) | 19.6/25.5 | 0.017/1.78 |
| 2itq* | G719S | AFN941 | 25-2.7 | 0.050 (0.382) | 98.8 (94.2) | 19.7/26.3 | 0.019/1.87 |
| 2itt | L858R | AEE788 | 25-2.7 | 0.053 (0.367) | 99.8 (99.8) | 20.8/26.2 | 0.018/1.81 |
| 2itu | L858R | AFN941 | 25-2.8 | 0.066 (0.394) | 99.9 (99.7) | 19.8/25.8 | 0.019/1.89 |
| 2itv | L858R | AMP-PNP | 25-2.4 | 0.055 (0.390) | 99.7 (100.0) | 19.5/24.2 | 0.016/1.70 |
| 2itz | L858R | gefitinib | 25-2.8 | 0.055 (0.334) | 93.9 (85.3) | 20.2/25.5 | 0.015/1.63 |
These structures were determined in potassium sodium tartrate buffer rather than PEG 400 as for the other structures(See Material and Methods for details).
PDB refers to the Protein Data Bank (www.pdb.org).
Rsym=Σ|Ii − <Ii>|/ΣIi, where Ii is the average intensity of symmetry-equivalent reflections.
Rcryst=Σ|Fo−Fc|/ΣFo where Fo and Fc are observed and calculated structure factor amplitudes, respectively.
Rfree is the Rcryst for reflections excluded from the refinement.
Both the L858R and G719S mutants adopt overall conformations that are very similar to that of the wild-type kinase in the activated conformation. The structure of the wild-type kinase in complex with AMP-PNP is shown in Figure 1A; superpositions with the L858R and G719S mutant structures are shown in Figs. 1B and 1C. The L858R mutant superimposes on the wild-type enzyme with an RMSD of 0.33Å for 292 C-α atoms, while the G719S mutant superimposes on the wild-type structure with an RMSD of 0.37Å. The close correspondence of the mutant kinases with the active conformation of the wild-type enzyme is not unexpected, as the mutants presumably must retain catalytic activity in order to induce transformation.
Figure 1.
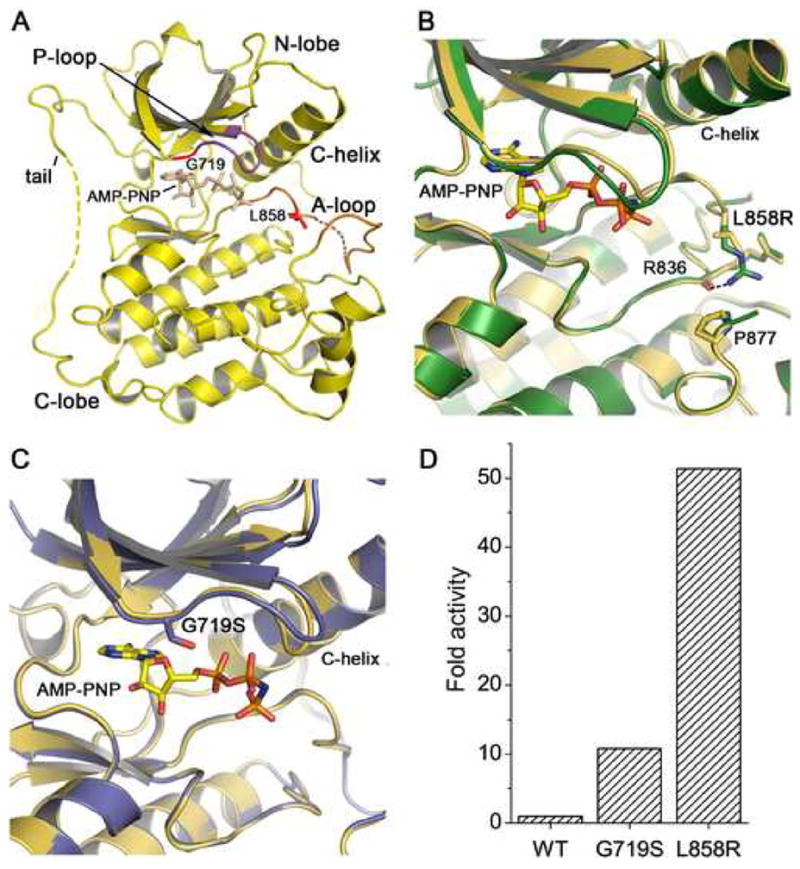
Structure and activity of mutant EGFR kinases. (A) Overview of the structure of the EGFR kinase. The structure of the wild-type kinase is shown in complex with the ATP analog AMP-PNP. The locations of the L858R and G719S mutations in the activation loop (A-loop) and P-loop, respectively, are indicated. Dashed lines indicate short segments of the activation loop and C-terminal tail that are disordered in the structures reported here. Due to a difference in crystallization buffers, we observe more of the C-terminal tail than reported previously (Stamos et al., 2002), allowing us to confirm that it makes an intra- rather than inter- molecular interaction with the N-lobe of the kinase in a manner similar to that described for the inactive kinase (Wood et al., 2004). (B) The structure of the active site region of the L858R mutant (green) superimposed on the wild-type kinase (yellow). (C) The structure of the active site region of the G719S mutant (blue) superimposed on the wild-type kinase (yellow). (D) Comparison of the activity of the wild-type, G719S and L858R kinases. The fold activity of wild-type and mutant enzymes was calculated by determining the kcat for each protein with saturating ATP and poly-[Glu4Tyr1] as peptide substrate and dividing by the kcat for the wild-type enzyme.
The L858R mutation lies in the N-terminal portion of the activation loop. The substitution of the larger, positively charged arginine side chain for the hydrophobic leucine side chain is readily accomodated in this active conformation of the kinase (Figure 1B). We note that there is no shift in the protein backbone around Arg 858, nor at residue Pro 877, which is opposite Arg 858 on the C-terminal lobe of the kinase. The side chain of Arg858 is well-ordered, and forms a hydrogen bond with the mainchain carbonyl of Arg 836.
The G719S substitution is located in the N-terminal lobe of the kinase, within the phosphate-binding “P-loop”. Gly 719 is the first glycine in the ‘GXGXXG’ sequence motif in the P-loop, which arches over the triphosphate moeity of the ATP substrate and participates in its coordination. In all structures described here (both wild-type and mutant), this loop appears to be loosely ordered as the corresponding electron density is weak. In the context of the active kinase, the substitution of Gly719 with serine is readily accomodated – the mainchain is not in a conformation that favors glycine, and the serine sidechain extends toward the β-phosphate of the bound ATP analog (Fig. 1C). Comparison of the G719S, L858R and wild-type kinases reveals an identical AMP-PNP binding mode in all three structures. Thus the G719S mutant retains catalytic competence, despite substitution of this conserved residue.
Because the activating mutations are found in regions critical for binding of substrates, we characterized the catalytic activity of the wild-type and mutant enzymes. The kinetic parameters for ATP and a peptide substrate (poly-Glu4Tyr1) were determined using a continuous, colorimetric in vitro kinase assay and are summarized in Table 2. The activity (kcat) of the wild-type and mutant kinases is also plotted in Figure 1D. The L858R mutant is approximately 50-fold more active than the wild-type enzyme, and the G719S mutant is about 10 times more active than wild-type. We measured similar increases in catalytic rate for both mutants using a physiologic substrate peptide derived from the Tyr 1197 autophosphorylation site in place of poly- Glu4Tyr1 (Supplemental Table S1). The increased catalytic activity of the mutants, as compared with the wild-type enzyme, likely results from a shift of the equilibrium toward the active conformation of the enzyme (see below). As previously observed for the wild-type kinase (Tice et al., 1999), autophosphorylation of the kinase activation loop does not appreciably alter the catalytic rate in our experiments with the wild-type or mutant enzymes (supplemental Figure S1).
Table 2.
Kinetic parameters of wild-type, G719S and L858R EGFR kinases
| ATPa |
Peptideb |
|||||
|---|---|---|---|---|---|---|
| Km μM | kcat s−1 | kcat/Km μM−1s−1 | Km μM | kcat s−1 | kcat/Km μM−1s−1 | |
| WT | 6.9±0.9 | 0.013 | 1.88E-3 | 949±66 | 0.026 | 2.73E-5 |
| G719S | 94.7±1.8 | 0.143 | 1.51E-3 | 1037±81 | 0.280 | 2.70E-4 |
| L858R | 31.5±1.7 | 0.234 | 7.43E-3 | 443±41 | 1.335 | 3.01E-3 |
parameters for ATP were determined using a synthesized peptide with the sequence RAHEEIYHFFFAKKK
parameters were determined using poly-Glu4Tyr as the peptide substrate
The G719S mutant has a Km for peptide substrate that is very close to that of the wild-type enzyme, while the Km of the L858R mutant for peptide is about half that of the wild-type kinase. The effect of the L858R mutation on the Km for peptide substrate is not surprising, given its proximity to the expected binding site for peptide substrates. Likewise, the ~14-fold increase in the Km of the G719S mutant for ATP is consistent with the modest structural effects of this mutation in the ATP binding pocket. The L858R mutation also affects the affinity for ATP, but to a lesser extent (approximately 5-fold higher Km than the wild-type kinase). These modest changes in affinity for ATP are likely to be irrelevant in vivo, given that intracellular ATP concentrations are in the millimolar range. The kinetic parameters we determined for the wild-type kinase are very close to those previously reported for EGFR using a different assay system (Brignola et al., 2002). Additionally, while the present work was in review, Zhang et al. reported a ~20 fold (kcat/Km) activation of the L858R mutant kinase in vitro (Zhang et al., 2006), and in a separate study Carey et al. found a ~2-fold increase it its Km for ATP (Carey et al., 2006).
Mechanism of Activation
Comparison of the structures of the mutant kinases with the previously determined structure of the wild-type kinase in an inactive conformation (Fig. 2A) shows that the mutations are expected to destabilize the inactive conformation, and therefore to promote the active conformation of the kinase (Fig. 2B). In particular, the L858R mutation is clearly incompatible with the inactive conformation. This structure was determined in complex with the small molecule inhibitor Lapatinib (Wood et al., 2004), and is very similar to that recently reported for an activation-resistant mutant of the EGFR (Zhang et al., 2006). In this inactive conformation, the C-helix is rotated outward and displaced from the active site, and the N-terminal portion of the activation loop forms a helical turn that locks the C-helix in the inactive position. Leucine 858 is within this helical turn, and forms key hydrophobic interactions with other residues in the N-lobe (Fig. 2A). Substitution of this residue with arginine, which has a much larger charged side chain, cannot be accomodated in this inactive conformation. In contrast, the L858R substitution is readily accomodated in the active form of the enzyme as demonstrated by the present structure (Figs. 1B, 2B). Thus we conclude that the L858R mutation locks the kinase in a constitutively active state because it prevents this activation loop segment (residue 858 and flanking residues) from adopting the inactive, helical conformation. It is likely that mutation of the adjacent leucine 861 to glutamine, which has also been observed in gefitinib and erlotinib responsive NSCLCs (Shigematsu and Gazdar, 2006; Chan et al., 2006), activates the kinase for the same reason (see Fig 2).
Figure 2.
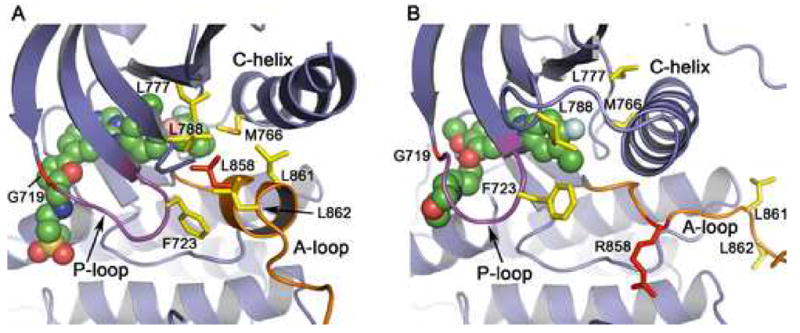
Mechanism of activation of the L858R and G719S mutants. The structure of the inactive, wild-type enzyme in complex with lapatinib (panel A) is compared with that of the active, L858R mutant in complex with gefitinib (panel B). (A) In the inactive state, the N-terminal portion of the activation loop (shown in orange) forms a short helix that displaces the regulatory C-helix from the active site. A cluster of hydrophobic residues (shown in yellow), including Leu 858 (red), stabilize the inactive conformation. Lapatinib (shown as CPK spheres) extends into the space created by the displaced C-helix and appears to have allowed “trapping” of the inactive conformation in the crystal structure. Substitution of Leu858 with arginine is expected to destabilize this conformation, as arginine cannot be favorably accommodated in the hydrophobic pocket occupied by Leu858. Similarly, substitution of G719S with serine may destabilize the inactive conformation of the P-loop (which has a conformation favoring glycine at this position), and therefore activate the kinase. (B) In the active conformation, the activation loop (orange) is reorganized and the C-helix rotates into its active position. Note that the hydrophobic cluster (yellow) is dismantled, and Arg858 (red) is readily accomodated (see also Fig. 1B). Also, note the difference in conformation of the P-loop (purple) and orientation of Phe 723 in the inactive vs. active structures.
We hypothesize that the G719S mutation also activates the kinase by destabilization of the inactive conformation. The P-loop contributes to the set of interactions that hold the C-helix in the inactive conformation, and a glycine residue at position 719 in the P-loop is favored for its proper conformation in the inactive state. Phe 723, which lies at the end of the P-loop, packs together with residues Leu 747 and Leu 862 – part of the same hydrophobic cluster surrounding Leu 858 in the inactive conformation. In this conformation, the mainchain of Gly 719 is in a conformation in which glycine is favored. Thus substitution with serine (or any non-glycine residue) destabilizes the P-loop and in turn weakens the set of hydrophobic interactions that locks the kinase in the inactive conformation (Figure 2A). We note that substitutions of G719 to alanine or cysteine also occur in NCSLC (Chan et al., 2006; Shigematsu and Gazdar, 2006).
Drug binding modes and affinities
Gefitinib (Iressa)
Gefitinib is a 4-anilinoquinazoline inhibitor that is structurally similar to erlotinib (Figure 3). The interactions of gefitinib in the active site of the wild-type EGFR are shown in Figure 4A. The quinazoline ring is oriented with the 1-N in the back of the ATP binding pocket, where it hydrogen bonds with the mainchain amide of Met 793. Met 793 lies in the so-called “hinge” region of the kinase, which connect the N- and C- lobes. Unlike most kinase inhibitors, gefitinib forms only a single hydrogen bond to the hinge. The 3-chloro-4-fluoro aniline substituent extends into the hydrophobic pocket in the back of the ATP binding cleft. The aniline ring forms approximately a 45° angle with the plane of the quinazoline, and the chloro group orients “up” and is surrounded by the sidechains of residues Lys 745, Leu 788 and Thr 790. The fluorine substituent in the para position extends toward the side chains of Leu 788, Met 766 and Glu762. The methoxy group in the 7 position of the quinazoline is in van der Waals contact with Gly 796, but the orientation of the methyl group is not well defined in the density. The 6-propylmorpholino group extends into solvent and is poorly ordered as the electron density for this portion of the inhibitor is weak. This is consistent with the reported structure-activity data for the compound, as this substituent was added to improve the pharmacokinetic properties of the inhibitor (Barker et al., 2001). The binding mode of gefitinib is strikingly similar to that previously reported for erlotinib (Stamos et al., 2002) see supplemental Figure S2.
Figure 3.
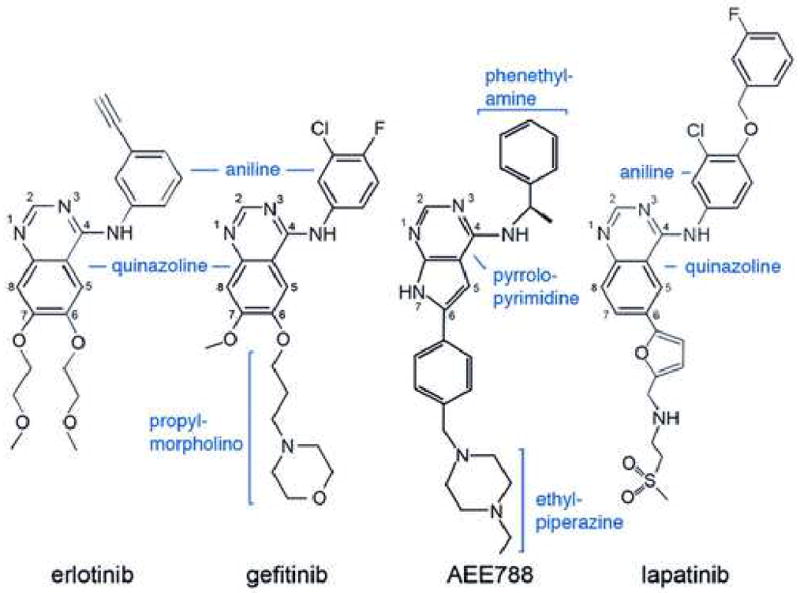
Schematic drawings of the EGFR inhibitors discussed here. Inhibitors are drawn in a consistent orientation approximately reflecting their conformations when bound to the EGFR kinase (see Figure 4).
Figure 4.
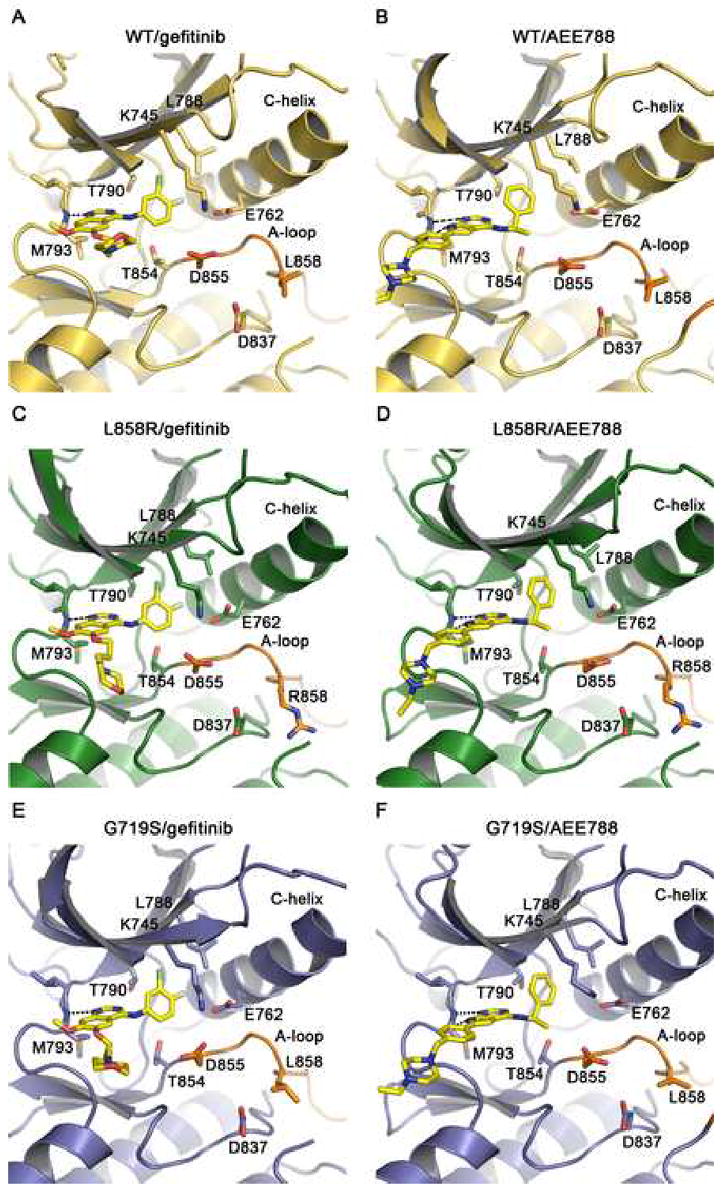
Drug binding modes in the wild-type and mutant EGFR kinase. The binding modes of gefitinib (panels A, C and E) and AEE788 (panels B, D and F) are compared in the wild-type (yellow), L858R (green), and G719S (blue) kinases. Key sidechains are labeled, the inhibitors are shown in stick form with carbons colored yellow, and hydrogen bonds are indicated with dashed lines. Compare binding of different inhibitors to the same mutant within rows, and binding of the same inhibitor among wild-type and mutants within columns. Binding modes of both compounds are essentially the same in all three structures. Note also the closely corresponding orientations of the pyrrolopyrimidine scaffold in the AEE788 complexes and the quinazoline core in the gefitinib complexes. Additionally, the phenylethyl amine moeity in AEE788 occupies the same space as the aniline substituent in the gefitinib and erlotinib complexes.
In order to address the question of whether the activating mutations might affect the binding mode of gefitinib, we also determined structures of gefitinib in complex with the L858R and G719S mutants. The active site regions of the wild-type, L858R, and G719S structures in complex with gefitinib can be compared in Figures 4A, C, and E, respectively. The binding mode is the same in all three structures; small differences in the propylmorpholino “tail” of the compound are not significant as this substituent is poorly defined in all three structures (see supplemental Figure S3). The gefitinib-bound structures are also very similar to the corresponding AMP-PNP complexes overall, but a clear shift in the protein backbone is noted at Arg 776 and Leu 777 in the back of the inhibitor binding pocket (by 1.7 and 0.6 Å, respectively). This shift is accompanied by a side chain rotation of Thr 790 (such that it hydrogen bonds with the mainchain carbonyl of Arg 776), and appears to be induced by the bulky chloroaniline group (not shown). A similar shift is observed in complex with AEE788.
Despite the close structural correspondence of the wild-type and mutant proteins, we find marked differences in their affinity for gefitinib. We measured binding of gefitinib and AEE788 to the wild-type, L858R and G719S mutants using a fluorescence quenching assay (see methods). As shown in Table 3, gefitinib binds the wild-type enzyme with Kd = 53.5 nM, but binds the L858R mutant 20-fold more tightly (Kd= 2.6 nM). In contrast, binding to the G719S mutant is somewhat weaker (Kd=123.6 nM) than to the wild-type kinase. Because the inhibitor must compete with ATP for binding in vivo, the ratio of the inhibitor Kd to the Km for ATP is a better estimate of potency (Table 3). These ratios show that gefitinib is ~100-fold and ~6-fold more potent against the L858R and G719S mutants, respectively, than against the wild-type kinase.
Table 3.
Inhibitor binding constants
| Kd (nM) | Kd/Km, ATP (×10−3) | |||||
|---|---|---|---|---|---|---|
| wt | G719S | L858R | wt | G719S | L858R | |
| gefitinib | 53.5±1.8 | 123.6±5.9 | 2.6±0.2 | 7.75 | 1.31 | 0.08 |
| AEE788 | 10.9±3.3 | 11.3±1.5 | 1.7±0.4 | 1.57 | .12 | 0.05 |
The higher affinity for the L858R mutant may be explained largely by tighter binding to the active conformation of the kinase than to the inactive conformation. Simple modeling of gefitinib in the inactive kinase (based on superposition with lapatinib) shows that favorable van der Waals interactions in the back of the ATP binding cleft that we observe in the active structures may be lost in the inactive state (not shown). It is possible that unique interactions with the L858R mutant also contribute to the enhanced potency of gefitinib. In this regard, we note that in a second crystal structure of the L858R mutant in complex with gefitinib (determined in the same crystal lattice and with similar unit cell dimensions) we observe an alternate conformation in which the aniline ring is rotated by 180° such that the meta- chlorine substituent points down, and is positioned to form a “halogen bond” (Auffinger et al., 2004) with the sidechain of Asp855 (supplemental Figure S4). This alternate binding mode may be possible only in the L858R mutant because the concomitant reorientation of Asp 855 and its coordination of a water molecule appear to be sterically hindered in the context of a leucine residue at position 858.
The markedly tighter binding of gefitinib to the L858R mutant likely explains its potency against cells bearing this mutation; L858R transformed Ba/F3 cells are significantly more gefitinib- sensitive than G719S transformed cells (Jiang et al., 2005). Additionally, NIH-3T3 cells transformed by the L858R mutant are much more sensitive to gefitinib than cells transformed by wild-type receptor with the addition of exogenous EGF (Greulich et al., 2005). The similar compound erlotinib is also reported to be a more potent inhibitor of the L858R mutant than of the wild-type or exon 19 deletion mutants in a cellular context (Pao et al., 2004).
AEE788
The pyrrolopyrimidine compound AEE788 (Novartis Pharmaceuticals) is a potent inhibitor of VEGFR and ErbB-family kinases (Traxler et al., 2004). It has low nanomolar potency against the EGFR, and is currently in Phase 1 clinical trials in oncology. Structures of the wild-type, L858R and G719S mutants in complex with AEE788 are shown in Fig. 4, panels B, D and F, respectively. The pyrrolopyrimidine core of AEE788 hydrogen bonds with the hinge, and the N3 atom interacts with the hydroxyl of Thr854 via a bridging water molecule. The 4-phenylethylamine moeity extends into the hydrophobic pocket defined by Thr790, Leu788, Lys745 and Met766. The 6-phenyl substituent is sandwiched between Leu718 above and Gly796 below. Finally, the ethylpiperazine group extends toward solvent near Asp800 and Glu804 at the edge of the active site. The space occupied by AEE788 is very similar to that exploited by gefitinib and other anilinoquinazolines – the pyrrolopyrimidine moeity superimposes with the quinazoline core and the phenylethylamine group superimposes with the aniline ring in gefitinib. The binding mode of AEE788 is the same in the wild-type and both the G719S and L858R mutants (Fig. 4B, D and F). AEE788 is also a more effective inhibitor of the L858R and G719S mutants than of the wild-type kinase (Table 3).
G719S alters binding mode of staurosporine analog AFN941
In contrast to the compounds described above, in which coordination of the quinazoline or pyrrolopyrimidine cores of the inhibitors remains unchanged in the mutant kinases, the overall binding mode of AFN941 is different in the G719S mutant as compared with the wild-type EGFR. AFN941 is a derivative of staurosporine in which one indole ring system has been hydrogenated to improve solubility (Boggon et al., 2005). In the wild-type kinase, the compound binds with lactam ring in the back of the ATP binding pocket, where the keto-oxygen hydrogen bonds with the backbone amide of Met 793 in the hinge (Figure 5A). The inhibitor is closely associated with the roof of the ATP binding cleft and the P-loop, and it is positioned to form a hydrogen bond between the ether oxygen of the glycosidic ring and the backbone amide of Gly 719 in the P-loop. In the G719S mutant, this close association with the P-loop is not possible and this hydrogen bond is not formed. Instead, the inhibitor is rotated downward (toward the C-lobe) by ~15° and rotated by ~30° deeper into the hydrophobic cleft in the back of the binding pocket (Figure 5B). The inhibitor pivots about the hydrogen bond with the hinge region such that this interaction is maintained. Additionally, hydrogen bonds are formed between the lactam ring amide and the carbonyl of Gln 791 in the hinge region, and between the methylamino nitrogen of the glycosidic ring in the inhibitor and the carbonyl of Arg 841. Curiously, the binding mode we observe in the G719S mutant is closely similar to the binding mode of staurosporine in other tyrosine kinases, including Jak3 (Boggon et al., 2005) and Lck (Zhu et al., 1999). We find that AFN941 binds to the L858R mutant in precisely the same manner that it binds the wild-type kinase (not illustrated). Although the staurosporine compound is not an EGFR-specific inhibitor, its divergent binding modes in the wild-type and L858R vs G719S kinases structurally demonstrate that the lung-cancer derived mutations can disrupt or alter inhibitor interactions.
Figure 5.
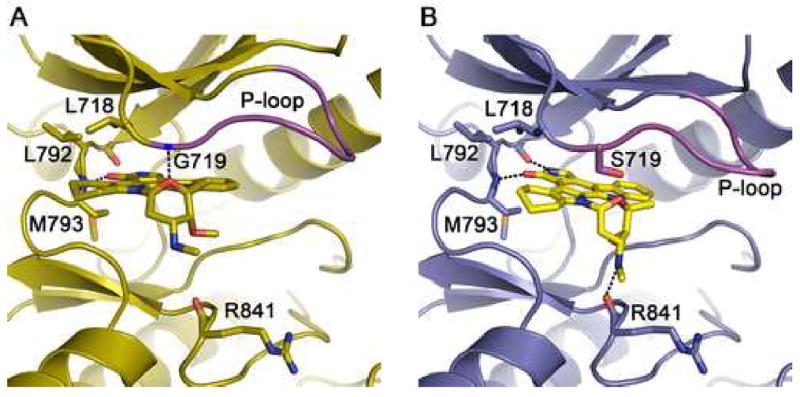
The G719S mutation alters the binding mode of staurosporine analog AFN941. (A) The wild-type EGFR in complex with AFN941. Hydrogen bonds (dashed lines) are formed with the backbone amides of Met 793 in the hinge region and Gly 719 in the P-loop. (B) Structure of the G719S mutant in complex with AFN941. The serine substitution displaces and rotates the inhibitor, disrupting the hydrogen bond with the P-loop and promoting additional interactions with the backbone carbonyls of Gln 791 and Arg 841.
Discussion
The successful development and clinical use of imatinib (Gleevec) as an inhibitor of the BCR-Abl kinase in chronic myeloid leukemia established a paradigm for molecularly-targeted therapy in oncology (Capdeville et al., 2002). The somatic mutations in the EGFR kinase that drive lung and other cancers present an analogous opportunity for intervention in these cancers, but with a critical distinction. In BCR-Abl, the mechanism of activation of the kinase is allosteric and the kinase domain proper is unchanged from the wild-type kinase (apart from emergent resistance mutations). It therefore presents a single molecular target for drug development. In contrast, the mutations in the EGFR activate the kinase by disrupting autoinhibitory interactions proximal to the ATP binding cleft. As we show here, these mutations alter the kinase domain in a manner that dramatically affects inhibitor binding and they therefore represent distinct targets for inhibitor development.
Our direct demonstration that gefitinib binds the L858R mutant 20-fold more tightly than the wild-type enzyme can explain, at least in part, its efficacy in treatment of NSCLCs bearing this substitution. Furthermore, the 50-fold weaker affinity of gefitinib for the G719S mutant (as compared with the L858R protein) shows that the distinct EGFR mutations can differ markedly in their inhibitor susceptibilities. A recent study of erlotinib showed it to be more potent against both the L858R mutant and an exon 19 deletion, but less active against the L861Q mutant (Carey et al., 2006). Cell-based studies of differential inhibitor sensitivity further highlight the potential clinical relevance of this issue – the L858R mutant is exquisitely sensitive to erlotinib and gefitinib, but the exon 20 insertion mutant is not (Greulich et al., 2005). Importantly, our data show that intrinsic differences in the inhibitor binding affinity of the altered EGFR kinases can explain the differential sensitivity of cell lines and tumor cells bearing the wild-type, L858R and G719S proteins. Of course we cannot exclude the possibility that mutation-specific differences in signaling or other effects in the cellular or tumor milieu may also play a role.
Although the structural divergence in the EGFR mutants may complicate pharmacologic intervention by “fragmenting” the disease as described above, it may also present an advantage in that it introduces the possibility of developing inhibitors even more potently selective for specific mutants over the wild-type EGFR. The therapeutic window for such inhibitors could in theory be even wider than that of current agents, which were developed to inhibit the wild-type enzyme. Inhibitors designed specifically to target mutants such as L858R should in principle be less toxic due to reduced inhibition of the wild-type kinase.
Exon 19 deletions and exon 20 insertions account for ~44% and 5%, respectively, of EGFR-mutant lung cancers (Shigematsu and Gazdar, 2006). The exon 19 deletions map to one end of the conformationally sensitive C-helix, the exon 20 insertions to the other; thus these lesions may also induce the active position of this regulatory element. Structural study of one or more of each of these mutations will be required to confirm this hypothesis, and to illuminate their unique inhibitor-binding characteristics.
Development of resistance to TKIs is a major clinical problem. Patients initially responsive to erlotinib and gefitinib become resistant through second-site mutations in the EGFR kinase, most notably in the “gatekeeper” residue threonine 790 (Kobayashi et al., 2005; Kwak et al., 2005; Pao et al., 2005). The T790M mutation in the EGFR is structurally analogous to the T315I resistance mutation in BCR-Abl, and is expected to sterically interfere with drug binding (Kobayashi et al., 2005). In gefitinib and AEE788, replacement of threonine with the larger methionine may impinge on the aniline and phenylethylamine substituents, respectively (Figures 4A and B). Irreversible inhibitors of the EGFR kinase, including HKI-272, EKB-569 and CL-387785, are reported to maintain activity against the T790M mutation (Kwak et al., 2005; Kobayashi et al., 2005; Yuza et al., 2006). These compounds are structurally similar to reversible anilinoquinazoline inhibitors such as gefitinib, but contain a reactive Michael-acceptor that forms a covalent bond with Cys 797 at the edge of the ATP cleft (Tsou et al., 2005). They are thought to react specifically with this cysteine because their potent binding to the EGFR –presumably in an orientation analogous to that of gefitinib – brings the reactive group into apposition with the target thiol of Cys 797. Their potency against the T790M mutation is therefore somewhat paradoxical, as the T790M substitution can be expected to clash with their aniline substituents as in other anilinoquinazolines. A better understanding of the activity of these compounds against the T790M resistance mutant will require structural and biochemical study of the T790M mutant with these inhibitors. As with rapidly mutating infectious agents such as the human immunodeficiency virus, a successful strategy to counter resistance mutations that emerge in the course of cancer chemotherapy with EGFR inhibitors may involve simultaneous treatment with inhibitors that have different susceptibilities to resistance mutations. The irreversible inhibitors noted above may offer promise in this respect. However, the close correspondence in the overall binding modes of the inhibitors studied here highlights the potential need for development of structurally diverse EGFR inhibitors that may differ more in their susceptibilities to resistance mutations.
Experimental procedures
Cloning, expression and purification of EGFR kinase domain
Constructs spanning residues 696–1022 of the human EGFR and bearing the wild-type sequence, or the L858R or G719S mutations were prepared as previously described (Greulich et al., 2005) and were expressed as GST-fusion proteins in Sf9 insect cells using the Baculogold system (Pharmingen).
Cells pellets were lysed by sonication in Tris-buffered saline (TBS) supplemented with 2 mM TCEP (tris(2-carboxyethyl) phosphine hydrochloride) and complete protease inhibitor mixture (Roche). After centrifugation (40000 g, 1 hour, 4°C), the supernatant was incubated with glutathione Sepharose beads (Amersham). The beads were washed with wash buffer (40mM Tris, 500mM NaCl, 1% Glycerol, 2mM TCEP, pH8.0) and then incubated overnight with 4mL wash buffer supplemented with 10 μL of 3 mg/mL α-thrombin to remove the GST-fusion. The eluted EGFR kinases were further purified by size-exclusion chromatography (Superdex 200) in wash buffer, were concentrated to 4mg/ml, and aliquots were flash-frozen in liquid nitrogen and stored at −80°C.
Crystallization, diffraction data collection, structure determination and refinement
All crystals were obtained by hanging-drop method. Well-ordered crystals of the L858R mutant were only obtained in 40% PEG400, 150mM NaCl, 0.1M Hepes pH8.0, 5mM TCEP, 0.1M NDSB-211 (referred as PEG buffer), while the wild-type and G719S proteins produced the best crystals in 1.2M Potassium Sodium Tartrate, 0.1M Hepes pH7.5, 5mM TCEP (referred to as tartrate buffer), but could also be crystallized in PEG buffer. Complex crystals were made by soaking crystals of the wild-type or mutant kinases in either PEG buffer or tartrate buffer (as indicated in Table 1) containing 400 μM inhibitor overnight. Analysis of wild-type crystals revealed that crystallization and/or soaking of crystals in PEG rather than tartrate buffer produces small conformational differences in some loop regions of the kinase, and induced disorder in residues 867–875 in the activation loop. These buffer-related differences do not appear to be relevant to inhibitor binding. Nevertheless, we compare structures obtained under identical conditions when possible. All crystals belong to space group I23 with unit cell parameters a=b=c≈145Å, α=β=γ=90º.
Diffraction data were collected at the Brookhaven NSLS X25 or X29 beamlines or at Argonne National Laboratory APS ID19 or ID24 beamlines at 100K. The data were processed with HKL2000 (Otwinowski and Minor, 1997). All structures were solved by molecular replacement with PHASER (Read, 2001) using the approximately isomorphous apo-EGFR structure as the search model (PDB code 1M14 or 1M17) (Stamos et al., 2002). The programs Arp/Warp (Perrakis et al., 1999) and CNS (Brünger et al., 1998) were then used to obtain less biased 2Fo-Fc and Fo-Fc maps for manual inspection and adjustment of the model. Repeated rounds of manual refitting and crystallographic refinement were performed using O(Jones et al., 1991), COOT(Emsley and Cowtan, 2004) and refmac5(Murshudov et al., 1997). Inhibitors were modeled into the closely fitting positive Fo-Fc electron density and then included in following refinement and fitting cycles. Topology and parameter files for the inhibitors were generated using PRODRG (Schuttelkopf and van Aalten, 2004).
Enzyme kinetic assays and data analysis
The ATP/NADH coupled assay system in a 96-well format was used to determine the initial velocity of EGFR tyrosine kinase catalyzed peptide phosphorylation (Barker et al., 1995). The reaction mixture contained 5mg/mL BSA, 2mM MnCl2, 1mM PEP (2-(Phosphonooxy)-2-propenoic acid, Sigma-Aldrich, Cat. P-7002), 1mM TCEP, 0.1M MOPS 7.5, fixed (to determine ATP kinetic parameters) or varied (to determine peptide kinetic parameters) concentration of peptide substrate, 1/50 of the final reaction mixture volume of PK/LDH enzyme (Pyruvate Kinase/Lactic Dehydrogenase enzymes from rabbit muscle, Sigma-Aldrich, Cat. P-0294), 0.5mM NADH and 1 μM EGFRK, and ATP was added last to start the reaction. The two substrate kinase reaction was simplified to two one substrate reactions to determine ATP kinetic parameters and peptide parameters separately. When determining ATP parameters, the peptide concentration was kept the same and in excess (more than 5 times the Km value). When determining peptide kinetic parameters the ATP concentration was kept the same and in excess in all reactions (more than 5 times as its Km value). Steady state initial velocity data were drawn from the slopes of the A340 curves and fit to the Michaelis-Menten Equation to determine Vm and Km values. The substrate peptide was either a synthetic peptide with the sequence RAHEEIYHFFFAKKK (Brignola et al., 2002; Wood et al., 2004) or the widely used tyrosine kinase substrate poly-[Glu4Tyr1] as indicated in Table 2. Due to the limited solubility of the synthetic peptide in the coupled enzyme assay system, we used poly-[Glu4Tyr1] as a peptide substrate in determining peptide kinetic parameters. To assure that our derived kcat parameters reflected concentrations of active enzyme, we treated parallel aliquots of the wild-type and mutant kinases with the EGFR-specific covalent inhibitor PD168393 (Fry et al., 1998) to determine the fraction of catalytically competent molecules. Mass spectrometry analysis of the each sample showed essentially complete labeling with this ATP-competitive compound, thus our Kcat values are based on 100% competent enzyme.
Binding constant assay and data analysis
The inhibitors and kinases were separately diluted to 10μM and 50nM respectively in fluorescence buffer containing 20mM Tris, 0.5% Glycerol, 250mM NaCl, 1mM TCEP. The buffer was degassed and aerated with pure nitrogen gas to remove dissolved oxygen. The assay was carried out on a FluoroMax-2 fluorometer using a 1.0 cm pathlength quartz cuvette with micro stirrer. Excitation and emission slits were set to 3nM and 5nM respectively. The excitation and emission wavelengths were 284nm and 341nm, respectively. The inhibitor solution was titrated into an aliquot of 2.5mL EGFR kinase solution in the cuvette to obtain the indicated total concentration (total volume increase was <200 μl). The emission fluorescence intensity was read 25s after addition of inhibitor and the average of 5 measurements was recorded. A blank assay was performed in exactly the same manner except that the buffer without inhibitor was used for the titration. Dissociation constants (Kd) were determined by non-linear fitting of the fluorescence data using a modified static quenching model (see supplementary information for details).
Supplementary Material
Acknowledgments
We thank Bruce Johnson and Nathanael Gray for helpful discussions. We thank the staff of the X25 and X29 beamlines at Brookhaven National labs and the ID-19 and ID-24 (NE-CAT) beamlines at Argonne National labs for facilitating synchrotron data collection. We thank Kevan Shokat and Jimmy Blair for the compound PD168393. This work was supported in part by Novartis (MJE and MM) and by NIH grants CA080942 (MJE) and CA116020 (MM). MJE is the recipient of a Scholar award from the Leukemia and Lymphoma Society. TJB is the recipient of an American Society of Hematology Basic Research Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, Peyton M, Juroske D, Huang Y, Stuart SJ, Kim YH, Pollack JR, Yanagisawa K, Gazdar A, Minna JD, Kurie JM, Carbone DP. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–235. [PubMed] [Google Scholar]
- Arao T, Fukumoto H, Takeda M, Tamura T, Saijo N, Nishio K. Small in-frame deletion in the epidermal growth factor receptor as a target for ZD6474. Cancer Res. 2004;64:9101–9104. doi: 10.1158/0008-5472.CAN-04-2360. [DOI] [PubMed] [Google Scholar]
- Auffinger P, Hays FA, Westhof E, Ho PS. Halogen bonds in biological molecules. Proc Natl Acad Sci U S A. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker AJ, Gibson KH, Grundy W, Godfrey AA, Barlow JJ, Healy MP, Woodburn JR, Ashton SE, Curry BJ, Scarlett L, Henthorn L, Richards L. Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001;11:1911–1914. doi: 10.1016/s0960-894x(01)00344-4. [DOI] [PubMed] [Google Scholar]
- Barker SC, Kassel DB, Weigl D, Huang X, Luther MA, Knight WB. Characterization of pp60c-src tyrosine kinase activities using a continuous assay: autoactivation of the enzyme is an intermolecular autophosphorylation process. Biochemistry. 1995;34:14843–14851. doi: 10.1021/bi00045a027. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Li Y, Manley PW, Eck MJ. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106:996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignola PS, Lackey K, Kadwell SH, Hoffman C, Horne E, Carter HL, Stuart JD, Blackburn K, Moyer MB, Alligood KJ, Knight WB, Wood ER. Comparison of the biochemical and kinetic properties of the type 1 receptor tyrosine kinase intracellular domains. Demonstration of differential sensitivity to kinase inhibitors. J Biol Chem. 2002;277:1576–1585. doi: 10.1074/jbc.M105907200. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr . 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Capdeville R, Silberman S, Dimitrijevic S. Imatinib: the first 3 years. Eur J Cancer . 2002;38(Suppl 5):S77–S82. doi: 10.1016/s0959-8049(02)80607-4. [DOI] [PubMed] [Google Scholar]
- Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, Park F, Haley JD, Gibson N, Sliwkowski MX. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66:8163–8171. doi: 10.1158/0008-5472.CAN-06-0453. [DOI] [PubMed] [Google Scholar]
- Chan SK, Gullick WJ, Hill ME. Mutations of the epidermal growth factor receptor in non-small cell lung cancer--search and destroy. Eur J Cancer. 2006;42:17–23. doi: 10.1016/j.ejca.2005.07.031. [DOI] [PubMed] [Google Scholar]
- De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim SH. Crystal structure of cyclin-dependent kinase 2. Nature. 1993;363:595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1992;89:4309–4313. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Janne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc Natl Acad Sci U S A. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, Shigematsu H, Herz J, Minna JD. Mutations and addiction to EGFR: the Achilles ‘heal’ of lung cancers? Trends Mol Med. 2004;10:481–486. doi: 10.1016/j.molmed.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. Oncogenic Transformation by Inhibitor-Sensitive and-Resistant EGFR Mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65:8968–8974. doi: 10.1158/0008-5472.CAN-05-1829. [DOI] [PubMed] [Google Scholar]
- Johnson BE, Janne PA. Epidermal growth factor receptor mutations in patients with non-small cell lung cancer. Cancer Res. 2005;65:7525–7529. doi: 10.1158/0008-5472.CAN-05-1257. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A . 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma SV, Isselbacher KJ, Settleman J, Haber DA. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, Halmos B, Pearlberg J, Tsuchihashi Z, Cantley LC, Tenen DG, Johnson BE, Janne PA. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–1194. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology, Volume 276: Macromolecular Crystallography, part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA, Davis JA, Vaidya MP, Arnold LD, Doty JL, Iwata KK, Morin MJ. Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999;291:739–748. [PubMed] [Google Scholar]
- Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, Gilmer TM. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306:1506–1507. doi: 10.1126/science.1105396. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler P, Allegrini PR, Brandt R, Brueggen J, Cozens R, Fabbro D, Grosios K, Lane HA, McSheehy P, Mestan J, Meyer T, Tang C, Wartmann M, Wood J, Caravatti G. AEE788: a dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2004;64:4931–4941. doi: 10.1158/0008-5472.CAN-03-3681. [DOI] [PubMed] [Google Scholar]
- Tsou HR, Overbeek-Klumpers EG, Hallett WA, Reich MF, Floyd MB, Johnson BD, Michalak RS, Nilakantan R, Discafani C, Golas J, Rabindran SK, Shen R, Shi X, Wang YF, Upeslacis J, Wissner A. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J Med Chem. 2005;48:1107–1131. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]
- Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–5754. [PubMed] [Google Scholar]
- Williams JC, Weijland A, Gonfloni S, Thompson A, Courtneidge SA, Superti-Furga G, Wierenga RK. The 2.35 A crystal structure of the inactivated form of chicken Src: a dynamic molecule with multiple regulatory interactions. J Mol Biol. 1997;274:757–775. doi: 10.1006/jmbi.1997.1426. [DOI] [PubMed] [Google Scholar]
- Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, Alligood KJ, Rusnak DW, Gilmer TM, Shewchuk L. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Yuza Y, Glatt KA, Jiang J, Greulich H, Woo MS, Shimamura T, Shapiro G, Lee JC, Ji H, Feng W, Chen TH, Minami Y, Yanagisawa H, Wong KK, Meyerson M. Mutation-dependent variation in the relative cellular potency of distinct EGFR inhibitors. 2006 doi: 10.4161/cbt.6.5.4003. submitted. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Zhu X, Kim JL, Newcomb JR, Rose PE, Stover DR, Toledo LM, Zhao H, Morgenstern KA. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure. 1999;7:651–661. doi: 10.1016/s0969-2126(99)80086-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.