

Abstract
An improved synthesis for the (Z)-14-methyl-9-pentadecenoic acid was developed based on the appropriate use of (trimethylsilyl)acetylene as the key reagent in the synthesis. The reported synthesis started with commercially available 8-bromo-1-octanol and furnished the desired acid in seven steps and in a 16% overall yield, a significant improvement over the previous reported synthesis for this fatty acid. The synthesis reported herein afforded sufficient amounts to study the acid topoisomerase I inhibitory potential and it was found that the title acid inhibits the human placenta DNA topoisomerase I enzyme at concentrations of 500 μM.
Keywords: DNA, fatty acids, inhibitors, synthesis, topoisomerase I
Introduction
There are several literature reports describing the inhibitory effects of fatty acids towards the topoisomerase I enzyme.1–9 Topoisomerases I is a key enzyme in the breaking and fixing of DNA strands and is involved in making the necessary topological changes to DNA for key cellular processes such as replication, transcription, and recombination.1 Topoisomerases have also evolved as key cellular targets for the development of effective anticancer drugs. Fatty acids are known inhibitors of topoisomerase I, but most of the reported work with fatty acids deals with straight-chain fatty acids. For example, work by Suzuki et. al. has shown that while saturated fatty acids (C6–C22) display no inhibition of the topo-I enzyme (even at concentrations of the acid as high as 2000 μM), cis-monounsaturated fatty acids (C14–C22) do exhibit inhibition of the enzyme. However, the geometry and position of the double bonds as well as the carbon chain lengths are critical for the inhibitory process.1 For example, while oleic acid (cis-18:1 Δ9) inhibits topoisomerase I with an IC50 of 31 μM, the trans-isomer, namely elaidic acid (trans-18:1 Δ9) does not inhibit the enzyme (IC50 > 1000 μM).1 Other interesting topo-I lipid inhibitors include conjugated C18 and eicosapentaenoic fatty acids,2,4 very-long chain (C26–C30) Δ5,9 fatty acids,5,6 and phospholipids containing unsaturated fatty acids.7
While most of the fatty acid topoisomerase-I inhibitory work has been performed with straight-chain fatty acids, there is just one report dealing with the topoisomerase I inhibitory activity of methyl-branched iso and anteiso fatty acids.8 J.H. Jung and collaborators reported on the topoisomerase I inhibitory activity of compounds isolated from the Streptomyces sp. strain KM86-9B, and observed that the compounds responsible for the activity were a series of saturated iso and anteiso fatty acids with chain lengths between 15 and 17 carbon atoms.8 The best topo-I inhibitions were reported at concentrations of the acids of 100 μg/ml.8 Therefore, we became interested in studying the topoisomerase I inhibitory activity of an iso fatty acid with a cis double bond at C-9 based on these preliminary findings.1,8 For this purpose, the (Z)-14-methyl-9-pentadecenoic acid (1), a known bacterial fatty acid, was needed for this study.10
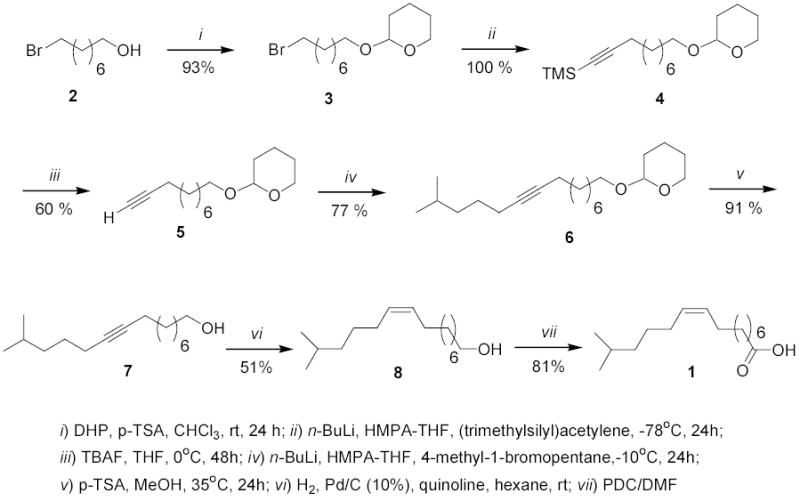
Acid 1 is not commercially available; therefore a good synthesis is required. The first total synthesis for 1 was recently reported by us.10 In this first generation synthesis the synthetic approach involved the initial coupling of (trimethylsilyl)acetylene to 4-methyl-1-bromopentane resulting in a volatile iso-alkyne followed, after removal of the silyl protecting group, by a second acetylide coupling to a longer-chain bromoalkane bearing a functional group (e.g., the protected alcohol 3 in Scheme 1) that could later be transformed into the carboxylic acid. The advantage of this strategy is that, after Lindlar hydrogenation of the resulting internal alkyne, a 100% Z stereoselectivity for the C-9 double bond was obtained.10 However, still a synthetic limitation to this approach was that the 6-methyl-1-heptyne is quite volatile, and the yields for the second alkyne-bromide coupling reaction were rather low (33% yields).10 Therefore, we envisaged that by reversing the coupling order, i.e., initial coupling of (trimethylsilyl)acetylene to 3 (Scheme 1) followed by a second acetylide coupling to 4-methyl-1-bromopentane would result in higher overall yields. This is the synthetic strategy we employed herein for the second generation synthesis of the required (Z)-14-methyl-9-pentadecenoic acid (1) (Scheme 1). We are also reporting that acid 1 inhibits human DNA topoisomerase I at concentrations of 500 μM.
Scheme 1.
Results and Discussion
Our second generation synthesis for 1 started with commercially available (Aldrich) 8-bromo-1-octanol (2), which was protected with 3,4-dihydro-2H-pyran (DHP) in the presence of p-toluenesulfonic acid (PTSA) affording the corresponding dihydropyranyl protected alcohol 3 in a 93% isolated yield (Scheme 1). The bromo dihydropyranyl protected alcohol 3 was then submitted to the first acetylide coupling reaction with the versatile reagent (trimethylsilyl)acetylene using n-BuLi in THF-HMPA affording the trimethylsilyl acetylenic derivative 4 in a 100% yield. Deprotection of the terminal trimethylsilyl group with tetrabutylammonium fluoride (TBAF) afforded the terminal alkyne 5 in a 60% yield. A second acetylide coupling with 4-methyl-1-bromopentane using again n-BuLi in THF-HMPA at −60° C resulted in the iso-branched alkyne 6 in a 77% isolated yield (Scheme 1). Deprotection of the dihydropyranyl group with PTSA in methanol at 45° C for 24 h afforded 14-methyl-9-pentadecyn-1-ol (7) in a 91% isolated yield. Catalytic hydrogenation of 7, under Lindlar’s conditions, afforded the cis-alkenol 14-methyl-9Z-pentadecen-1-ol (8) in a 51% yield. Final oxidation of alcohol 8 with pyridinium dichromate in dimethylformamide (DMF) afforded the desired acid 1, in an 81% yield (Scheme 1). The overall total yield for the seven steps was 16% and this synthesis afforded enough acid 1 to study its topo-I inhibitory activity.
Aimed at broadening the scope of the biological potential of the (Z)-14-methyl-9-pentadecenoic acid (1), we studied its inhibitory activity against human topoisomerase I (from freshly extracted human placenta) and compared it with the optimized concentration of camptothecin as shown in Figure 1. We found that 1 completely inhibits DNA topoisomerase I at concentrations of 500 μM (Figure 1). We should mention that palmitic acid does not inhibit topoisomerase I, even at concentrations > 1000 μM.1,9 Therefore, the inhibitory activity displayed by 1 confirms that the cis double bond geometry in 1, as well as the iso ramification, is important for the inhibition process. Our results thus confirm the preliminary findings by Jung et. al.8, that methyl-branched iso fatty acids are good inhibitors of the human DNA topoisomerase I. The topoisomerase I activity displayed by 1 is important since it opens the possibility that 1 might also display cytotoxicity towards cancer cell lines. In fact, we have previously shown that similar methyl-branched monounsaturated fatty acids, such as the (Z)-15-methylhexadec-11-enoic acid, are cytotoxic towards carcinoma cell lines.11
Figure 1.
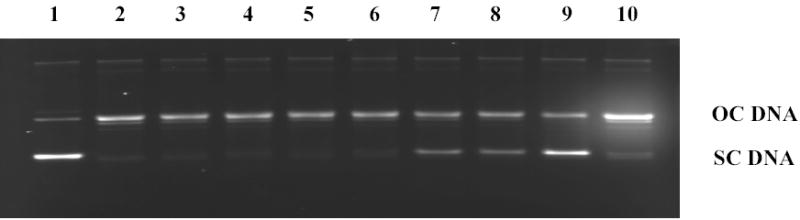
Agarose gel stained with ethidium bromide showing the inhibitory effect of (Z)-14-methyl-9-pentadecenoic acid (1) starting at 500 μM. OC DNA stands for open circular DNA and SC DNA for supercoiled DNA: lane 1, SC DNA; lane 2 SC DNA + topo-I; lane 3 acid 1 at 25 μM; lane 4 acid 1 at 50 μM; lane 5 acid 1 at 150 μM; lane 6 acid 1 at 250 μM; lane 7 acid 1 at 500 μM; lane 8 acid 1 at 750 μM; lane 9 acid 1 at 1000 μM; lane 10 camptothecin at 250 μM (optimized concentration).
In summary, we have shown that a higher yield is indeed obtained in the synthesis of monounsaturated iso methyl-branched fatty acids with double bonds close to the ωend of the acyl chain when (trimethylsilyl)acetylene is first coupled to the long-chain bifunctional bromoalkane (100 % yield) followed by a second acetylide coupling to the short-chain iso-bromoalkane (77% yield). The DNA topoisomerase I inhibitory results obtained for 1 should open the door to the synthesis of other structurally related analogs.
Experimental Section
General Procedures
Tetrahydrofuran (THF) was dried over Na and benzophenone prior to use. N,N-Dimethyl formamide (DMF) and hexamethylphosphoramide (HMPA) were dried over calcium hydride and distilled prior to use. (Trimethylsilyl)acetylene and other reagents used were purchased from Aldrich Chemical company. All compounds were analyzed by 1H and 13C Nuclear Magnetic Resonance (NMR) using a Bruker Advance DPX-300 or DRX-500 and the samples were measured in chloroform-d (CDCl3). The chloroform-d signals at 7.26 and 77.0 ppm were used as internal standard for proton and carbon, respectively. IR spectra were recorded on a Nicolet 600 FT-IR spectrophotometer. Mass spectral data was acquired using a GC-MS (Hewlett-Packard 5972A MS Chem-Station; Hewlett-Packard, Palo Alto, CA, USA) at 70 eV equipped with a 30 m x 0.25 mm special performance capillary column (HP-5MS) of polymethylsiloxane cross-linked with 5 % phenyl methylpolysiloxane. The high resolution mass spectral data was performed at the Emory University Mass Spectrometry Center.
2-[(8-Bromooctyl)oxy]tetrahydro-2H-pyran (3)
12 A solution of 8-bromo-1-octanol (1.6 mL, 9.6 mmol) in chloroform (20 mL) was placed in a 100-mL round bottom flask equipped with a magnetic stirrer and catalytic amounts of p-toluenesulfonic acid (PTSA). Dihydropyran (1.7 mL, 19.1 mmol) was added dropwise at room temperature. The reaction mixture was stirred for 3 h and then a saturated NaHCO3 solution was added. The organic layer was washed with water (2 x 20 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified using silica gel column chromatography and hexane/ether (9:1 v/v) as the mobile phase affording 3 (2.60 g) as a colorless oil for a 93 % yield, with spectral data comparable to the one previously reported in the literature.12 IR(neat) νmax 2935, 2856, 1454, 1440, 1383, 1200, 1135, 1120, 1077, 1034, 970, 905, 869, 814, 644–490 cm−1; 1H-NMR (500 MHz, CDCl3) δ 4.56 (1H, m), 3.88–3.70 (2H, m), 3.51–3.34 (2H, m), 3.39 (2H, t, J = 6.8 Hz, H-8), 1.48 (4H, m), 1.56 (6H, m), 1.41–1.32 (8H, m, -CH2-); 13C-NMR (125 MHz, CDCl3) δ 98.8 (d), 67.6 (t), 62.3 (t), 33.9 (t, C-8), 32.8 (t, C-7), 30.8 (t), 29.7 (t), 29.2 (t), 28.7 (t), 28.1 (t), 26.1 (t), 25.5 (t), 19.7 (t); GC-MS m/z (% rel. int.): [M-1]+ 291 (1), 148 (1), 135 (1), 111 (3), 101 (14), 85 (100), 69 (23), 55 (27).
Trimethyl [10-(tetrahydro-2H-pyran-2-yloxy)-1-decynyl] silane (4)
13 To a stirred solution of (trimethylsilyl)acetylene (2.9 mL, 20.4 mmol), in dry THF (20.0 mL), n-BuLi (2.5 M, 17.0 mmol) in dry hexane (6.8 mL) was added dropwise while keeping the temperature approximately at −78° C. After 45 min, HMPA (6.8 mL) and 2 (2.00 g, 6.8 mmol) were added dropwise to the reaction mixture while maintaining the temperature approximately at −78° C. After 24 h at rt, the reaction mixture was worked up by pouring into a large volume of water, and extracting with diethylether (2 x 20 mL). The organic layer was washed with brine (1 x 20 mL) before drying over MgSO4. Filtration, rotoevaporation of the solvent and fractional distillation of the impurities by Kugelrohr distillation (110–120° C/3 mm Hg) afforded 4 (2.11 g) as a colorless oil for a 100 % yield, with spectral data comparable to the one previously reported in the literature.13
2-[(9-Decynyl)oxy]tetrahydro-2H-pyran (5)
14 A mixture of 4 (2.11 g, 6.8 mmol) in 20.0 mL of dry THF was stirred at 0° C and then 2.0 mL (6.8 mmol) of tetrabutylammonium fluoride (1 M) in THF was added dropwise to the stirred solution. After 2 h, the reaction mixture was quenched with a 2M HCl solution and extracted with diethyl ether (2 x 20 mL). The organic extracts were dried over MgSO4, and concentrated in vacuo. The product was purified by fractional distillation of the impurities by Kugelrohr distillation (110–120° C/3 mmHg), affording 5 (0.97 g) for a 60 % yield as a colorless oil, with spectral data comparable to the one previously reported in the literature.14
2-[(14-Methyl-9-pentadecynyl)oxy]tetrahydro-2H-pyran (6)
To a stirred solution of 5 (0.97 g, 4.8 mmol), in dry THF (20.0 mL), n-BuLi (2.5 M, 10.2 mmol) in dry hexane (4.1 mL) was added dropwise at room temperature. After 45 min, HMPA (4.1 mL) and 4-methyl-1-bromopentane (1.8 mL, 12.2 mmol) were added dropwise to the reaction mixture while maintaining the temperature approximately at −60°C. After 24 h at rt, the reaction mixture was worked up by pouring into a large volume of water, and extracting with hexane (2 x 20 mL). The organic layer was washed with brine (1 x 20 mL) before drying over MgSO4. Filtration, rotoevaporation of the solvent and fractional distillation of the impurities by Kugelrohr distillation (110–120 °C/3 mm Hg) furnished 6 in a 77 % yield (1.10 g) as a colorless oil: IR (neat) νmax 2933, 2856, 2118, 1466, 1366, 1352, 1136, 1034 cm−1; 1H-NMR (300 MHz, CDCl3) δ 4.57 (1H, m), 3.90–3.68 (2H, m), 3.53–3.33 (2H, m), 2.10 (4H, m, H-8, H-11), 1.84–1.31 (23H, m), 0.87 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C-NMR (75 MHz, CDCl3) δ 98.8 (d), 80.3 (s), 80.2 (s), 67.6 (t, C-1), 62.3 (t), 38.2 (t), 30.7 (t), 29.7 (t), 29.4 (t), 29.1 (t) x 2, 28.8 (t), 27.6 (d), 27.0 (t), 26.2 (t), 25.5 (t), 22.6 (q, C-15, C-16), 19.7 (t), 19.0 (t), 18.7 (t); GC-MS m/z (% rel. int.): [M]+ 322 (1), 279 (1), 251 (3), 237 (4), 193 (3), 149 (1), 135 (3), 121 (4), 109 (13), 101 (19), 95 (18), 85 (100), 67 (26), 55 (27). HRMS (APCI) Calcd for C21H39O2 [M + H]+ 323.2944, Found 323.2945.
14-Methyl-9-pentadecyn-1-ol (7)
Compound 6 (1.01 g, 3.1 mmol) in methanol (15.0 mL), and catalytic amounts of PTSA were stirred at 45°C for 24 h. The solvent was rotoevaporated, hexane (10 mL) and then diethyl ether (10 mL) were added to crystallize excess PTSA, the solution was filtered, and rotoevaporated under high vacuum affording 0.68 g (91 % yield) of 7 as a colorless oil. This product was used in the next step without further purification: IR (neat) νmax 3345 (br, -OH), 2931, 2856, 1956, 1466, 1384, 1366, 1058 cm−1; 1H-NMR (300 MHz, CDCl3) δ 3.63 (2H, t, J = 6.6 Hz, H-1), 2.12 (4H, m, H-8, H-11), 1.63–1.20 (18H, m), 0.87 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C-NMR (75 MHz, CDCl3) δ 80.3 (s), 80.2 (s), 63.0 (t, C-1), 38.2 (t), 30.7 (t, C-13), 32.7 (t), 29.3 (t), 29.1 (t) x 2, 28.7 (t), 27.6 (d), 27.0 (t), 25.7 (t), 22.5 (q, C-15), 19.0 (t), 18.7 (t); GC-MS m/z (% rel. int.): [M-15]+ 223 (1), 164 (2), 135 (9), 121 (14), 109 (59), 95 (63), 81 (73), 69 (100), 55 (73). HRMS (APCI) Calcd for C16H31O [M + H]+ 239.2369, Found 239.2369.
14-Methyl-9Z-pentadecen-1-ol (8)
Into a 25-mL two-necked round-bottomed flask were placed 1.9 mL of dry hexane, 7 (0.30 g, 1.3 mmol), quinoline (1 mL), and palladium in activated carbon (0.17 g). One of the necks was capped with a rubber septum and the other was connected via tygon tubing to a 25-mL graduated pipet ending in a 150-mL beaker with distilled water.15 While stirring at room temperature a 20-mL syringe with needle was used to withdraw air from the system and to draw water up into the graduated pipet to the 0.0-mL mark. Hydrogen was then introduced into the system using a balloon filled with hydrogen attached to a hose barb-to-luer lock adapter with a stopcock and a needle. The reaction mixture consumed 31 mL of hydrogen during 1 h. The reaction mixture was filtered and the solvent was removed in vacuo affording 0.15 g (51 % yield) of 8 as a colorless oil: IR (neat) νmax 3332 (br, -OH), 3004, 2927, 2854, 1656, 1466, 1384, 1366, 1057, 723 cm−1; 1H-NMR (300 MHz, CDCl3) δ 5.35 (2H, m, H-9, H-10), 3.64 (2H, t, J = 6.6 Hz, H-1), 2.00 (4H, m, H-8, H-11), 1.57–1.15 (18H, m), 0.87 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C-NMR (75 MHz, CDCl3) δ 130.0 (d), 129.8 (d), 63.1 (t, C-1), 38.6 (t), 32.8 (t, C-2), 29.7 (t), 29.5 (t), 29.4 (t), 29.2 (t), 27.9 (d), 27.5 (t), 27.4 (t), 27.2 (t), 25.7 (t), 22.6 (q, C-15, C-16); GC-MS m/z (% rel. int.): [M]+ 240 (1), 222 (3), 166 (2), 151 (2), 123 (14), 109 (28), 95 (58), 82 (92), 69 (86), 55 (100). HRMS (APCI) Calcd for C16H31O [M - H]+ 239.2369, Found 239.2370.
14-Methyl-9Z-pentadecenoic acid (1)
10 To a stirred solution of 8 (0.15 g, 0.64 mmol) in 5.0 mL of DMF was slowly added pyridinium dichromate (1.00g, 2.6 mmol) at room temperature. After 48 h at rt, the reaction mixture was worked up by pouring 18 mL of water and extracting with hexane (3 x 12 mL). Once the solvent was evaporated and dried in vacuo, 95 mg of 8 were obtained, resulting in an 81 % yield of 1 as a colorless oil with spectral data comparable to the one previously reported in the literature as follows: IR (neat) νmax 3000–2500, 2927, 2855, 1722 (C=O), 1462, 1384, 1413, 1366, 1284, 1260, 1093, 940, 807, 724 cm−1; 1H-NMR (300 MHz, CDCl3) δ 5.35 (2H, m, H-9, H-10), 2.31 (2H, t, J = 7.5 Hz, H-2), 1.98 (2H, m, H-8, H-11), 1.30 (17H, m, -CH2-), 0.86 (6H, d, J = 6.6 Hz, H-15); 13C-NMR (75 MHz, CDCl3) δ 179.4 (s, C-1), 130.0 (d), 129.8 (d), 38.6 (t, C-13), 34.5 (t, C-2), 29.7 (t), 29.2 (t), 29.13 (t), 29.10 (t), 27.9 (d, C-14), 27.5 (t), 27.4 (t), 27.2 (t), 24.9 (t), 22.6 (q, C-15, C-16); ); GC-MS m/z (% rel. int.): [M]+ 254 (6), 246 (2), 236 (3), 221 (1), 193 (3), 181 (6), 173 (13), 163 (5), 151 (3), 137 (4), 123 (6), 111 (12), 97 (24), 83 (38), 69 (89), 55 (100); HRMS (APCI) Calcd for C16H31O2 [M + H]+ 255.2318, Found 255.2318.
DNA Topoisomerase I assay
Eukaryotic DNA Topoisomerase I Drug Screening Kit and purified human DNA Topoisomerase I was purchased from TopoGEN, Inc., Columbus OH. Proteinase K obtained from Tritirachium album was purchased from Sigma-Aldrich, Inc., Saint Louis, MI. The assay was carried out according to the procedure described by Carballeira et. al. [9]. Reactions (20 μL) that contained a mixture of sterile water, TGS buffer (10 mM Tris-HCl at pH 7.9, mM EDTA, 0.15 M NaCl, 0.1 % BSA, 0.1 mM Spermidine, 5 % glycerol), supercoiled pHOT1 plasmid DNA (0.25 μg/μL in 10 mM Tris-HCl at pH 7.5, 1 mM EDTA), 14-methyl-9Z-pentadecenoic acid (dissolved in DMSO and tested at final concentrations of 1000 μM, 750 μM, 500 μM, 250 μM, 150 μM, 50 μM and 25 μM), and DNA topoisomerase I obtained from human placenta (1 unit can relax 0.25 μg of supercoiled DNA in 30 min at 37°C) were assembled in sterile microcentrifuge tubes on ice. After the topoisomerase I enzyme addition, the tubes were transferred to a 37°C heating block, incubated for 30 minutes to relax supercoiled DNA and terminated by rapid addition of 2 μL of 10 % sodium dodecyl sulfate (SDS). Bound protein was digested by incubation of proteinase K (final concentration 0.05 mg/ml) at 37°C for 30 min. After the incubation, the digestion was terminated transferring the microcentrifuge tubes to an ice bath. Prior to the loading of the gel, 2.3 μL of loading buffer (0.25 % bromophenol blue, 50 % glycerol) and 23.2 μL of steril water were added to the tubes. A 20 μL of reaction was electrophoresed in a 1 % agarose gel in TBE 1X at 70 V for 120 min. Then, the gel was stained with 3 μL ethidium bromide (10 mg/mL) and 60 mL of distilled water for 60 min and destained with 60 mL of distilled water for 30 min in order to visualize the reaction products. DNA topoisomers bands were detected and quantified using a Versa Doc imaging system (model 1000, Bio Rad).
Acknowledgments
This work was supported by a grant from the SCORE program of the National Institutes of Health (Grant No. S06GM08102). D. Sanabria thanks the NSF-AGEP (Alliance for Graduate Education and the Professoriate) program for a graduate fellowship while D. Oyola thanks the NIH-MARC program for an undergraduate fellowship. We thank Elsie A. Orellano and Dr. Fernando González for their help in the topoisomerase I inhibitory bioassays. We thank Dr. Fred Strobel (Emory University) for the high resolution mass spectral data.
Footnotes
This paper is dedicated to Professor Waldemar Adam on the occasion of his 70th birthday
References
- 1.Suzuki K, Shono F, Kai H, Uno T, Uyeda M. J Enzyme Inhib. 2000;15:357. doi: 10.1080/14756360009040693. [DOI] [PubMed] [Google Scholar]
- 2.Mizushina Y, Tsuzuki T, Eitsuka T, Miyazawa T, Kobayashi K, Ikawa H, Kuriyama I, Yonezawa Y, Takemura M, Yoshida H, Sakaguchi K. Lipids. 2004;39:977. doi: 10.1007/s11745-004-1319-y. [DOI] [PubMed] [Google Scholar]
- 3.Harada H, Yamashita U, Kurihara H, Fukushi E, Kawabata J, Kamei Y. Anticancer Res. 2002;22:2587. [PubMed] [Google Scholar]
- 4.Yonezawa Y, Tsuzuki T, Eitsuka T, Miyazawa T, Hada T, Uryu K, Murakami-Nakai C, Ikawa H, Kuriyama I, Takemura M, Oshige M, Yoshida H, Sakaguchi K, Mizushina Y. Arch Biochem Biophys. 2005;435:197. doi: 10.1016/j.abb.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Nemoto T, Ojika M, Sakagami Y. Tetrahedron Lett. 1997;38:5667. [Google Scholar]
- 6.Remoto T, Yoshino G, Ojika M, Sakagami Y. Tetrahedron. 1997;53:16699. [Google Scholar]
- 7.Mizushima T, Natori S, Sekimizu K. Biochem J. 1992;285:503. doi: 10.1042/bj2850503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee HK, Lee DS, Lim J, Kim JS, Im KS, Jung JH. Arch Pharm Res. 1998;21:729. doi: 10.1007/BF02976766. [DOI] [PubMed] [Google Scholar]
- 9.Carballeira NM, Betancourt JE, Orellano EA, González FA. J Nat Prod. 2002;65:1715. doi: 10.1021/np0202576. [DOI] [PubMed] [Google Scholar]
- 10.Carballeira NM, Sanabria D, Ayala NL, Cruz C. Tetrahedron Lett. 2004;45:3761. [Google Scholar]
- 11.Reyes ED, Carballeira NM. Synthesis. 1996:693. [Google Scholar]
- 12.Pöhler, T. Johann Wolfgang Goethe-Universität in Frankfurt am Main, pg. 185. 2003. Ph.D. thesis. [Google Scholar]
- 13.Bjoerkling F, Norin T, Unelius CR, Miller RB. J Org Chem. 1987;52:292. [Google Scholar]
- 14.Santangelo EM, Coracini M, Witzgall P, Correa AG, Unelius CR. J Nat Prod. 2002;65:909. doi: 10.1021/np010551i. [DOI] [PubMed] [Google Scholar]
- 15.Blanchard DE. J Chem Educ. 2003;80:544. [Google Scholar]