

Abstract
Antiproliferative factor (APF) is a sialoglycopeptide elevated in the urine of patients with interstitial cystitis, a urinary bladder disorder of unknown etiology that is characterized by chronic pelvic pain. The present study was directed toward uncovering a pathway through which APF signals. Treatment of human urothelial cells with native APF resulted in growth inhibition accompanied by blockade of cell cycle transit and increased p53. Reduced expression of p53 by RNA interference diminished, while ectopic expression of p53 mimicked, the effects of APF. These are the first findings implicating the network of p53 target genes in urothelial defects associated with interstitial cystitis.
Keywords: Antiproliferative factor, Interstitial cystitis, Human urothelial cell, p53, p21Cip1/Waf1
1. Introduction
Interstitial cystitis (IC) is a chronic bladder disorder with long-term symptoms of pelvic and or perineal pain, thinning or ulceration of the bladder epithelial lining, urinary frequency, and urgency. IC patients are generally diagnosed by the presence of certain clinical criteria in the absence of other identifiable causes for the symptoms (e.g., urinary tract infection). Although, there are multiple hypotheses about the primary cause of IC, the underlying molecular mechanism of IC remains completely undefined [1].
Three urine biomarkers for IC have been identified in published reports: the EGFR/ErbB1 peptide ligands, heparin-binding epidermal growth factor-like growth factor (HB-EGF), and epidermal growth factor (EGF) [2-4], and a novel sialoglycopeptide, named antiproliferative factor (APF) [3,4]. HB-EGF and EGF are present under normal conditions in human urine, but their concentration varies in a statistically significant and inverse manner (EGF levels are higher and HB-EGF levels are lower) in patients with IC as compared to their age-, race- and gender-matched controls [4]. APF, a low molecular weight, glycosylated peptide related to the membrane receptor frizzled-8, was purified from the urine of IC patients [5]. Bioactivities attributed to APF include: suppression of urothelial cell proliferation; increases in transcellular permeability; lowering of the expression of proteins that form intercellular junctional complexes; and reduction in the production of HB-EGF from urothelial cells [2,4,6-9]. APF/antiproliferative activity is detectable in the urine of approximately 95% of IC patients (as compared to approximately 9% of controls) [2-4]. This accumulation in urine of a bioactive factor, capable of altering the behavior of urothelial cells, is consistent with the clinical observation of epithelial thinning and denudation observed in IC bladder tissue [10,11].
The cell cycle regulatory protein, p53, is an essential mediator of cell cycle transit, apoptotic cell death, and cellular responses to stress [12,13]. p53 is a transcription factor that controls the expression of numerous genes in a network that is presently being defined using multiple approaches. In response to stressful environmental conditions, p53 protein levels rise, leading to cell cycle arrest or apoptosis. Loss of p53 function by mutation has been found to be the most common genetic alteration observed in human cancer cells, implying p53 as a suppressor of hyper-proliferation [12,13].
Because of its atypical features, the signaling mechanisms by which APF induces its anti-proliferative activity are of considerable interest. We show here that treatment of normal human urothelial cells, and T24 human bladder carcinoma cells, with APF increases p53 levels and that experimentally induced changes in p53 levels alter the APF effect on cell growth, indicating that p53 is involved in the mechanism of APF-induced growth suppression observed in the context of IC.
2. Materials and methods
APF was purified from the supernatant of bladder epithelial cells explanted from one IC patient [6]. Mock APF was prepared using the supernatant of bladder epithelial cells from a normal control and the same purification procedure. Small interfering RNAs (siRNAs) and Abs against p53 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
2.1. Cell culture and transfection
Human urothelial cells derived from bladder biopsies and specific growth medium were obtained from Oligene (Berlin, Germany). T24 cells (ATCC HTB4) were cultured in McCoy’s 5A containing 10% fetal bovine serum (FBS), 1% l-glutamine, and 1% antibiotic/antimycotic solution (all from Sigma) at 37 °C/5% CO2 condition. T24 cells were transiently transfected with expression plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA).
2.2. Construction of wild type p53 plasmid
For expression of p53 with C-terminal HA-tag, pCMV-Tag4B (Stratagene) was used after modification. Oligonucleotides containing HA-tag encoding sequences and overhangs compatible with the XhoI and KpnI restriction sites (sense; 5′-tcgagTACCCATACGACGTCCCAGACTACGCTTAGtac, antisense; 5′-taAGCGTAGTCTGGGACGTCGTATGGGTAc) were used for pCHAb construct. The coding region of p53 was amplified by PCR using 5′ sense primer containing a NotI restriction site (5′-gatcgcggccgccATGGAGGAGCCGCAGTCA) and 3′ antisense primer containing the XhoI restriction site (5′-gatcctcgagGTCTGAGTCAGGCCCTTC), and pGBKT7-53 (BD-Clontech) as template was then inserted into the NotI and XhoI restriction sites of pCHAb (pCHAb-p53).
2.3. Cell proliferation assay
Cells were plated onto 24-well tissue culture plates in standard growth medium as described above at a density of 1 × 104 cells/well. After 12 h serum starvation, APF or Mock APF was added to the medium, and cells were incubated for the additional time indicated. Viable cell number was determined by uptake of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) as described [14].
2.4. Cell cycle analysis by FACS
Cells were treated with purified APF or Mock APF for the indicated times. After fixing and staining with propidium iodide, they were visualized by flow cytometry [14].
3. Indirect immunofluorescence microscopy
Cells were treated with purified APF or Mock APF for 7 d, fixed with 3% paraformaldehyde and incubated with anti-p53 or anti-GAPDH Abs in 1% BSA/PBS solution, followed by species-specific secondary antibodies conjugated to Texas-Red fluorophores. Slides were mounted in Vectashield mounting medium containing DAPI (Vector Laboratories, Inc., Burlingame, CA) and analyzed with oil immersion objectives using a LSM 510 META NLO laser scanning confocal microscope (Carl Zeiss MicroImaging, Inc. Thornwood, NY).
3.1. Western blot analysis
Cells were solubilized with lysis buffer [1% Nonidet P-40; 50 mM Tris, pH 7.4; 10 mM NaCl; 1 mM NaF; 5 mM MgCl2; 0.1 mM EDTA; 1 mM PMSF; and COMPLETE protease inhibitor cocktail tablet (Roche Diagnostocs Corp.)] and centrifuged at 12000 × g for 15 min. The supernatant was assayed for protein concentration determination and equal amounts of protein were used for Western blotting.
3.2. Statistical analysis
Data were compared using a paired Student’s t-test. P-values <0.05 were considered significant.
4. Results
4.1. APF suppresses human urothelial cell proliferation and cell cycle transit
APF was previously demonstrated to inhibit cell proliferation and arrest the cell cycle of explanted primary normal human bladder cells [6]. We confirmed the inhibitory effect of APF on cell growth using early passage (<6 passages) urothelial cells derived from human bladder tissues obtained from surgical biopsies. Proliferation rate was significantly decreased in the presence of APF as compared to Mock APF (Fig. 1A). Similarly, treatment of urothelial cells with APF increased the G2/M cell number as measured by FACS analysis (Fig. 1B), demonstrating that APF appears to block cell cycle transit predominantly at the G2/M boundary of primary urothelial cells. Mock APF did not show anti-proliferative activity.
Fig. 1.
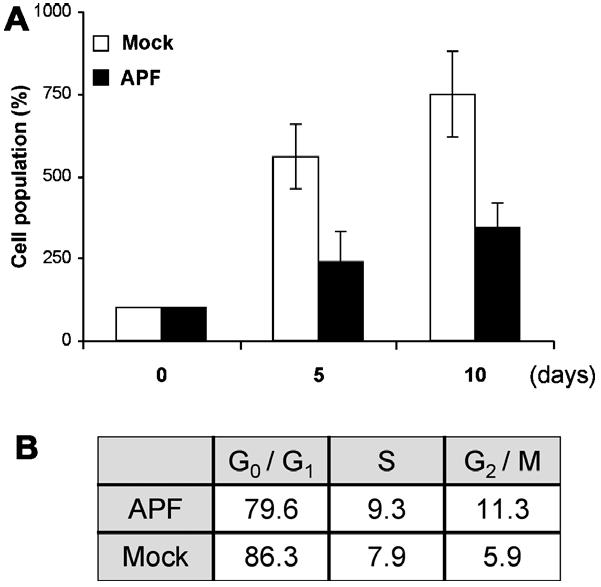
APF effect on cell proliferation and cell cycle progression. (A) Human urothelial cells were incubated in media containing 10 ng/ml APF or Mock APF control. At the indicated time points, cell number was counted by MTT assay. (B) FACS analysis was performed to determine the cell cycle transit 48 h after treatment.
4.2. p53 mediates APF effect
The identification of p53 as a downstream mediator of the effects of APF provides support for the hypothesis that cell cycle modulation may be a factor in IC. While the p53 protein expression level increased following APF treatment (Fig. 2A), the level of GAPDH was not changed (data not shown). Consistent with these data, immunofluorescence (IF) staining demonstrated increased p53 levels following treatment with APF as compared to Mock APF (Fig. 2B).
Fig. 2.
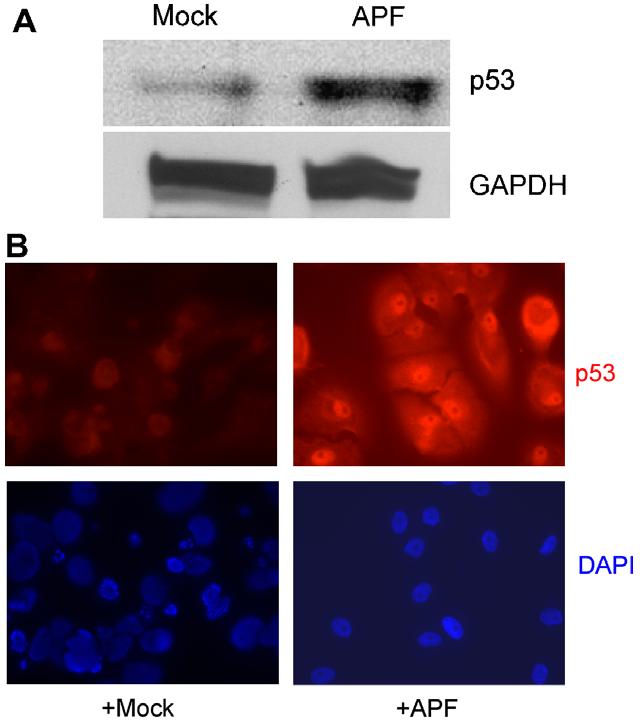
Increased p53 protein expression in response to APF. (A) Whole cell lysates were prepared from urothelial cells treated with 10 ng/ml APF or Mock APF for 7 d. Western blot analysis was performed using anti-p53 and GAPDH Abs. (B) Cells were stained with anti-p53 Ab (red), and nuclei were counterstained with DAPI (blue).
In subsequent experiments, we assessed whether a high p53 level is associated with biological functions of the APF peptide using RNA interference to lower p53 expression in urothelial cells. When p53 protein expression was decreased experimentally by transfection with p53 small inhibitory RNA (siRNA) duplexes, the inhibitory effect of purified native APF on cell growth was partially reversed (Fig. 3). APF exposure increased expression of p53 and its target, p21Cip1/WAF1, a cyclin-dependent kinase (CDK) inhibitor [15]. Down-regulation of p53 eliminated the induction in p21Cip1/WAF1 protein level even in the presence of APF.
Fig. 3.
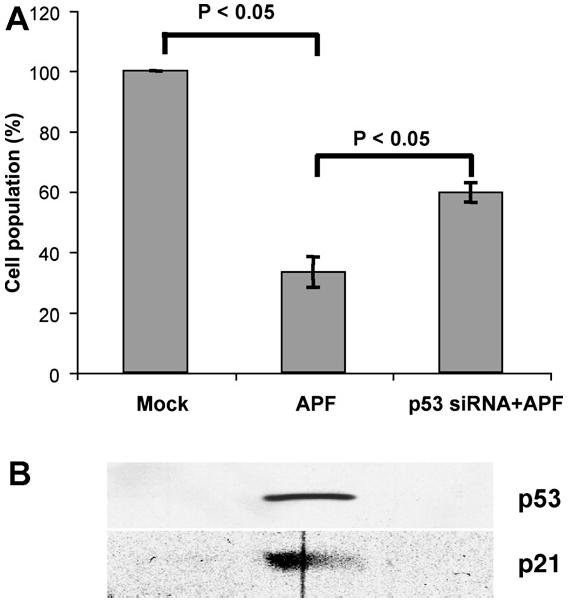
Reversal of the APF effect by p53 knockdown. Urothelial cells were transiently transfected with p53 siRNA duplex and incubated in media containing 10 ng/ml APF or Mock APF (control) for 7 days. Cell number was determined by MTT analysis. The protein levels of p53 and p21Cip1/WAF1 under individual experimental conditions were determined by Western blot.
4.3. APF peptide inhibits growth of T24 bladder cells
To expand our observations to another bladder cell type, and to avoid the limitations associated with primary cell cultures, we performed similar experiments to assess the effect of APF on cell growth using T24 bladder cancer cells. This cell line was chosen because of its human urothelial origin and because it responds to the antiproliferative effect of APF in a similar fashion to primary bladder urothelial cells [6]. As shown in Fig. 4A, APF treatment of serum-starved T24 cells led to growth suppression by about 40% in comparison to treatment with the Mock control (P < 0.05). In addition, there was a significant accumulation of the G2/M cell number in APF-treated cells (Fig. 4B).
Fig. 4.
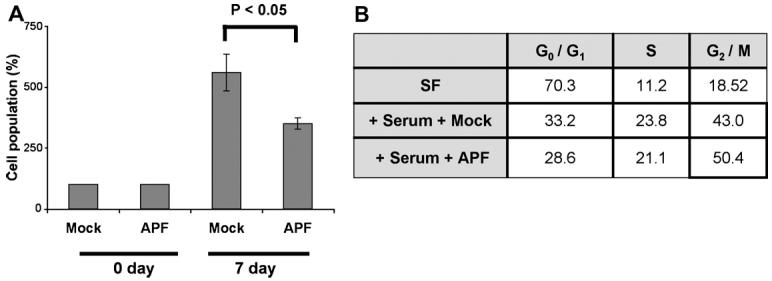
Inhibition of T24 bladder cancer cell growth by APF. (A) T24 cells were serum-starved and treated with 10 ng/ml APF or Mock APF (control), followed by cell number determination by MTT assay at the indicated time points. (B) Serum-starved T24 cells were stimulated with APF or Mock APF in the presence of serum. Three days later, FACS analysis was performed to assess the cell cycle.
4.4. Over expression of p53 inhibits cell proliferation in Mock APF-treated cells
To investigate whether p53 mediates the APF effect on cell growth, we attempted to alter intracellular p53 protein levels. T24 cells were transfected with vector only or vector containing a p53 expression construct and incubated with purified APF or Mock APF-containing serum free medium. Fig. 5A shows that incubation with APF suppressed the proliferation of cells transfected with vector alone (“APF”), compared to Mock APF treatment of the same cells. While p53 overexpression inhibited the proliferation of Mock APF-treated cells, there was no significant difference in growth inhibition when p53 and APF were used together. Western blotting assessment of p53 levels under the different conditions showed that, as anticipated, p53 accumulated with both APF treatment and p53 over-expression. However, ectopic expression of p53 did not significantly elevate the p53 level beyond that induced by APF in T24-p53 cells.
Fig. 5.
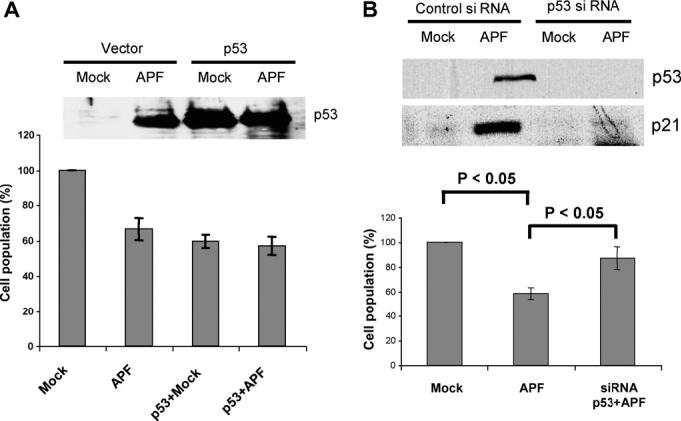
Effects of altered p53 level. (A) T24 cells were transiently transfected with p53 or vector only. Cell counting was performed 7 days after treatment by MTT assay. p53 levels were determined by Western blotting. (B) p53 in T24 cells was knocked down using siRNA. Following 10 ng/ml APF or Mock APF treatment for 7 d, the cells were counted, and p53 and p21Cip1/WAF1 protein levels were assessed.
To test directly whether p53 contributes to APF-induced growth arrest in T24 cells, we examined whether siRNA knockdown of p53 expression affected the response of T24 cells to APF. p53 levels were assessed in cell lysates collected after purified APF or Mock APF treatment in the presence of control siRNA or p53 siRNA. As shown in Fig. 5B, knock down of p53 using interfering RNA significantly attenuated the growth arrest elicited by APF.
5. Discussion
In this study, we identified p53 as an important mediator of APF-induced effects on bladder epithelial cells. Several lines of evidence support this conclusion: (1) APF treatment suppressed cell proliferation by cell cycle arrest of human bladder urothelial cells; (2) p53 levels increased significantly following APF stimulation; (3) p53 down-regulation enhanced the suppressive effect of APF on cell growth; and (4) over-expression of p53 itself induced cell cycle arrest in the absence of APF. These observations indicate that p53 can function as a cell cycle regulator of human urothelial cells of bladder origin.
The finding that p53 is involved in the mechanism of action of APF will now allow detailed mechanistic studies of the effects of APF on cells. Several avenues of investigation are warranted based on our present results. The activated proteasome degrades several proteins involved in cell cycle control, including p53. Thus, it is possible that stabilization of p53 levels following APF exposure results in transcriptional activation of proteins involved in cell cycle control. The stability of p53 is also influenced by a number of p53-interacting proteins, including the p53-associated factor, HAUSP [16,17], the homedomain-interacting protein kinase 2 (HIPK2) [18], kinases such as ataxia-telangiectasia mutated (ATM) [19-21] and ATR (ATM and Rad3-related) [22,23], and the nuclear ribonucleoprotein K (hnRNP K), an Mdm2 target and p53 transcriptional co-activator [24,25]. Phosphorylation of p53 is known to protect the protein from degradation via Mdm2 ubiquitin ligase-dependent mechanisms [13,26-31]. Although these and other aspects of the p53 regulatory network have been described, the molecular mechanisms linked to p53 in the context of IC have not been determined and additional studies on the p53 signaling pathway and of the p53 modification status in response to APF remain to be investigated.
A chronic bladder disorder, IC affects more than 1 million Americans. Although medical treatments and procedures, such as bladder instillation, hydrodistension, and the pharmaceutical drugs are prescribed for those afflicted with the disease, these treatments are effective in only a small percentage of patients with undesirable side effects. We have described in this paper the novel finding that exposure of bladder urothelial cells to APF, a urinary glycopeptide biomarker of IC, results in increased p53 protein expression and that p53 is a mediator of cell cycle arrest induced by APF. These data suggest the possibility of targeting p53, or p53 signaling network proteins, as a means of abrogating the pathologic effects of urinary APF, and new options for therapeutic intervention in IC seen clinically.
Acknowledgements
We thank Dr. Jee-Yeong Jeong (Harvard Medical School) for providing p53 expression constructs, and Drs. Galit Lahav (Harvard Medical School) and Yeoung-Rok Seo (Kyung Hee University) for providing advice and for critical reading of the manuscript. This research was supported by NIH Grants: R37 DK47556, R01 DK 57691, P50 DK65298 (to M.R.F.), R01 DK52596 (to S.K.K.) and R01 DK065990 (to J.D.D.). J.K. is an American Urological Association Foundation Research Scholar.
Abbreviations
- APF
antiproliferative factor
- EGF
epidermal growth factor
- FBS
fetal bovine serum
- HB-EGF
heparin-binding epidermal growth factor-like growth factor
- IC
interstitial cystitis
- MTT
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide
References
- [1].Bogart LM, Berry SH, Clemens JQ. Symptoms of interstitial cystitis, painful bladder syndrome and similar diseases in women: a systematic review. J. Urol. 2007;177:450–456. doi: 10.1016/j.juro.2006.09.032. [DOI] [PubMed] [Google Scholar]
- [2].Keay S, et al. Concentrations of specific epithelial growth factors in the urine of interstitial cystitis patients and controls. J. Urol. 1997;158:1983–1988. doi: 10.1016/s0022-5347(01)64198-3. [DOI] [PubMed] [Google Scholar]
- [3].Keay S, et al. A diagnostic in vitro urine assay for interstitial cystitis. Urology. 1998;52:974–978. doi: 10.1016/s0090-4295(98)00488-9. [DOI] [PubMed] [Google Scholar]
- [4].Keay S, Zhang CO, Marvel R, Chai T. Antiproliferative factor, heparin-binding epidermal growth factor-like growth factor, and epidermal growth factor: sensitive and specific urine markers for interstitial cystitis. Urology. 2001;57:104. doi: 10.1016/s0090-4295(01)01028-7. [DOI] [PubMed] [Google Scholar]
- [5].Keay SK, Szekely Z, Conrads TP, Veenstra TD, Barchi JJ, Jr., Zhang CO, Koch KR, Michejda CJ. An antiproliferative factor from interstitial cystitis patients is a frizzled 8 protein-related sialoglycopeptide. Proc. Natl. Acad. Sci. USA. 2004;101:11803–11808. doi: 10.1073/pnas.0404509101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rashid HH, Reeder JE, O’Connell MJ, Zhang CO, Messing EM, Keay SK. Interstitial cystitis antiproliferative factor (APF) as a cell-cycle modulator. BMC Urol. 2004;4:3. doi: 10.1186/1471-2490-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Keay S, Zhang CO, Shoenfelt JL, Chai TC. Decreased in vitro proliferation of bladder epithelial cells from patients with interstitial cystitis. Urology. 2003;61:1278–1284. doi: 10.1016/s0090-4295(03)00005-0. [DOI] [PubMed] [Google Scholar]
- [8].Keay S, Seillier-Moiseiwitsch F, Zhang CO, Chai TC, Zhang J. Changes in human bladder epithelial cell gene expression associated with interstitial cystitis or antiproliferative factor treatment. Physiol. Genom. 2003;14:107–115. doi: 10.1152/physiolgenomics.00055.2003. [DOI] [PubMed] [Google Scholar]
- [9].Zhang CO, Wang JY, Koch KR, Keay S. Regulation of tight junction proteins and bladder epithelial paracellular permeability by an antiproliferative factor from patients with interstitial cystitis. J. Urol. 2005;174:2382–2387. doi: 10.1097/01.ju.0000180417.11976.99. [DOI] [PubMed] [Google Scholar]
- [10].Skoluda D, Wegner K, Lemmel EM. [Critical notes respective immune pathogenesis of interstitial cystitis (author’s transl)] Urologe A. 1974;13:15–23. [PubMed] [Google Scholar]
- [11].Tomaszewski JE, et al. Biopsy features are associated with primary symptoms in interstitial cystitis: results from the interstitial cystitis database study. Urology. 2001;57:67–81. doi: 10.1016/s0090-4295(01)01166-9. [DOI] [PubMed] [Google Scholar]
- [12].Lane DP. Exploiting the p53 pathway for the diagnosis and therapy of human cancer. Cold Spring Harb. Symp. Quant. Biol. 2005;70:489–497. doi: 10.1101/sqb.2005.70.049. [DOI] [PubMed] [Google Scholar]
- [13].Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- [14].Kim J, Adam RM, Freeman MR. Trafficking of nuclear heparin-binding epidermal growth factor-like growth factor into an epidermal growth factor receptor-dependent autocrine loop in response to oxidative stress. Cancer Res. 2005;65:8242–8249. doi: 10.1158/0008-5472.CAN-05-0942. [DOI] [PubMed] [Google Scholar]
- [15].Bunz F, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- [16].Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell. 2004;13:879–886. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- [17].Cummins JM, Vogelstein B. HAUSP is required for p53 destabilization. Cell Cycle. 2004;3:689–692. [PubMed] [Google Scholar]
- [18].Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, Sun Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61:8211–8217. [PubMed] [Google Scholar]
- [19].Canman CE, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- [20].Khanna KK, et al. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat. Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- [21].Banin S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- [22].Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. Embo J. 2005;24:3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pereg Y, et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc. Natl. Acad. Sci. USA. 2005;102:5056–5061. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Meulmeester E, Pereg Y, Shiloh Y, Jochemsen AG. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle. 2005;4:1166–1170. doi: 10.4161/cc.4.9.1981. [DOI] [PubMed] [Google Scholar]
- [25].Moumen A, Masterson P, O’Connor MJ, Jackson SP. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005;123:1065–1078. doi: 10.1016/j.cell.2005.09.032. [DOI] [PubMed] [Google Scholar]
- [26].Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999;447:5–9. doi: 10.1016/s0014-5793(99)00254-9. [DOI] [PubMed] [Google Scholar]
- [27].Asher G, Lotem J, Sachs L, Kahana C, Shaul Y. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc. Natl. Acad. Sci. USA. 2002;99:13125–13130. doi: 10.1073/pnas.202480499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. Embo. J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- [30].Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- [31].Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]