

Abstract
While there are major advances made in the treatment of recurrent stenosis (restenosis) often resulting from percutaneous coronary and peripheral interventions, the persistent complications of acute thrombosis secondary to intimal hyperplasia and restenosis remain a mainstay for repeat hospitalizations in this patient population. For many years, a ubiquitous cell surface receptor called the αvβ3 integrin was the target of many investigators in the prevention of intimal hyperplasia and restenosis as its interaction with the extracellular matrix was believed to coordinate the migration of smooth muscle cells from the media to the intima, the seminal event in the formation of intimal lesion. After the publication of uniformly positive studies demonstrating that αvβ3 integrin blockade led to a significant reduction in new intimal (neointimal) lesion formation in a variety of animal models of balloon angioplasty, early clinical trials supported the association of decreased target lesion revascularization and the use of antagonists to the SMC integrin αvβ3 and its related platelet integrin αIIbβ3. However, a series of clinical trials subsequently demonstrated that these antagonists did not necessarily prevent revascularizations by inhibiting intimal hyperplasia. Additional animal studies subsequently showed that indeed in the setting of pre-existing smooth muscle cells in the intimal lesion (i.e., atherosclerotic plaque, fatty streaks), inhibiting smooth muscle cell migration by way of β3 integrin blockade was an ineffective approach in the prevention of intimal hyperplasia and restenosis as demonstrated in the clinical trials. However, given the wealth of basic and clinical information on the αvβ3 integrin and the use of its antagonists in the vasculature, we discuss in this manuscript our new approach to an old solution by targeting a new clinical problem of early failure arteriovenous access for hemodialysis. Given the uniqueness of arteriovenous access in that there are essentially no significant atherosclerotic lesions in the artery and vein prior to the anastomosis, the seminal event of the coordinated migration of smooth muscle cells from the media to the neointima could by targeted once again with β3 integrin antagonists.
Introduction
While there are significant advances made in the primary (e.g., surgical bypass and angioplasty and stenting) and secondary treatments (e.g., drug-eluding stents) for coronary and peripheral arterial occlusive disease, the ultimate solution to the persistent problems of anastomotic and in-stent narrowing (or restenosis) and the resulting acute thrombosis remains elusive.1,2 Restenosis is the reduction of the arterial luminal size due to loss in lumen size following the percutaneous and open arterial intervention, and its pathogenesis is thought to be multifactoral with a complex orchestrating of a number of biochemical and cellular events.3,4 The initial response to injury of the arterial wall during the formation of an anastomosis or overstretching by balloon catheter is elastic recoil, responsible for loss of initial luminal gain (constrictive remodeling), which characterizes the early and late phases of restenosis. The endothelial disruption and the exposure of subintimal components initiate the middle phase with platelet adherence and aggregation, fibrinogen binding, and thrombus formation. The thrombus, in turn, creates a scaffold into which smooth muscle cells (SMC) can migrate, synthesize matrix, and reorganize the thrombus, providing the substrate for intimal growth or intimal hyperplasia. Moreover, inflammatory mediators and cellular elements contribute to trigger a complex array of events that modulate matrix production and intimal cellular proliferation. The present manuscript focuses on the αvβ3 integrin, a cell surface receptor, as a potential therapeutic target for the prevention of SMC migration and restenosis.
αvβ3 integrin structure, function and distribution
Integrins are a family of transmembrane glycoproteins that mediate cell-cell and cell-matrix interaction.5 All known members of this superfamily are noncovalently associated heterodimers composed of an α and a β subunit. At present, at least 8 β and 18 α subunits have been characterized, and these subunits associate to generate at least 24 different integrins.5 For instance, subunit β3 associates with subunits αIIb and αv to generate integrins αIIbβ3 and αvβ3. Integrins are type I membrane proteins with a large extracellular, a transmembrane and a short cytoplasmic domains. The interaction between integrins and their ligands, besides mediating cell adhesion, plays a role in a number of cellular processes.6
αvβ3 integrin is one of the most prevalent integrins - expressed on almost all the cells originating from the mesenchyme and on a variety of cell types in the blood vessel (e.g., endothelial cells, SMCs, fibroblasts, macrophage, and platelets). It is known to mediate many biological events (e.g., migration of vascular SMCs, adhesion of osteoclasts to the bone matrix and angiogenesis). It is the most promiscuous integrin for it binds to many different ligands including a number of extracellular matrix proteins (e.g., vitronectin, fibronectin, osteopontin, fibrinogen and von Willebrand factor) via the interaction with the Arg-Gly-Asp (RGD) motif.5,7 On the other hand, a related integrin, αIIbβ3 is exclusively expressed on platelets and is largely responsible for the final cohesive phase of platelet activation in vivo, such as platelet aggregation supported by the binding of adhesive protein.8 Interestingly, the αIIbβ3 integrin recognizes the same RGD motif and binds to the same extracellular matrix proteins.9,10
Osteopontin (OPN), one of the ligands for αvβ3, contains the canonical integrin recognition sequence, RGD and bind to αvβ3 integrin through the sequence.11 In vitro studies have demonstrated that OPN promotes the migration of cultured rat arterial SMC12 and human coronary artery SMC.13 Previous data showed that OPN was coordinately expressed with β3 integrins in the vessel wall and that a blockade of αvβ3 resulted in a reduction of neointimal formation in animal models following vascular injury.14 These data suggest that αvβ3 binding to OPN are important in mediating SMC migration from the media to the neointima in vivo.
The integrin-mediated adhesion of cells to extracellular matrix leads bidirectional intracellular signaling events that regulate cell migration, as well as survival and proliferation. In outside-in signaling, ligand binding activates intracellular signaling pathways. In inside-out signaling, signals received by other receptors activate intracellular signaling pathways that impinge on integrin cytoplasmic domains, and change the extracellular domain conformation for binding to ligands.5 Recent studies have shown that αvβ3 expression on SMC is subject to regulation and is increased by treatment with thrombin,15 transforming growth factor-β (TGF-β) and platelet-derived growth factor-BB (PDGF-BB).16 In endothelial cells, vascular endothelial growth factor (VEGF) can induce activation of αvβ3 and NF-κB3 which leads to suppression of p53 and p21WAF1/CIP1 is an important transcription factor in αvβ3-dependent signals for endothelial cells.17,18 Moreover αvβ3, along with membrane type 1 matrix metalloproteinase-1 (MT1-MMP), is associated with matrix metalloproteinase-2 (MMP-2) at the cell surface.19,20
MMPs belong to a family of zinc-dependent endopeptidases that degrade many components of the extracellular matrix. Most MMPs are secreted in a latent form (pro-MMP), and a specific multistep activation process is required to convert pro-MMP to proteolytic active forms. Localization of functionally active MMP on the cell surface is essential and tightly regulated elements during a variety of normal and disease processes, such as tumor cell invasion.21 For instance, MMP-2 is activated at the cell surface of invasive cells by a multimeric receptor/activation complex consisting of the tissue inhibitor of metalloproteinase 2 (TIMP2), and the membrane type 1 MMP (MT1-MMP).22 In line with the theory of cellular invasion requiring a coordinated expression of proteolytic enzymes and adhesion molecules, Hofmann and colleagues23 suggested that functional cooperation of MT1-MMP and αvβ3 is critical for spatial and temporal control of extracellular matrix proteolysis in human melanoma cells. They indicated that joint MT1-MMP and αvβ3 might enforce most efficient docking, and activation of MMP-2 and, in turn, facilitate cellular locomotion. Furthermore, Brooks and colleagues19 demonstrated that the functionally active form of MMP-2 on the cell surface seems to predominantly involve αvβ3 in angiogenesis and concomitant melanoma growth.
αvβ3 integrin and animal models of arterial injury
Our group first reported the potential therapeutic benefit of αvβ3 blockade in the prevention of intimal hyperplasia and restenosis.24 We demonstrated that a potent chemotactic agent present in the arterial wall following injury, PDGF, regulates the surface distribution of αvβ3 on the SMC surface in cell culture. Using indirect immunofluorescence, focal adhesions containing αvβ3 were localized to the leading edge of migrating cells when stimulated with PDGF. In contrast, αvβ3 was evenly distributed on the surface of SMC grown in the absence of PDGF. These results suggest that a redistribution of αvβ3 in focal adhesion is necessary for SMC motility.
In an in vitro assay, we determined that PDGF-induced human SMC migration is mediated by αvβ3 by using a blocking antibody to αvβ3 (LM609). This PDGF-mediated migration was also attenuated with an αvβ3-blocking RGD peptide (GpenGRGDSPCA) demonstrating that the RGD sequence is the binding site in the extracellular matrix proteins. We also tested the effects of the local administration of this RGD peptide in a rabbit model of carotid balloon angioplasty injury. This RGD antagonist was delivered to the adventitia of the injured artery and inhibited the new intimal (neointimal) lesion formation by 70%. Neointimal hyperplasia seen in an animal model should be distinguished from intimal hyperplasia seen in humans as there are no inherent SMC in the non-injured intimal layer in most, normocholesterolemic animals. Subsequently, the same peptide locally applied to the carotid artery through an adventitial pluronic gel in rats led to a 92% reduction in neointimal hyperplasia after a similar balloon angioplasty injury.25
αvβ3 is present both in normal artery and in site of SMC accumulation and angiogenesis in atherosclerotic plaques in humans.26 In normal artery, αvβ3 is generally detectable only along the luminal surface with minimal expression in the media.26,27 Several studies in animal models have shown that arterial injury is a stimulus for expression of αvβ3 by endothelial cells and medial SMC.24,27 For instance, Srivatsa and colleagues28 showed in the pig coronary stent model that there is early upregulation of αvβ3 at sites of cell accumulation within the neointima and adventitia at 7 days after arterial injury, followed by persistent high levels of αvβ3 expression within the media and neointima up to 21 days, decreasing towards baseline by 28 days. Indeed, Table 1 depicts a number of reports demonstrating the efficacy of αvβ3 and αIIbβ3 antagonists in the reduction of neointimal hyperplasia in a variety of species.
Table 1.
Scientific investigations on the effectiveness of the β3 integrin antagonists in the inhibition of intimal lesion formation in various animal models of arterial injury.
| Species | Antagonist against | β3 integrin | Artery | Type of injury | IH lesion reduction? | Reference |
|---|---|---|---|---|---|---|
| Rat | ReoPro | αvβ3, αIIbβ3 | Carotid | Angioplasty | Yes | 25,44 |
| Rat | Gpen | αvβ3 | Carotid | Angioplasty | Yes | 45 |
| Hamster | Gpen | αvβ3 | Carotid | Angioplasty | Yes | 46 |
| Hamster | FK633 | αIIbβ3 | Carotid | Angioplasty | Yes | 47 |
| Rabbit | Gpen | αvβ3 | Carotid | Angioplasty | Yes | 24 |
| Rabbit | Vitaxin | αvβ3 | Carotid | Angioplasty | Yes | 48 |
| Rabbit | AZ-1 | αIIbβ3 | Femoral | Angioplasty | No | 33 |
| Pig | XJ 735 | αvβ3 | Coronary | Stenting | Yes | 28 |
| Monkey | ReoPro | αvβ3, αIIbβ3 | Iliac | Angioplasty | No | 34 |
| Monkey | ReoPro | αvβ3, αIIbβ3 | Subclavian | Stenting | No | 34 |
IH (intimal hyperplastic).
αvβ3 integrin and the clinical trials of restenosis
The results from the animal studies were consistent with findings from the early clinical trials examining the effect of various antagonists to platelet integrin αIIbβ3 and SMC integrin αvβ3 on the issue of long-term benefit of reduced target lesion revascularization (Table 2). In the Evaluation of Platelet IIb/IIIa Inhibition for Prevention of Ischemic Complications (EPIC) trial, ReoPro (abciximab, an monoclonal antibody fragment directed against the β3 integrin) was effective in limiting the need for late coronary revascularization after coronary angioplasty for at least 3 years after treatment.29 A subsequent study confirmed that ReoPro treatment reduced ischemic complications and late mortality, particularly in the diabetic population.30
Table 2.
Clinical trials on the effectiveness of the β3 integrin antagonists in the inhibition of restenosis in the coronary arteries.
| Study | Antagonist against | β3 integrin | Type of injury | TLR | Reference |
|---|---|---|---|---|---|
| EPIC | ReoPro | αvβ3, αIIbβ3 | Angioplasty, atherectomy | Reduced | 49 |
| Lincoff et al. | ReoPro | αvβ3, αIIbβ3 | Stenting | Reduced | 30 |
| IMPACT II | Integrilin | αIIbβ3 | Angioplasty | No difference | 31 |
| ERASER | ReoPro | αvβ3, αIIbβ3 | Stenting | No difference | 32 |
| CAPTURE | ReoPro | αvβ3, αIIbβ3 | Angioplasty | No difference | 50 |
TLR (targeted lesion revascularization).
However, the Integrilin to Minimize Platelet Aggregation and Coronary Thrombosis (IMPACT) II trial, which used Integrilin (an agent with anti-αIIbβ3 activity but without specific αvβ3 inhibitory activity) was ineffective in the reduction of coronary revascularizations in the same clinical setting as the EPIC trial.31 As in the animal studies, these clinical results suggest that αIIbβ3 integrin inhibition had no place in the treatment of coronary restenosis. However, a more detailed clinical study (ERASER trial) revealed that ReoPro given at the time of or a short duration after coronary angioplasty and stenting had little or no effect on the size of the intimal hyperplastic lesion as measured by intravascular ultrasound.32 Still, these clinical trials did not adequately address the role of αvβ3 in restenosis since short-term infusions of Integrilin and ReoPro would not be expected to block αvβ3 during crucial periods of vascular repair. Bleeding complications limited the long-term administration of these antagonists during percutaneous intervention to humans. Indeed, there is no certainty the local concentrations of these antagonists in the vessel wall are sufficient to inhibit αvβ3 integrin clinically.
αvβ3 integrin and the clinical significance based on animal models
As the exact role of αvβ3 in intimal hyperplastic lesion formation and restenosis remains unknown, it became critical to re-examine the precise mechanism of action of αvβ3 in cell culture and in animal models. Indeed, Azrin and colleagues33 tested in a hypercholesterolemic rabbit model of balloon angioplasty with pre-existing atherosclerotic lesions an antibody (AZ-1) that binds to the rabbit platelet αIIbβ3 and inhibits platelet function in vivo. There were no significant differences in intimal hyperplastic lesion formation between the AZ-1 antibody-treated and control groups 4 weeks after angioplasty. In this case, the αIIbβ3 antagonist failed to inhibit restenosis in the setting of pre-existing intimal lesion, similar to the human clinical situation where SMC migration is not a requisite for intimal lesion generation. While the case could made simply against the αIIbβ3 antagonists in the treatment of intimal hyperplasia, Deitch and coworkers34 reported that ReoPro failed to reduce the intimal hyperplastic lesion formation in atherosclerotic nonhuman primates after angioplasty and stenting in separate arteries, suggesting that in the setting of pre-existing intimal SMC, blockade of SMC migration is not critical.
Since the publication of the reports on the ineffectiveness of αvβ3 blockade on the intimal hyperplasia development in complex animal models, Smyth and colleague35 used a combination of “guidewire-induced endothelial denudation and arterial ligation” and demonstrated that (β3-integrin deficiency (β3−/−) did not have a role in intimal lesion formation.
However, our group subsequently classified the injury methodology by creating 3 distinct injury patterns that differed in the extent of medial injury induced in these β3−/− mice: (1) guidewire probe–induced transmural injury with medial disruption; (2) nonmedial disruptive ligation injury; and (3) eccentric medial disruptive injury followed by arterial ligation.36 We believed that guidewire probe injury generated more transmural mechanical damage to the media over a longer segment of the vessel compared with the ligation injury, which generates a more modest, focal lesion with stagnant flow and thrombosis.
As before, we showed that β3-integrin deficiency did not protect against neointimal lesion formation after a significant medial disruption seen with guidewire probe injury. In contrast, in the setting of arterial ligation injury, β3-integrin deficiency protected against neointimal lesion formation at 1, 2, and 3 weeks and 3 months after injury. When the combination of medial disruption and arterial ligation was used in β3−/− mice, there was eccentric neointimal lesion formation only at the site of disruption. The lack of neointimal lesion formation on the opposite, nondisrupted section is consistent with the dependence of neointimal formation on the mechanical disruption of the internal elastic lamina and media as described by others.37
One satisfactory explanation for these discrepancies based on the arterial injury patterns in β3−/− mice is that different models and/or methodologies accentuate the various, distinct functions of β3 integrins. For instance, Carmeliet and colleagues38 compared mechanical injury–induced intima formation in plasminogen activator inhibitor 1 (PAI-1)–deficient and wild-type mice and demonstrated that PAI-1 blocks intimal thickening by inhibiting the migration of SMCs. In contrast, Peng and colleagues39 demonstrated that when ligation-induced intima formation was examined in PAI-1+/+ and PAI-1−/− mice, PAI-1 promoted neointimal thickening. One can conclude that these injury models emphasize a contrasting cascade of events despite the apparently simple injuries. Moreover, Tanaka and colleagues40 showed in a carotid ligation injury model, there is minimal bone marrow-derived cell contribution to the neointimal lesion development. Therefore, in the carotid ligation model of intimal hyperplasia, the seminal event appears to be a directional cellular migration from the media to the neointima with the aid of αvβ3 with little or no contribution from the bone marrow (Figure 1B). Hence, although β3-integrin blockade effectively reduces neointimal hyperplasia in animal models, this blockade may not be effective for prevention of neointimal lesion formation in the less defined, more disruptive injury induced by percutaneous transluminal coronary angioplasty in human coronary arteries (Figure 1A).
Figure 1.
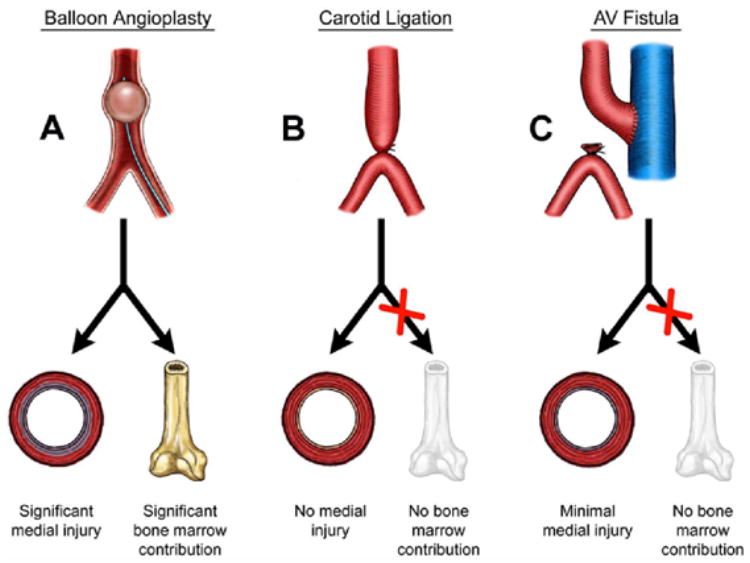
Leading animal models of arterial injury. A. The balloon angioplasty model results in a disruptive injury to the intima and media leading to neointimal hyperplastic lesion formation with a significant SMC contribution from the bone marrow. B. The carotid ligation (flow cessation) model results in little or no disruptive injury to the media. The seminal event appears to be a directional SMC migration from the media to the neointima with little or no contribution from the bone marrow. C. Like the carotid ligation model, the critical event appears to be SMC migration with minimal or no medial disruption and bone marrow contribution.
Future directions
The complications with the hemodialysis access constitute a major cause of morbidity for patients with end-stage kidney disease. In the United States alone, approximately 70% of the 250,000 patients on hemodialysis use expanded polytetrafluroethylene (ePTFE) grafts for permanent vascular access.41 Currently, the 1 and 2-year primary patency rates of these ePTFE grafts are 50% and 25%, respectively, while hemodialysis access related hospitalizations cost well over 1 billion dollars per annum.42 The failure of hemodialysis access grafts is predominantly due to a neointimal hyperplastic response in the region of the venous anastomosis resulting in reduction of shunt flow and ineffective hemodialysis. Subsequently, the access needs to be revised by an open surgical revision or a percutaneous angioplasty and/or stenting. By then, placement of another hemodialysis access at a difference site is not far off quickly exhausting the available sites predominantly in the upper extremities.
Castier and colleagues43 created recently an arteriovenous fistula (AVF) model in mice that demonstrated a rapid neointimal hyperplasia development at the anastomosis, the site most relevant to the clinical problem of venous neointimal hyperplasia and acute thrombosis. In this AVF model, there are traumatic injury to the blood vessels involved and turbulent blood flow near the anastomosis along with a compliance mismatch between artery and vein that believed to be factors that produce the rapid neointimal lesion formation. They further demonstrated that like the arterial ligation injury model, the neointimal SMC of the AVF anastomosis do not originate from bone marrow stem cells. Hence, they have demonstrated that this animal model and the clinical situation of arteriovenous access surgery for hemodialysis are uniquely suited to target the seminal event in the formation of the neointimal lesion formation, the SMC migration (Figure 1C). Unlike intimal hyperplasia seen with preocclusive atherosclerotic arteries after angioplasty and stenting, neointimal hyperplasia is seen with an anastomosis involving a synthetic graft (e.g., ePTFE, Dacron) and a relatively disease-free segment of vein or artery. Hence, there is no pre-procedural, stenotic intimal plaque with abundant resident SMCs. Therefore, adhesion and directional migration (relocation) of SMCs into the provisional matrix on the luminal surface are indeed the seminal events, not unlike the invasive tumor cells (metastasis). In this setting of ESKD and AV access, targeting the αvβ3 integrin could have a significant impact on the prevention of neointimal hyperplasia. Indeed, animal studies examining the role of the αvβ3 antagonists on the long-term patency of the AV accesses need to be performed in the setting of uremia, and then perhaps properly designed clinical trials in this ESKD patient population might ultimately provide a clinical problem to this old solution.
Acknowledgments
This research was supported in part by National Institutes of Health grant HL-68119 (to E.T.C.) and by Grant-in-Aid from AHA Heartland Affiliate, Inc. (to E.T.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schwartz RS, Henry TD. Pathophysiology of coronary artery restenosis. Rev Cardiovasc Med. 2002;3 (Suppl 5):S4–9. [PubMed] [Google Scholar]
- 2.Kotani J, Awata M, Nanto S, Uematsu M, Oshima F, Minamiguchi H, Mintz GS, Nagata S. Incomplete neointimal coverage of sirolimus-eluting stents: angioscopic findings. J Am Coll Cardiol. 2006;47:2108–11. doi: 10.1016/j.jacc.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz RS. The vessel wall reaction in restenosis. Semin Interv Cardiol. 1997;2:83–8. [PubMed] [Google Scholar]
- 4.Sarkar K, Sharma SK, Sachdeva R, Romeo F, Garza L, Mehta JL. Coronary artery restenosis: vascular biology and emerging therapeutic strategies. Expert Rev Cardiovasc Ther. 2006;4:543–56. doi: 10.1586/14779072.4.4.543. [DOI] [PubMed] [Google Scholar]
- 5.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 6.Hynes RO. Metastatic potential: generic predisposition of the primary tumor or rare, metastatic variants-or both? Cell. 2003;113:821–3. doi: 10.1016/s0092-8674(03)00468-9. [DOI] [PubMed] [Google Scholar]
- 7.Ruoslahti E, Pierschbacher MD. Arg-Gly-Asp: a versatile cell recognition signal. Cell. 1986;44:517–8. doi: 10.1016/0092-8674(86)90259-x. [DOI] [PubMed] [Google Scholar]
- 8.Topol EJ, Byzova TV, Plow EF. Platelet GPIIb-IIIa blockers. Lancet. 1999;353:227–31. doi: 10.1016/S0140-6736(98)11086-3. [DOI] [PubMed] [Google Scholar]
- 9.Pytela R, Pierschbacher MD, Ginsberg MH, Plow EF, Ruoslahti E. Platelet membrane glycoprotein IIb/IIIa: member of a family of Arg-Gly-Asp--specific adhesion receptors. Science. 1986;231:1559–62. doi: 10.1126/science.2420006. [DOI] [PubMed] [Google Scholar]
- 10.Barker PL, Bullens S, Bunting S, Burdick DJ, Chan KS, Deisher T, Eigenbrot C, Gadek TR, Gantzos R, Lipari MT, et al. Cyclic RGD peptide analogues as antiplatelet antithrombotics. J Med Chem. 1992;35:2040–8. doi: 10.1021/jm00089a014. [DOI] [PubMed] [Google Scholar]
- 11.Liaw L, Skinner MP, Raines EW, Ross R, Cheresh DA, Schwartz SM, Giachelli CM. The adhesive and migratory effects of osteopontin are mediated via distinct cell surface integrins. Role of alpha v beta 3 in smooth muscle cell migration to osteopontin in vitro. J Clin Invest. 1995;95:713–24. doi: 10.1172/JCI117718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gadeau AP, Campan M, Millet D, Candresse T, Desgranges C. Osteopontin overexpression is associated with arterial smooth muscle cell proliferation in vitro. Arterioscler Thromb. 1993;13:120–5. doi: 10.1161/01.atv.13.1.120. [DOI] [PubMed] [Google Scholar]
- 13.Panda D, Kundu GC, Lee BI, Peri A, Fohl D, Chackalaparampil I, Mukherjee BB, Li XD, Mukherjee DC, Seides S, Rosenberg J, Stark K, Mukherjee AB. Potential roles of osteopontin and alphaVbeta3 integrin in the development of coronary artery restenosis after angioplasty. Proc Natl Acad Sci U S A. 1997;94:9308–13. doi: 10.1073/pnas.94.17.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liaw L, Lindner V, Schwartz SM, Chambers AF, Giachelli CM. Osteopontin and beta 3 integrin are coordinately expressed in regenerating endothelium in vivo and stimulate Arg-Gly-Asp-dependent endothelial migration in vitro. Circ Res. 1995;77:665–72. doi: 10.1161/01.res.77.4.665. [DOI] [PubMed] [Google Scholar]
- 15.Brown SL, Lundgren CH, Nordt T, Fujii S. Stimulation of migration of human aortic smooth muscle cells by vitronectin: implications for atherosclerosis. Cardiovasc Res. 1994;28:1815–20. doi: 10.1093/cvr/28.12.1815. [DOI] [PubMed] [Google Scholar]
- 16.Janat MF, Argraves WS, Liau G. Regulation of vascular smooth muscle cell integrin expression by transforming growth factor beta1 and by platelet-derived growth factor-BB. J Cell Physiol. 1992;151:588–95. doi: 10.1002/jcp.1041510319. [DOI] [PubMed] [Google Scholar]
- 17.Soldi R, Mitola S, Strasly M, Defilippi P, Tarone G, Bussolino F. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. Embo J. 1999;18:882–92. doi: 10.1093/emboj/18.4.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stromblad S, Becker JC, Yebra M, Brooks PC, Cheresh DA. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin alphaVbeta3 during angiogenesis. J Clin Invest. 1996;98:426–33. doi: 10.1172/JCI118808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996;85:683–93. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- 20.Deryugina EI, Bourdon MA, Jungwirth K, Smith JW, Strongin AY. Functional activation of integrin alpha V beta 3 in tumor cells expressing membrane-type 1 matrix metalloproteinase. Int J Cancer. 2000;86:15–23. doi: 10.1002/(sici)1097-0215(20000401)86:1<15::aid-ijc3>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 21.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–5. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 22.Kinoh H, Sato H, Tsunezuka Y, Takino T, Kawashima A, Okada Y, Seiki M. MT-MMP, the cell surface activator of proMMP-2 (pro-gelatinase A), is expressed with its substrate in mouse tissue during embryogenesis. J Cell Sci. 1996;109 ( Pt 5):953–9. doi: 10.1242/jcs.109.5.953. [DOI] [PubMed] [Google Scholar]
- 23.Hofmann UB, Westphal JR, Waas ET, Becker JC, Ruiter DJ, van Muijen GN. Coexpression of integrin alpha(v)beta3 and matrix metalloproteinase-2 (MMP-2) coincides with MMP-2 activation: correlation with melanoma progression. J Invest Dermatol. 2000;115:625–32. doi: 10.1046/j.1523-1747.2000.00114.x. [DOI] [PubMed] [Google Scholar]
- 24.Choi ET, Engel L, Callow AD, Sun S, Trachtenberg J, Santoro S, Ryan US. Inhibition of neointimal hyperplasia by blocking alpha V beta 3 integrin with a small peptide antagonist GpenGRGDSPCA. J Vasc Surg. 1994;19:125–34. doi: 10.1016/s0741-5214(94)70127-x. [DOI] [PubMed] [Google Scholar]
- 25.Sheu JR, Wu CH, Chen YC, Hsiao G, Lin CH. Mechanisms in the inhibition of neointimal hyperplasia with triflavin in a rat model of balloon angioplasty. J Lab Clin Med. 2001;137:270–8. doi: 10.1067/mlc.2001.114065. [DOI] [PubMed] [Google Scholar]
- 26.Hoshiga M, Alpers CE, Smith LL, Giachelli CM, Schwartz SM. Alpha-v beta-3 integrin expression in normal and atherosclerotic artery. Circ Res. 1995;77:1129–35. doi: 10.1161/01.res.77.6.1129. [DOI] [PubMed] [Google Scholar]
- 27.van der Zee R, Murohara T, Passeri J, Kearney M, Cheresh DA, Isner JM. Reduced intimal thickening following alpha(v)beta3 blockade is associated with smooth muscle cell apoptosis. Cell Adhes Commun. 1998;6:371–9. doi: 10.3109/15419069809109146. [DOI] [PubMed] [Google Scholar]
- 28.Srivatsa SS, Fitzpatrick LA, Tsao PW, Reilly TM, Holmes DR, Jr, Schwartz RS, Mousa SA. Selective alpha v beta 3 integrin blockade potently limits neointimal hyperplasia and lumen stenosis following deep coronary arterial stent injury: evidence for the functional importance of integrin alpha v beta 3 and osteopontin expression during neointima formation. Cardiovasc Res. 1997;36:408–28. doi: 10.1016/s0008-6363(97)00184-3. [DOI] [PubMed] [Google Scholar]
- 29.Topol EJ, Califf RM, Weisman HF, Ellis SG, Tcheng JE, Worley S, Ivanhoe R, George BS, Fintel D, Weston M, et al. Randomised trial of coronary intervention with antibody against platelet IIb/IIIa integrin for reduction of clinical restenosis: results at six months. The EPIC Investigators. Lancet. 1994;343:881–6. doi: 10.1016/s0140-6736(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 30.Lincoff AM, Califf RM, Moliterno DJ, Ellis SG, Ducas J, Kramer JH, Kleiman NS, Cohen EA, Booth JE, Sapp SK, Cabot CF, Topol EJ. Complementary clinical benefits of coronary-artery stenting and blockade of platelet glycoprotein IIb/IIIa receptors. Evaluation of Platelet IIb/IIIa Inhibition in Stenting Investigators. N Engl J Med. 1999;341:319–27. doi: 10.1056/NEJM199907293410503. [DOI] [PubMed] [Google Scholar]
- 31.Randomised placebo-controlled trial of effect of eptifibatide on complications of percutaneous coronary intervention: IMPACT-II. Integrilin to Minimise Platelet Aggregation and Coronary Thrombosis-II. Lancet. 1997;349:1422–8. [PubMed] [Google Scholar]
- 32.Acute platelet inhibition with abciximab does not reduce in-stent restenosis (ERASER study). The ERASER Investigators. Circulation. 1999;100:799–806. doi: 10.1161/01.cir.100.8.799. [DOI] [PubMed] [Google Scholar]
- 33.Azrin MA, Ling FS, Chen Q, Pawashe A, Migliaccio F, Homer R, Todd M, Ezekowitz MD. Preparation, characterization, and evaluation of a monoclonal antibody against the rabbit platelet glycoprotein IIb/IIIa in an experimental angioplasty model. Circ Res. 1994;75:268–77. doi: 10.1161/01.res.75.2.268. [DOI] [PubMed] [Google Scholar]
- 34.Deitch JS, Williams JK, Adams MR, Fly CA, Herrington DM, Jordan RE, Nakada MT, Jakubowski JA, Geary RL. Effects of beta3-integrin blockade (c7E3) on the response to angioplasty and intra-arterial stenting in atherosclerotic nonhuman primates. Arterioscler Thromb Vasc Biol. 1998;18:1730–7. doi: 10.1161/01.atv.18.11.1730. [DOI] [PubMed] [Google Scholar]
- 35.Smyth SS, Reis ED, Zhang W, Fallon JT, Gordon RE, Coller BS. Beta(3)-integrin-deficient mice but not P-selectin-deficient mice develop intimal hyperplasia after vascular injury: correlation with leukocyte recruitment to adherent platelets 1 hour after injury. Circulation. 2001;103:2501–7. doi: 10.1161/01.cir.103.20.2501. [DOI] [PubMed] [Google Scholar]
- 36.Choi ET, Khan MF, Leidenfrost JE, Collins ET, Boc KP, Villa BR, Novack DV, Parks WC, Abendschein DR. Beta3-integrin mediates smooth muscle cell accumulation in neointima after carotid ligation in mice. Circulation. 2004;109:1564–9. doi: 10.1161/01.CIR.0000121733.68724.FF. [DOI] [PubMed] [Google Scholar]
- 37.Clowes AW, Schwartz SM. Significance of quiescent smooth muscle migration in the injured rat carotid artery. Circ Res. 1985;56:139–45. doi: 10.1161/01.res.56.1.139. [DOI] [PubMed] [Google Scholar]
- 38.Carmeliet P, Moons L, Lijnen R, Janssens S, Lupu F, Collen D, Gerard RD. Inhibitory role of plasminogen activator inhibitor-1 in arterial wound healing and neointima formation: a gene targeting and gene transfer study in mice. Circulation. 1997;96:3180–91. doi: 10.1161/01.cir.96.9.3180. [DOI] [PubMed] [Google Scholar]
- 39.Peng L, Bhatia N, Parker AC, Zhu Y, Fay WP. Endogenous vitronectin and plasminogen activator inhibitor-1 promote neointima formation in murine carotid arteries. Arterioscler Thromb Vasc Biol. 2002;22:934–9. doi: 10.1161/01.atv.0000019360.14554.53. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka K, Sata M, Hirata Y, Nagai R. Diverse contribution of bone marrow cells to neointimal hyperplasia after mechanical vascular injuries. Circ Res. 2003;93:783–90. doi: 10.1161/01.RES.0000096651.13001.B4. [DOI] [PubMed] [Google Scholar]
- 41.Schwab SJ, Harrington JT, Singh A, Roher R, Shohaib SA, Perrone RD, Meyer K, Beasley D. Vascular access for hemodialysis. Kidney Int. 1999;55:2078–90. doi: 10.1046/j.1523-1755.1999.00409.x. [DOI] [PubMed] [Google Scholar]
- 42.Lee H, Manns B, Taub K, Ghali WA, Dean S, Johnson D, Donaldson C. Cost analysis of ongoing care of patients with end-stage renal disease: the impact of dialysis modality and dialysis access. Am J Kidney Dis. 2002;40:611–22. doi: 10.1053/ajkd.2002.34924. [DOI] [PubMed] [Google Scholar]
- 43.Castier Y, Lehoux S, Hu Y, Foteinos G, Tedgui A, Xu Q. Characterization of neointima lesions associated with arteriovenous fistulas in a mouse model. Kidney Int. 2006;70:315–20. doi: 10.1038/sj.ki.5001569. [DOI] [PubMed] [Google Scholar]
- 44.Bendeck MP, Nakada MT. The beta3 integrin antagonist m7E3 reduces matrix metalloproteinase activity and smooth muscle cell migration. J Vasc Res. 2001;38:590–9. doi: 10.1159/000051095. [DOI] [PubMed] [Google Scholar]
- 45.Margolin L, Fishbein I, Banai S, Golomb G, Reich R, Perez LS, Gertz SD. Metalloproteinase inhibitor attenuates neointima formation and constrictive remodeling after angioplasty in rats: augmentative effect of alpha(v)beta(3) receptor blockade. Atherosclerosis. 2002;163:269–77. doi: 10.1016/s0021-9150(02)00035-7. [DOI] [PubMed] [Google Scholar]
- 46.Matsuno H, Stassen JM, Vermylen J, Deckmyn H. Inhibition of integrin function by a cyclic RGD-containing peptide prevents neointima formation. Circulation. 1994;90:2203–6. doi: 10.1161/01.cir.90.5.2203. [DOI] [PubMed] [Google Scholar]
- 47.Kaida T, Matsuno H, Niwa M, Kozawa O, Miyata H, Uematsu T. Antiplatelet effect of FK633, a platelet glycoprotein IIb/IIIa antagonist, on thrombus formation and vascular patency after thrombolysis in the injured hamster carotid artery. Thromb Haemost. 1997;77:562–7. [PubMed] [Google Scholar]
- 48.Bishop GG, McPherson JA, Sanders JM, Hesselbacher SE, Feldman MJ, McNamara CA, Gimple LW, Powers ER, Mousa SA, Sarembock IJ. Selective alpha(v)beta(3)-receptor blockade reduces macrophage infiltration and restenosis after balloon angioplasty in the atherosclerotic rabbit. Circulation. 2001;103:1906–11. doi: 10.1161/01.cir.103.14.1906. [DOI] [PubMed] [Google Scholar]
- 49.Use of a monoclonal antibody directed against the platelet glycoprotein IIb/IIIa receptor in high-risk coronary angioplasty. The EPIC Investigation. N Engl J Med. 1994;330:956–61. doi: 10.1056/NEJM199404073301402. [DOI] [PubMed] [Google Scholar]
- 50.Randomised placebo-controlled trial of abciximab before and during coronary intervention in refractory unstable angina: the CAPTURE Study. Lancet. 1997;349:1429–35. [PubMed] [Google Scholar]