

Abstract
Mutations in Leucine Rich Repeat Kinase 2 (LRRK2) are the leading genetic cause of Parkinson’s disease (PD). LRRK2 is predicted to contain kinase and GTPase enzymatic domains, with recent evidence suggesting that the kinase activity of LRRK2 is central to the pathogenic process associated with this protein. The GTPase domain of LRRK2 plays an important role in the regulation of kinase activity. To investigate the how the GTPase domain might be related to disease, we examined the GTP binding and hydrolysis properties of wild type and a mutant LRRK2. We show that LRRK2 immunoprecipitated from cells has a detectable GTPase activity that is disrupted by a familial mutation associated with PD located within the GTPase domain, R1441C.
Keywords: LRRK2, Parkinson’s disease, GTPase, kinase
Introduction
LRRK2 is a 2527 amino acid protein of unknown function predicted to contain a kinase domain, a ROC (Ras of Complex Proteins) domain and several protein/protein interaction domains [1]. Mutations in LRRK2 result in autosomal dominant Parkinson disease (PD) [2; 3; 4; 5] and there is a great deal of interest in both the normal biology of LRRK2 and its role in the pathogenesis of PD.
Pathogenic mutations are scattered throughout the predicted domains of LRRK2. Several mutations have been described in the kinase domain of LRRK2 and two of these, I2020T and G2019S, increase the kinase activity of the protein in autophosphorylation assays [6; 7]. Furthermore, whilst expression of familial mutants in cultured cells leads to cell death, expression of the mutants on a kinase dead background substantially reduces toxicity [8; 9], implying that the kinase activity of LRRK2 is required for pathogenesis. However, mutations outside of the kinase domain have variable effects on kinase activity, at least as measured by autophophorylation [9; 10], leaving open the question of how mutations cause toxicity.
The ROC domain of LRRK2 is predicted to bind GTP and may have GTPase activity, similar to other large proteins containing this domain [11]. However, sequence alignment of the ROC domain with small GTPases reveals key differences between the two groups that imply that LRRK2 would have lower intrinsic GTPase activity than small GTPases [12]. This is supported by experiments that report very low GTPase activity for LRRK2 compared to ras [12]. West et al used GTP binding as a surrogate measure of GTPase activity and concluded that mutations in LRRK2 increased activity [10]. Several previous studies have shown that GTP binding increases kinase activity for both LRRK2 and the homologous kinase LRRK1 [8; 12; 13].
In this study, we established conditions where the low GTPase activity of LRRK2 could be measured. We show that mutation in the ROC domain, R1441C, does not increase GTP binding to any significant extent but instead decrease GTPase activity. We also determine the effects of GTP binding mutations on the related kinase activity.
Materials and Methods
Vectors and cell lines
Wild type and pathogenic LRRK2 variants were cloned as previously described [9] and expressed from the pDEST51 vector (Invitrogen, Carlsbad, CA) containing an EF1α promoter and a C-terminal V5 epitope tag. The K1347A mutant in the GTP γ phosphate coordinating motif, predicted to interfere with GTP binding [ 8 ; 13 ], was also made by mutagenesis (primer sequences are available on request from the authors).
GTP Pulldown Assay
Pulldowns were carried out using GTP conjugated to sepharose beads as previously described [13; 14]. Competition experiments were performed by adding 10mM GTP to the beads after pulldown. Amounts of V5 tagged LRRK2 were measured by densitometry using a STORM 840 scanner (Amersham) and results are expressed as the amount of protein in the pulldown relative to the input.
GTPase Assay
Cells were lysed as described previously [14] and centrifuged to remove insoluble material. Supernatants were incubated with anti-V5 agarose beads (Sigma) for 2 hrs at 4°C then washed 5 times with 1ml PBS supplemented with 300 mM NaCl and 1% Triton X-100. The beads were washed once in 1mL of assay buffer (20mM HEPES pH 7.2, 2mM MgCl2, 1mM DTT, 0.005% BSA), re-suspended in 40μL of the same buffer and α32P-GTP (5μCi; GE healthcare) was added to each reaction. Samples were incubated at room temperature with vigorous shaking and 1μL aliquots removed at time points from 0-60 minutes and spotted onto TLC plates (Sigma). Samples were then subjected to rising thin layer chromatography under 1M formic acid, 1.2M LiCl for two hours. Plates were dried for 5min and radioactive bands were detected by autoradiography using a phosphoscreen. The precipitated V5 beads were washed with 1mL of high salt PBS with 1% Triton X-100 and incubated with an equal volume of 2X Laemmli sample buffer (Bio-rad) with 10% β Mercaptoethanol at 100°C for 10 min and run on SDS PAGE. Gels were washed once and stained with coomassie blue to visualize isolated protein.
In vitro Kinase Assays
Autophosphorylation assays using immunoprecipitated LRRK2 were carried out as previously described [9; 14] and separated on 5% SDS-PAGE gels. Myelin basic protein (MBP) assays were performed with the same kinase preparations and separated on 10-20% SDS-PAGE gels. Quantitation was performed by measuring the amount of incorporated radioactivity in LRRK2 or MBP, and was corrected for protein loading by densitometry of blots for V5-tagged LRRK2 or from Ponceau staining of membranes for MBP.
Results
GTP binding properties of LRRK2
LRRK2 variants were precipitated from whole cell lysates using immobilized GTP (figure 1). Wild type and R1441C LRRK2 both bound strongly to GTP, which was blocked by a molar excess of GTP. Quantitation of the amount of GTP bound LRRK2 compared to inputs did not reveal any differences between R1441C and wild type. LRRK2 (P=0.94 by t-test; n=4). K1347A could not be precipitated under the same conditions, confirming that this mutation disrupts GTP binding.
Figure 1. Interaction of LRRK2 with GTP.
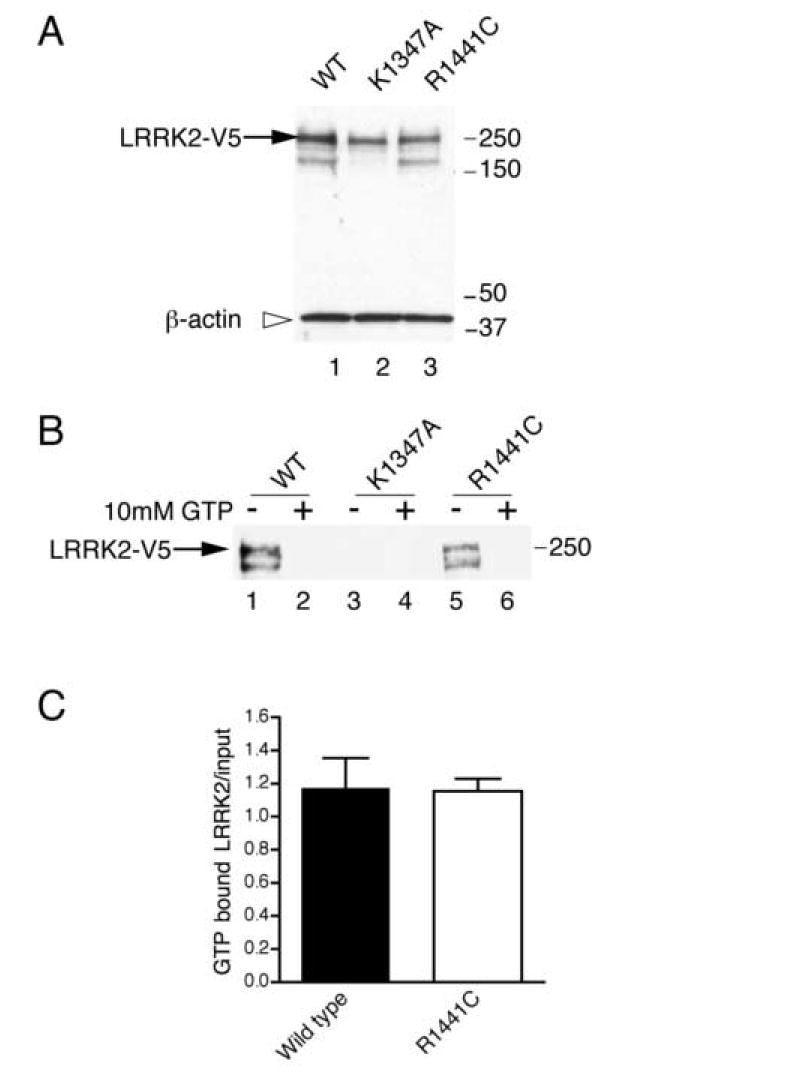
(A) Full length, C-terminal V5 tagged, wild type (lane 1), K1347A (lane 2) and R1441C (lane 3) LRRK2 were expressed in COS7 and blotted for V5 (arrow shows LRRK2-V5) and β-actin as a loading control (open arrowhead). Markers on the right of this and all blots are in kilodaltons. (B) LRRK2 could be precipitated from cell lysates with sepharose beads conjugated to GTP (lanes 1 and 5). Addition of 10mM GTP abolished this interaction (lanes 2 and 6). In contrast, the K1347A mutant could not be precipitated under the same conditions (lanes 3 and 4). (C) Quantitation (n=4, error bars show the SEM) of the amount of V5-LRRK2 precipitated on GTP beads, corrected for the amount of V5-LRRK2 in the inputs, showed that WT and R1441C were equivalent.
GTPase activity of LRRK2
Using an in vitro GTPase assay system, the ability of wild type, R1441C and K1347A LRRK2 to hydrolyze GTP to GDP was examined. Compared to the K1347A mutant, wild type LRRK2 displayed an increased ability to convert GTP to GDP, demonstrating that LRRK2 contains an active GTPase (figure 2). In contrast, the R1441C mutant had a much lower activity compared to wild type LRRK2 under conditions of equal protein loading.
Figure 2. LRRK2 GTPase activity.
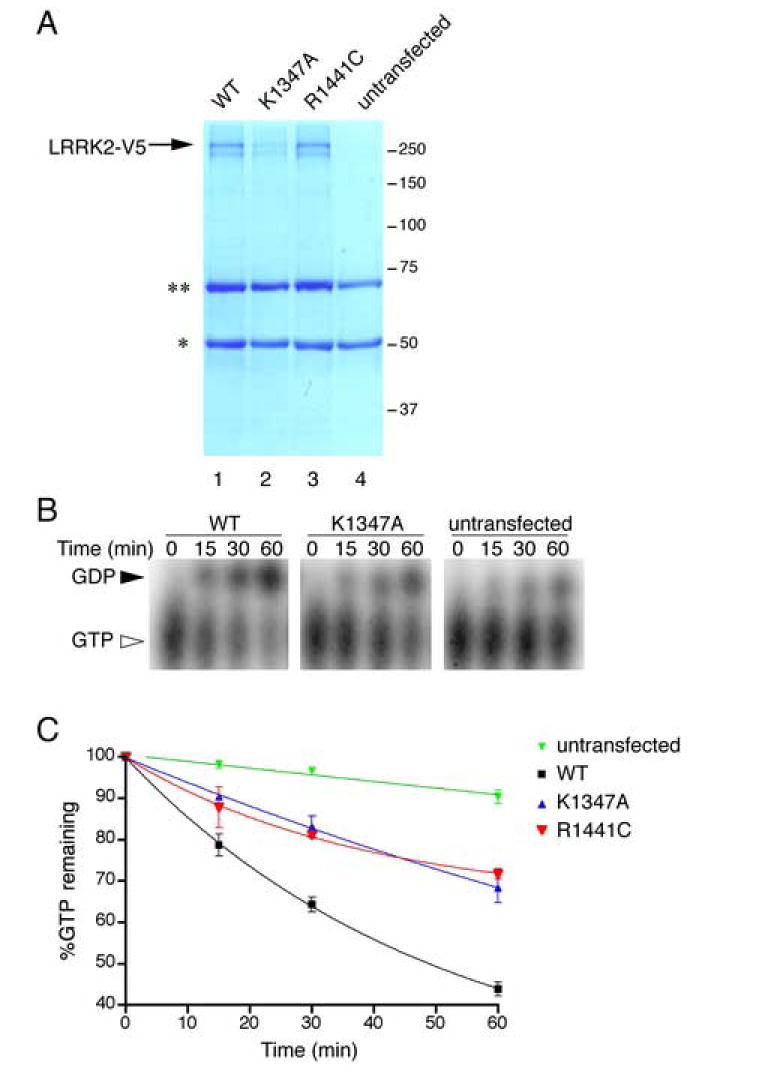
(A) LRRK2-V5 constructs (lane 1, wild type; lane 2, K1347A; lane 3, R1441C) were immunoprecipitated and analyzed by SDS PAGE with coomassie blue staining. Arrow shows LRRK2 protein, the asterisks are non-specific immunoglobulin bands also present in untransfected control samples (lane 4). (B) Over a 60-minute time course, wild type LRRK2 (left panel) hydrolyzes GTP (closed arrowhead) to GDP (open arrowhead) more readily than K1347A (middle panel). Background hydrolysis was measured using samples from untransfected cells (right panel). (C) Quantification of the percentage of GTP remaining (n=3; error bars indicate the SEM) shows that the R1441C mutant (red) has lower GTPase activity than wild type LRRK2 (black). K1347A (blue) also had lower activity. Background activity was measured using extracts from untransfected cells (green).
Kinase Analysis of LRRK2 GTPase mutants
The kinase activities of wild type, R1441C and K1347A LRRK2 were examined using an in vitro assay for autophosphorylation and for phosphorylation of the generic substrate, MBP (figure 3). As previously reported, the K1347A mutant displayed a decreased autophosphorylation ability compared to wild type and decreased ability to phosphorylate MBP. However, R1441C had activity indistinguishable from wild type in either assay.
Figure 3. Kinase assay of LRRK2 GTPase domain variants.

The kinase activities of wild type (lane 1), K1347A (lane 2) and R1441C (lane 3) LRRK2 were compared to untransfected cells (lane 4) for their ability to autophosphorylate (A, B) or to phosphorylate the generic kinase substrate MBP (C, D). Activity was measured as incorporated 32P into LRRK2 or MBP, corrected for LRRK2 protein in the assays by blotting for V5 or for MBP loading by Ponceau staining and is expressed in arbitrary units (au). Statistical significance was assessed with ANOVA with Newman-Kuells post-hoc tests (n=3; **P<0.01 compared to wild type LRRK2). Background activity for autophosphorylation was not detectable.
Discussion
In this study, the GTP binding and hydrolysis properties of the PD associated protein LRRK2 were examined. In our experiments, immunoprecipitated LRRK2 is capable of both binding and hydrolyzing GTP. We confirmed that the artificial K1347A mutant does not bind to GTP, and showed that it also has a greatly decreased GTPase activity. Ito and colleagues [12] have recently shown that the GTPase activity of LRRK2 is much less than that of ras, as expected from sequence homology, but in apparent contradiction to our results. However, conditions for immunoprecipitation differ slightly between the studies, suggesting that we may have co-preciptated an active GTPase activating protein (GAP) that increases activity in a complex with LRRK2. The low level hydrolysis of GTP in the K1347A mutant in our experiments would be consistent with a copurifying GAP. There is some experimental evidence that LRRK2 can be precipitated from cell lysates as a complex under conditions similar to those used for protein isolation used in this study [7]. Identification of LRRK2 GAPs is an important priority to resolve these difficulties. However, even in these conditions, ras is approximately 10-fold more active than LRRK2 (data not shown), demonstrating that the GTPase activity of LRRK2 is in fact very low and in agreement with previous studies [12].
Under these conditions, R1441C decreases GTPase activity. The R1441C mutation is predicted to lie outside the GTPase binding pocket in a region that is not conserved through the various GTPase families, so it is unlikely that it directly alters the GTPase activity of LRRK2. One mechanism whereby this amino acid substitution could impact GTPase activity is by disrupting an interaction between LRRK2 and a putative GAP or other co-factor. Whichever mechanisms are involved, the fact that R1441C has lower GTPase activity in immune precipitated complexes predicts that this mutation will increase the amount of time that LRRK2 spends in a high affinity, GTP bound state. Although this does not increase kinase activity in two assays, we suggest that it is likely to increase activity towards heterologous substrates, especially if LRRK2 functions as a scaffold for other signaling molecules. We did not find that R1441C increases binding to GTP per se, despite previous suggestions that it might using similar techniques [10]. Although we have validated (using K1347A) that these assays are capable of distinguishing binding versus non-binding variants, whether these techniques are sensitive enough to determine small differences in binding affinity is not yet clear.
In summary, this study represents the first demonstration that LRRK2 possesses GTPase activity. Furthermore, the introduction of a PD familial mutation into the ROC domain of LRRK2 disrupts GTPase activity, representing a mechanism whereby mutations in the ROC domain could exert a pathogenic affect. These data are consistent with evidence that the GTPase domain of LRRK2 is central to the regulation of the protein. To confirm this, investigation of other mutations in and around the ROC domain and their impact on both GTPase activity and the kinase activity of LRRK2 directed towards a physiologically relevant substrate is required. However, our results suggest that both the GTPase and kinase domains of LRRK2 represent tractable therapeutic targets for PD.
Acknowledgments
This work was funded by the Intramural Research Program of the NIH, National Institute on Aging. We would like to thank Dr Craig Blackstone, National Institute of Neurological Diseases and Stroke, for helpful advice on the GTPase assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marin I. The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol. 2006;23:2423–33. doi: 10.1093/molbev/msl114. [DOI] [PubMed] [Google Scholar]
- 2.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood JW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 3.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T, Wood NW. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365:415–6. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- 5.Nichols WC, Pankratz N, Hernandez D, Paisan-Ruiz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, Singleton A, Foroud T. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005;365:410–2. doi: 10.1016/S0140-6736(05)17828-3. [DOI] [PubMed] [Google Scholar]
- 6.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–7. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O′Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–32. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- 8.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–3. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 9.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006 doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 10.West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–32. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 11.Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 12.Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T. GTP Binding Is Essential to the Protein Kinase Activity of LRRK2, a Causative Gene Product for Familial Parkinson’s Disease. Biochemistry. 2007;46:1380–8. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- 13.Korr D, Toschi L, Donner P, Pohlenz HD, Kreft B, Weiss B. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–20. doi: 10.1016/j.cellsig.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Greggio E, Lewis PA, van der Brug MP, Ahmad R, Kaganovich A, Ding J, Beilina A, Baker AK, Cookson MR. Mutations in LRRK2/dardarin associated with Parkinson disease are more toxic than equivalent mutations in the homologous kinase LRRK1. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04523.x. In Press. [DOI] [PubMed] [Google Scholar]