

Summary
A specific complex of 5 S rRNA and several ribosomal proteins is an integral part of ribosomes in all living organisms. Here we studied the importance of Escherichia coli genes rplE, rplR and rplY, encoding 5 S rRNA-binding ribosomal proteins L5, L18 and L25, respectively, for cell growth, viability and translation. Using recombineering to create gene replacements in the E. coli chromosome, it was shown that rplE and rplR are essential for cell viability, whereas cells deleted for rplY are viable, but grow noticeably slower than the parental strain. The slow growth of these L25-defective cells can be stimulated by a plasmid expressing the rplY gene and also by a plasmid bearing the gene for homologous to L25 general stress protein CTC from Bacillus subtilis. The rplY mutant ribosomes are physically normal and contain all ribosomal proteins except L25. The ribosomes from L25-defective and parental cells translate in vitro at the same rate either poly(U) or natural mRNA. The difference observed was that the mutant ribosomes synthesized less natural polypeptide, compared to wild type ribosomes both in vivo and in vitro. We speculate that the defect is at the ribosome recycling step.
Keywords: 5 S rRNA-binding proteins, ribosomal protein L25, ribosome, translation, Escherichia coli
INTRODUCTION
The 5 S rRNA-protein complex, which forms an autonomous structural domain of the ribosome, is essential for ribosome function. During the last 30 years a great body of experimental data has been accumulated on the importance of the complex and its formation as part of an active ribosomal particle. Thus, the total in vitro reconstitution of the 50 S ribosomal subunit performed in the absence of 5 S rRNA led to dramatic consequences:1,2 the ribosomes with such 50 S subunits were unable to synthesize a polypeptide chain and only retained EF-G-dependent GTPase activity. Removing more than one of the 5 S rRNA genes (out of eight) in Escherichia coli resulted in reduced cell growth.3 On the basis of data on cross-linking of the 5 S rRNA and the 23 S rRNA within the ribosome it was suggested that 5 S rRNA might link the peptidyl transferase and GTPase centres.4
Three 5 S rRNA-binding proteins have been found in the E. coli ribosome: L5, L18 and L25.5–7 Later, the E. coli 5S rRNA-binding ribosomal proteins L5 and L18 were shown to be important for incorporation of the 5S rRNA into the 50S ribosomal subunit in vitro.8 Many spontaneous E. coli mutants lacking one or two ribosomal proteins have been found and characterized (see for review).9 However, no mutant lacking any of the 5 S rRNA-binding proteins was found. This was indirect evidence that 5 S rRNA-binding proteins are important for ribosome functioning. Nevertheless, despite extensive in vitro studies, a specific role of the 5 S rRNA-protein complex in the ribosome remains unknown (see for review).10–12
Experiments on the binding of ribosomal proteins to isolated 5 S rRNA have identified two to three 5 S rRNA-binding proteins in Bacteria (homologues of E. coli L5 and L18 are found in all cases), two in Archaea and one in Eukarya (see for review).13 Two archaeal 5 S rRNA-binding proteins appeared to be homologous to E. coli ribosomal proteins L5 and L18.14 A protein homologous to L18 was the only 5 S RNA-binding protein isolated from eukaryal ribosomes.15 However, a gene encoding a protein homologous to the bacterial ribosomal protein L5 was also found in eukaryotic genomes.16 Thus, all three domains of life have the two conserved ribosomal proteins, L5 and L18.
Genes encoding proteins homologous to E. coli L25 were found in Bacteria only.17 At present, the ribosomes having such a protein were isolated and studied from three bacteria: E. coli (protein L25), Thermus thermophilus (protein TL5) and Deinococcus radiodurans (protein DraCTC).5,18,19 These three proteins have homologues in different bacteria that belong to a so-called CTC-family because of their homology to the general stress protein CTC of Bacillus subtilis (BsuCTC).20 The BsuCTC protein also specifically binds 5 S rRNA in vitro,21 it is accumulated in cells only under various stress conditions,22 and can be found in the ribosomal fraction.23 However, in vegetative B. subtilis cells this protein is almost absent.22,24 Although, the genome of another representative of Bacillaceae, Bacillus stearothermophilus, contains a gene encoding CTC protein,25 the protein was not found in ribosomes.5 Moreover, genomes of other bacterial families such as Streptococcaceae, Mycoplasmataceae and also some species of Bacillaceae are seemed to contain no corresponding gene.17,25 This rises a question whether the proteins of the CTC family, including L25, are essential.
In the present work we studied the importance of each of the three E. coli 5 S rRNA-binding proteins, L5, L18 and L25, for cell viability and growth. Our data indicate that knockouts of L5 and L18 genes are lethal, whereas cells lacking the gene for L25 protein are viable, but grow more slowly than the parental strain. Detailed analysis revealed that ribosomes from the rplY knockout contain a full set of ribosomal proteins except L25 and that these ribosomes, although physically normal, were impaired for protein biosynthesis.
RESULTS AND DISCUSSION
Disruption of rplE, rplR and rplY genes in E. coli using recombineering
The E. coli 5 S RNA-binding protein genes rplE, rplR, and rplY (encoding ribosomal proteins L5, L18 and L25, correspondingly), were disrupted by recombineering.26 This technique allows precise replacement of a target chromosomal gene open reading frame by a drug-resistance gene orf (Figure 1(a)) without nonspecific polar effects on the expression of genes flanking the replaced gene.27,28 Each of the three 5 S rRNA-binding protein genes has been inactivated by precisely replacing their orfs with the orf of chloramphenicol acetyltransferase cassette, cat (Figure 1). After recombination (described in Materials and Methods), two types of recombinants distinguished by the level of recombination were found. For one type, as represented by the rplY<>cat replacement, a high recombination efficiency was observed (~104 colonies/108 viable cells) similar to what is found for knocking out nonessential genes in E. coli.28 The PCR analysis of the rplY chromosomal region in recombinant cells confirmed the rplY<>cat replacement in numerous slow-growing CmR colonies (Figure 1(b), Table 1). This recombineering phenotype features a growth-impaired gene knockout (see Materials and Methods).
Figure 1.

(a) Recombineering, the strategy applied for in frame deletion of E. coli chromosomal genes. Chromosomal gene orf is replaced by a drug-resistance PCR cassette during recombination utilizing short (39–40nt) homologies (shown with striped boxes). Open arrows indicate position of checking primers used to verify gene replacements by PCR. (b) Gene essentiality assay. Nonessential gene such as rplY can readily be replaced with a drug-resistance cassette using recombineering (see scheme, plate and gel on the left column). In the case of rplY knockout, a few normally growing rplY<>cat/rplY+ diploids are seen among thousands of growth-impaired rplY<>cat knockouts on the LB-Cm plate. Only cat insert-related PCR product is synthesized when rplY configuration is tested in rplY<>cat CmR recombinants. At the same time, only gene orf<>cat orf/gene orf+ gene partial diploids are viable when essential gene such as rplE or rplR is replaced (right column: scheme, plate and gels). In these cases only rare gene<>cat/gene+ diploids survive on the selective LB-Cm plate. Agarose gels below the plate show that in the case of rplE and rplR gene replacements both, cat-insert and wild type gene-related PCR products are synthesized.
Table 1.
Results of gene knockouts
| Target | Type of knockout | Recombination efficiency a | Configuration of replaced gene b |
|---|---|---|---|
| rplY | rplY<>cat | ~104 | rplY<>cat |
| rplE | rplE<>cat | ~10 | rplE<>cat/rplE+ |
| rplE c | rplE<>cat | ~102 | rplE<>cat |
| rplR | rplR<>cat | ~10 | rplR<>cat/rplR+ |
Recombination efficiency was calculated as the number of CmR recombinant. colonies per 108 viable cells.
Determined by PCR analysis of chromosomal region with cat-replaced gene.
Recombination was done in the presence of the rplE-expressing plasmid.
For the second type, as represented by rplR<>cat and rplE<>cat replacements, only ~ 10 CmR colonies per 108 viable cells were observed. By PCR analysis, these recombinants carried the rplR<>cat and rplE<>cat replacements, but also contained the intact rplR or rplE allele, respectively (Figure 1(b), right panel). It is known that subpopulations within E. coli cultures contain cells with large regions of the chromosome duplicated.29 Such partial diploid cells occur at a frequency ~10−2 for any one region of the chromosome, and we have previously shown that essential genes in such diploid regions are targeted for gene replacement.27,28 Thus, two of the three 5 S rRNA-binding proteins, L5 and L18, appear to be essential.
By providing a cloned open reading frame of the rplE gene on an expression plasmid and repeating the recombination to replace the chromosomal rplE with cat (Figure 2), we found a higher recombination level and all recombinants tested by PCR analysis contained only the cat-replaced copy of rplE (Table 1). This confirmed that rplE was essential and also demonstrated that the spc operon control element,30 which is located within the 5’ proximal region of rplE gene (Figure 2(a)) is not essential.
Figure 2.

(a) The E. coli spc operon with rplE and rplR genes. The hairpin needed for feedback regulation of the operon is indicated with an asterisk. (b) The replacement of rplE orf with cat was made either in the absence or presence of an rplE-expressing plasmid. Only rplE<>cat/rplE+ diploids survive in the absence of the plasmid (left). The rplE<>cat haploids survive if cells carry an L5-expressing plasmid, showing that rplE is indeed essential while regulatory hairpin is not (right). (c) Agarose gel showing the configuration of rplE in viable CmR recombinants in the presence (+) or absence (−) of an rplE-expressing plasmid, as analyzed by PCR of rplE chromosomal region.
It has been shown that ribosomes lacking 5 S rRNA are unable to synthesize polypeptide.1,2 Moreover, the E. coli 5 S rRNA-binding ribosomal proteins L5 and L18 were shown to be important for incorporation of the 5 S rRNA into the 50 S ribosomal subunit in vitro.8 Our finding that E. coli L5 and L18 are essential for cell viability is in good agreement with those in vitro experiments. Most likely, these two ribosomal proteins, which are found in all three domains of life, are indispensable for in vivo assembly of the active ribosome. The third E. coli 5 S rRNA-binding ribosomal protein, L25, and its homologues are found only in bacteria and not even in all bacteria.17,25 Our experiments show that L25 is dispensable for viability, but nevertheless is required for normal E. coli cell growth.
Growth of rplY knockout and in vivo analysis of L25 function
It was observed that the rplY knockout grew but more slowly than the parental W3110 cells over a wide range of temperatures, from 20 to 42ºC. Under standard conditions of growth (LB broth, 37ºC), the generation time for these L25-defective cells was approximately 72 minutes while it was 32 minutes for W3110 (Table 2).
Table 2.
The growth rates of the parental strain (W3110) and rplY knockout (KNB800) alone and with plasmids constitutively expressing E. coli L25 (pKAB101), B. subtilis CTC (pKAB110) or N-terminal fragment of CTC protein (pKAB104)
| Strain (plasmid) | Protein expressed from the plasmid | Generation time, min |
|---|---|---|
| W3110 | - | 32 |
| KNB800 | - | 72 |
| W3110 (control plasmid) | - | 39 |
| KNB800 (control plasmid) | - | 120 |
| KNB800 (pKAB101) | L25 | 46 |
| KNB800 (pKAB104) | NfrCTC | 54 |
| KNB800 (pKAB110) | CTC | 54 |
Measured in LB medium at 37ºC by monitoring the optical density at 600 nm (cells transformed with plasmids were grown in the presence of kanamycin).
Growth of the rplY knockout was complemented by the expression of the plasmid-cloned rplY gene (Figure 3, Table 2). This shows that the growth defect of the rplY knockout is specifically caused by the absence of L25 function, which for the most part can be restored by in-trans expression of L25.
Figure 3.

Growth of wild type W3110 (WT) and L25-defective KNB800 (ΔL25) strains in the presence of plasmids providing expression of E. coli rplY for L25 protein (pKAB101), B. subtilis gene ctc for CTC protein (pKAB110) and 5’-terminal portion of ctc encoding for N-terminal fragment (NfrCTC) of CTC protein (pKAB104). WT + control plasmid (1), ΔL25 + control plasmid (2), ΔL25 + L25-expressing plasmid (3), ΔL25 + NfrCTC-expressing plasmid (4), ΔL25 + CTC-expressing plasmid (5). Growth medium was supplemented with kanamycin. Incubation at 37°C for 16 hours.
Interestingly, we have also been able to partially complement the growth defect of the rplY knockout by in-trans expression of B. subtilis general stress protein CTC. It has been shown before, that the L25 protein is homologous to the N-terminal domain of CTC protein of B. subtilis.20 The CTC protein specifically binds to bacterial 5 S rRNA21 and can be found in the ribosomal fraction of B. subtilis cells.23 The expression of either the entire CTC protein or its N-terminal domain stimulated the growth of the knockout mutant, albeit less efficiently than the E. coli L25 protein (Figure 3, Table 2). These data show for the first time that the general stress protein CTC of B. subtilis is a functional homologue of E. coli ribosomal protein L25.
L25 is a ribosomal protein, which, due to its functional homology to the general stress protein CTC of B. subtilis, might also be involved with the stress response of the cell. Both L25 activities, as a ribosomal protein and a potential stress protein, have been analyzed in vivo in this work.
The potential stress functions of L25 have been analyzed by growing the rplY knockout under a variety of stressful conditions for cell growth: cold, heat shock and oxidative stress. As these stressful conditions were shown to induce the synthesis of the CTC protein in B. subtilis cells22 one might expect that E. coli L25-defective cells would become less resistant to such growth conditions compared to the parental strain. It turned out, that there were no significant differences between the rplY knockout and the parent cells in response to any of these stressful conditions. Hence, it is likely, that L25 is not involved with a cell stress response, at least in E. coli.
We measured the in vivo protein-synthesizing ability of the rplY knockout, using the standard β-galactosidase assay. The rplY knockout accumulated β-galactosidase approximately 5 times slower than the parent strain (Figure 4). At the same time as expected, there was no defect noticed in the rplY knockout regarding the major metabolic pathways: the basic metabolic microarray analysis31 did not reveal a difference between the mutant and the wild type cells (data not shown).
Figure 4.

Protein-synthesizing activity of wild type (●) and ΔL25 (▲) cells determined by standard β-galactosidase assay. The β-galactosidase activity is normalized to the cell mass (OD600).
Thus, the lack of the ribosomal protein L25 decreases the protein-synthesizing capacity of the cell, which has been the only difference between L25 knockout and parental cells observed in vivo in our tests. We believe that this is the main reason for the slow growth of the L25-defective cells.
Analysis of L25-defective ribosomes
Ribosomes from the L25-defective strain were analyzed by sucrose density gradient centrifugation. The ribosomes retained compactness comparable to that of the wild-type ribosomes (Figure 5(a)). L25-defective 50 S ribosomal subunits were able to associate normally with 30 S ribosomal subunit to form a 70 S particle. Protein content of the mutant ribosomes was analyzed by two-dimensional gel-electrophoresis. Except for L25 protein, all other individual ribosomal proteins are present in 70 S ribosomes and found in similar amounts as in the wild-type ribosomes (Figure 5(b)). Thus, L25 is not an “assembly” protein and does not influence the incorporation of other proteins into the large ribosomal subunit. So, our data suggest that the ribosomes lacking L25 are physically normal, and L25 most probably is not required for either 50 S or 70 S ribosome formation.
Figure 5.
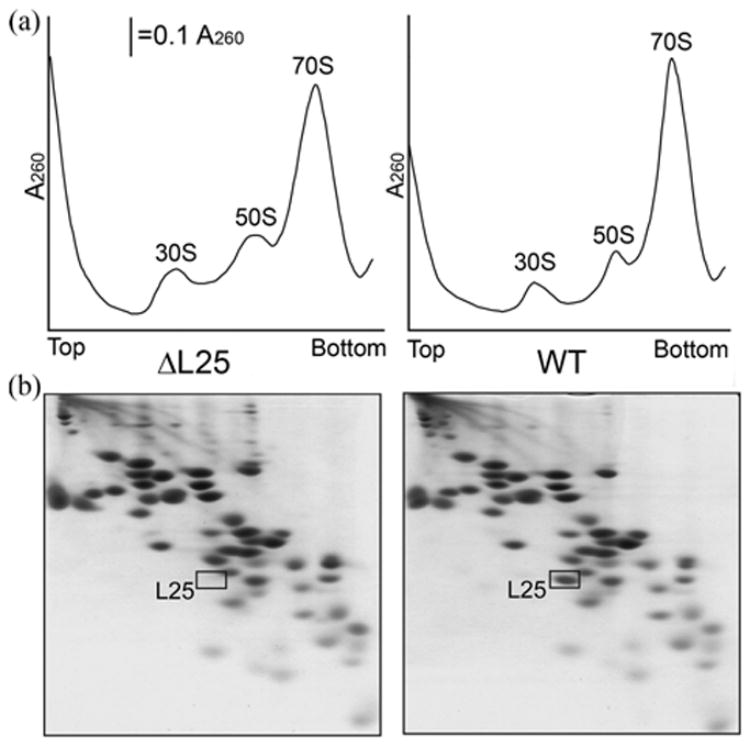
(a) A sucrose gradient analysis of ribosomes from L25-defective (ΔL25, left panel) and wild type (WT, right panel) cells. Crude cell lysates were fractionated by centrifugation on 15–30% sucrose gradients. (b) Two-dimensional polyacrylamide gel analysis of protein content of L25-defective (left panel) and wild type (right panel) ribosomes.
Protein-synthetic activity of L25-defective ribosomes
Two approaches were applied to analyze the protein-synthesizing activity of L25-defective ribosomes in vitro. First, translation of poly(U) template, which measures the rate of elongation, and second, translation of a natural mRNA, which involves an initiation, termination and ribosome recycling, as well as the elongation step of translation.
Ribosomes from both the L25-defective and wild type cells were able to translate poly(U) template at virtually the same rate (Figure 6). The data show that both, L25 mutant and wild type ribosomes perform polypeptide chain elongation at the same efficiency. Next, we performed in vitro translation experiments using natural mRNAs for green fluorescent protein (GFP, Figure 7(a)) and luciferase (Figure 7(b)). In these experiments, the luciferase activity began to appear at the same time point (approximately 3 min) in reaction mixtures, containing either the L25-defective or wild type ribosomes. Since luciferase becomes active only if it is completely synthesized and released from the ribosome.32, we may conclude, that the entire process of natural mRNA translation (i.e., initiation, elongation, and termination) proceeds at the same rate with the wild type and L25 mutant ribosomes. The only difference observed between the wild type and the mutant ribosomes was in the amount of synthesized protein: L25-defective ribosomes produced 1.5 to 2 times lower levels of both the luciferase and GFP proteins than the wild type ribosomes. This result is in good agreement with our in vivo data comparing β-galactosidase translation levels (Figure 4).
Figure 6.

Poly(U)-dependent polyphenylalanine synthesis by wild type (●) and L25-defective (▲) ribosomes.
Figure 7.

Time course of GFP (a) and luciferase (b) mRNA translation in E. coli cell-free translation system containing wild type (WT, ●) and L25-defective (ΔL25, ▲) ribosomes. In case of GFP synthesis, an aliquot was taken at 30 min time point from the wild type and L25-defective systems and analyzed by SDS-PAGE with subsequent autoradiography (right panel of section (a)).
One possible reason for the low yield of translation products in the L25-defective cells might be translation errors, leading to accumulation of inactive protein or abortive peptides. Monitoring the ratios of the active GFP (by fluorescence) to the total GFP polypeptide (by [14C]Leu incorporation) during linear synthesis (6–30 min) shows that the ratios for the mutant and wild ribosomes coincided. This means that specific activity of GFP synthesized in both cases is the same. It is seen in Figure 7(a) (right panel) that the majority of synthesized polypeptide is represented by the full-length GFP and that the distribution of translation products synthesized by the mutant and wild type ribosomes is the same. The main conclusion from these in vitro translation tests is that L25-defective ribosomes are able to synthesize active full-length protein with a rate similar to that of the wild type ribosomes.
Is the ribosome recycling step defective in the L25 mutant?
It is interesting that results similar to ours were obtained for an E. coli mutant having a thermosensitive ribosome recycling factor, RRF.33 When this RRF mutant is grown at non-permissive temperatures, decreasing the level of RRF, the protein synthesis in vivo is depressed and in vitro translation of natural mRNA by ribosomal fraction from such cells is reduced. However, poly(U) translation by the RRF mutant ribosomal fraction was unaltered; the poly(U) assay does not require ribosome recycling and only measures elongation rate.
It is commonly recognized that after protein release, 70S ribosomes normally dissociate into subunits and are freed from ligands (see for review).34, 35 This ribosome recycling provides the bacterial cell with 30S and 50S ribosomal subunits competent for initiating the synthesis of new protein molecules.36 Thus, defective ribosome recycling should decrease the productivity of protein synthesis. At this point, we have no direct evidence that L25-defective ribosomes are impaired in the recycling process. However, on the basis of the above-mentioned data we hypothesize that the defect in protein synthesis in cells lacking L25 might be at ribosome recycling at the post-termination stage.
MATERIALS AND METHODS
Strains, plasmids and bacteriological techniques
The strains and plasmids used in this work are listed in table S1. Standard techniques for P1 transduction and β-galactosidase assay were used.39 For β-galactosidase assay cells were grown to OD600=0.2 and production of the enzyme was induced by adding isopropyl-β-D- thiogalactoside to 1 mM concentration. The enzyme accumulation in the culture was monitored using color reaction of 2-nitrophenyl-β-D-galactopyranoside conversion. Enzyme activity was then normalized to the cell mass (OD600). Cells were grown in LB medium supplemented with 50μg/ml kanamycin (Kan) or 10 μg/ml chloramphenicol (Cm) when needed.
Inactivation of E. coli rplE, rplR and rplY genes
The E. coli genes rplE, rplR and rplY, were inactivated by recombineering28 (Figure 1(a)). Recombineering is based on the λ red-dependent recombination of DNA substrates into the chromosomes or plasmids of enteric bacteria40 using short 40–50nt flanking homologies. Briefly, E. coli DY330 cells were induced for Red functions at A600 ~ 0.5 and prepared for electroporation with 300 ng of appropriate linear DNA cassette. The cells were recovered in 0.9 ml LB, grown for 2–3 h at 32ºC, spread on LB agar plates supplemented with Cm and incubated for 1–3 days at 32–34ºC to reveal Cm-resistant recombinants. The cat cassette for gene disruption was made by PCR amplification of cat orf (originated from pCR-Script Cam (Stratagene) using Platinum Taq DNA Polymerase High Fidelity Kit (Invitrogen) with primers (Table S2) that contained a 39–40nt sequence homologous to chromosomal regions surrounding the gene orf targeted for disruption followed by a 22–25nt priming sequence complementary to the 5' and 3' regions of the drug orf. The resulting cassettes should have a cat orf flanked by the 39–40nt regions of homology (Figure 1) that are needed for its recombination into chromosomal regions of the respective gene, so that the chromosomal orf is replaced with the drug orf.
For rplE replacement, the cat cassette was made using primers, rplE-cat-F and rplE-cat-R. For rplR replacement, the cat cassette was made using primers, rplR-cat-F, rplR-cat-R. To replace rplY, the cat cassette was made using primers, rplY-cat-F and rplY-cat-R.
Analysis of gene replacements
Recombinant colonies were purified on LB-Cm plates at 34ºC. The individual colonies from the streaks were suspended in 30μl of sterile water and 1 μl used as a template for PCR with two check primers flanking the target gene (Table S2, Figure 1(a)). For rplE<>cat, the verifying forward primer was rplE-check-F, and reverse primer was rplE-check-R; to verify rplR<>cat replacement the pair of primers used was rplR-check-F and rplR-check-R; for examination of rplY<>cat replacement those were rplY-check-F and rplY-check-R.
Cloning genes by gap repair recombination
The E. coli rplE and rplY genes were cloned by gene retrieval to a pCR-Blunt plasmid (Invitrogen) (generating pKAB122 and pKAB101 plasmids, respectively) from the chromosome using the gap repair41 version of recombineering.28 The linear pCR plasmid, including kan and the origin, was amplified by PCR with primers bearing 39–40-nucleotide extensions homologous to the very 5'-and 3'-coding regions of the genes to be cloned (Table S2). This should allow the cloned orfs to start directly from the AUG initiation codon of the lacZ gene present in the pCR plasmid, thereby acquiring the promoter and translation elements of lacZ. Retrieval was carried out in E. coli strain DY330 by recombineering as described above. The desired plasmids were selected as KanR and were identified by restriction analysis of the insert and verified by DNA sequencing. The pairs of primers for rplE and rplY cloning were rplE-pCR-F with rplE-pCR-R and rplY-pCR-F with rplY-pCR-R, respectively.
We cloned the entire ctc gene, as well as its 5'-portion encoding N-terminal domain (1–94 amino acids) into pCR-Blunt by recombineering (plasmids pKAB110 and pKAB104, respectively). In this case, the linear pCR-Blunt was used directly from the company and coelectroporated with linear PCR products of the ctc gene fragments carrying flanking homologies to PCR-Blunt so as to allow end-joining of the linear vector in DY330. The pairs of primers for the entire ctc gene or its 5'-portion were ctc-pCR-F and ctc-pCR-R or ctc-pCR-F and ctcN-pCR-R, respectively. Cells were outgrown after electroporation in LB broth for 2 hrs at 30ºC, spread on LB plates supplemented with Kan and incubated at 34ºC.
Gene essentiality assay
Recombineering can be used for analysis of gene essentiality via direct selection27 (Figure 1). Briefly, the assay is based on the high efficiency of recombineering that allows selection for gene knockouts when the essential gene region in the chromosome is duplicated.27,28 These duplications occur with a frequency ~10−2 in a cell population.29 When an essential gene is replaced during recombineering (Figure 1(b)), only these rare partial diploids survive, reflecting the lowered recombination frequency (<102/108 viable cells). The duplication is maintained in these cells by positively selecting for the drug marker in the replaced essential gene and the functional wild type allele, gene orf<>drug orf/gene orf+(Figure 1(b), right column). Nonessential genes are replaced with standard high recombination efficiency (~104/108 viable cells) generating only haploid gene orf<>drug orf knockouts (Figure 1(b), left column). Consequently, three cell phenotypes can be detected featuring nonessential, essential and growth impaired gene knockouts. The essential and nonessential gene phenotypes are described above. They are characterized by the low and high recombination frequencies, as well as by the partial diploid and the haploid configurations of the replaced genes, respectively. The growth impaired cell gene knockout is represented by a few normally growing colonies amongst thousands of slow growing ones. The rarer normal colonies are duplicated for the replaced gene, whereas the small growth-impaired colonies contain just a single disrupted gene (Figure 1(b), left column).
To analyze the essentiality of the rplE, rplR, and rplY genes, their chromosomal orfs were replaced with a cat gene orf by recombineering, as described above and recombination frequency was scored. The diploid/haploid configuration of replaced genes was determined by the PCR analysis of the targeted chromosomal regions, as above.
Phenotypic analysis of rplY knockout
The rplY<>cat knockout was P1 transduced from KNB212 to W3110 selecting for CmR colonies at 37ºC. The resulting KNB800 strain contains a precise rplY orf deletion in the otherwise wild type background. This was confirmed again by PCR analysis of the KNB800 cells followed by 2-D electrophoretic analysis of the ribosomal proteins.
The growth of the rplY knockout was analyzed on LB and M63 agar plates without antibiotic in the 30–42ºC range of temperatures. The growth of cells under cold shock conditions was analyzed on LB plates at 15ºC for 10 days. The heat shock growth of cells was analyzed at 46ºC for 2 days. For oxidative stress, the cells were incubated in LB broth with 1–2 mM H202 at 37ºC, measuring their growth rate at A600, as recommended.39 The phenotypic microarray analysis31 for major metabolic functions was done as recommended by the manufacturer (Biolog, Inc.), growing cells in the respective microtiter plates at 37ºC. The parent wild type cells were always used as a control in these experiments.
Growth complementation of rplY knockout with plasmids expressing L25 or CTC
L25-defective cells were transformed by electroporation with plasmids expressing E. coli L25 (pKAB101), entire B. subtilis CTC protein or its N-terminal fragment (pKAB110 and pKAB104, respectively) and grown at 37ºC on LB plates containing Kan. Individual colonies were streaked on the LB-Kan plates and incubated at 30, 37 and 42ºC along with control cells, which were wild type and L25-defective strains carrying a blank pCR-Blunt vector (control plasmid). For estimating the growth rate, overnight cultures were obtained from individual colonies, diluted 1/50 into LB broth with Kan and grown at 37ºC with shaking at 220 rpm.
Preparation of ribosomes
Ribosomes were obtained according to published procedure42 with modifications. E. coli cells were grown on LB medium at 37°C in an orbital shaker at 200 rpm to A600 ~ 1 o.u., chilled on ice and collected by centrifugation at 14,000 x g and 4°C for 7 min. Cell pellet was resuspended in 1/4 of initial volume of cold buffer (10 mM Tris-HCl (pH 7.5), 200 mM NH4Cl), centrifuged and kept at −70°C if needed. Cells were disrupted by grinding with Al2O3 using 1 ml of cold buffer A (10 mM Tris-HCl (pH 7.5), 200 mM NH4Cl, 10 mM MgCl2, 3 mM β-mercaptoethanol) and 2 g of Al2O3 per g of cells (wet weight) at 4°C. The ground paste was then suspended in buffer A (~ 2 ml for each gram of cell paste). The cell debris and alumina were removed by double centrifugation at 15,000 × g and 4°C for 30 minutes. The resulting supernatant was loaded over 30% (w/w) sucrose cushion made in buffer A, containing 500 mM NH4Cl and centrifuged in a TL-100 ultracentrifuge (Beckman-Coulter, USA) in a TLA-100.3 rotor at 380,000 x g for 3 h at 4°C. Ribosomes were dissolved in cold buffer A containing 50 mM NH4Cl plus 10% glycerol to a approximate concentration of 20 mg/ml, frozen in liquid nitrogen and kept at −70°C.
Analysis of ribosomes and ribosomal proteins
The distribution of ribosomes and ribosomal particles in logarithmically growing cells was analyzed by sucrose gradient centrifugation as follows: cells were grown and harvested as described above. Cell pellet was suspended in 1 volume of cold PS buffer (20 mM HEPES-KOH (pH 7.5), 100 mM KCl, 10 mM MgCl2) and harvested again. Cells were then resuspended in 1/25 volume of cold PS buffer, containing 5 mM β-mercaptoethanol, 0.2 mM EDTA and 1 mg/ml lysozyme, put on ice for 10 minutes and then frozen at −70°C. After defrosting, debris was removed by centrifugation at 15,000 x g at 4°C for 10 min. 3 o.u. at 260 nm of supernatant (approximately 0.1 ml) were loaded onto a 15–30% (w/v) sucrose gradient in PS buffer, containing 5 mM β-mercaptoethanol, 0.2 mM EDTA. The gradients were centrifuged at 24900 rpm (Beckman SW-41 rotor) and 4°C for 14 h.
Proteins were isolated from 300 μg of 70 S ribosomes by acetic-acid extraction43 and fractionated by two-dimensional gel electrophoresis44 using acidic-acidic system IV. The gels were stained with Coomassie blue G250.
Preparation of ribosome-free extract and total enzyme fraction
To obtain the total enzyme fraction for cell-free translation of poly(U), the S100 extract was isolated from E. coli MRE600 strain and fractionated on DEAE-cellulose as described.45
To obtain ribosome-free S100 extract E. coli W3110 cells were grown and collected. The cell pellet was suspended in 1/10 of original culture volume of cold buffer A, containing 150 mM NH4Cl plus 0.1 mM EDTA, and centrifuged for another 20 min. The cells were resuspended in the same buffer (~1 ml for each gram of cells) and disrupted by ultrasonic treatment. Cell debris was removed by two centrifugations at 15,000 x g at 4°C for 15 min. The supernatant was supplemented with NH4Cl to final concentration of 500 mM, loaded on a sucrose cushion and centrifuged for 1 h as described in “Prepation of ribosomes”. The ribosome-free extract was then dialyzed against buffer (10 mM Tris-acetate (pH 8.2), 100 mM potassium acetate, 10 mM magnesium acetate, 3 mM β-mercaptoethanol, 0.1 mM EDTA), frozen in liquid nitrogen and kept at −70°C.
Cell-free translation of poly(U) (polyuridylic acid) and natural mRNA
Cell-free poly(U)-dependent synthesis of polyphenylalanine was carried out as described.46 10 pmol of 70 S ribosomes, 80 μg of total enzyme fraction protein, 70 μg of poly(U) (Sigma), 50 μg of total tRNA E. coli MRE600 (Sigma), 3 mM ATP (Sigma), 0.4 mM GTP (Sigma) and 500 pmol of [14C]Phe (450 mCi/mmol, Amersham) were incubated in 100 μl of buffer (20 mM Tris- HCl (pH 7.5), 100 mM NH4Cl, 10 mM MgCl2, 3 mM β-mercaptoethanol, 0.1 mM EDTA) at 37°C. The reaction was terminated by applying the mixture to a 3MM filter paper (Whatman) treated with 5% trichloroacetic acid (TCA). The filters were boiled for 5 min in 5% TCA, washed with acetone, dried at room temperature and counted for 14C in Supersolve X scintillation liquid (Koch-Light LTD) in a scintillation counter Beckman LS 1801 (Beckman).
Aequorea victoria green fluorescent protein (GFP cycle 3 mutant47) and Photinus pyralis luciferase mRNAs were synthesized in vitro using T7 RNA polymerase48 from pMGFP38 and pT7 luc32 plasmids, respectively. A published system32 was followed for in vitro translation of natural mRNAs.
30 pmol of 70 S ribosomes, 11 μl of ribosome-free S100 extract, 25 μg of total tRNA E. coli MRE600, 1.2 mM ATP, 0.8 mM GTP, 0.4 mM each L-amino acid (or except for Leu in case of GFP mRNA translation, when 0.225 mM Leu and 7.5 μM [14C]Leu were added), 0.1 mg/ml folinic acid, 0.02 mg/ml pyruvate kinase, 20 mM phosphoenolpyruvic acid and 20 mM acetyl phosphate were incubated in 50 μl of buffer (100 mM HEPES-KOH (pH 7.5), 60 mM potassium acetate, 13 mM magnesium acetate, 2% PEG 8000, 2 mM DTT, 0.1 mM EDTA) at 30°C for 5 min, then 5 μg of mRNA (and luciferin to 0.1 mM concentration in case of luciferase mRNA translation) were added at start point. The synthesis was carried out at 30°C.
Light emission by luciferase was measured in a thermostatic cell using a luminometer (Pribori Oy, Finland). In case of GFP mRNA translation, 3 μl aliquots were taken from the reaction at each time point, 50 fold diluted with cold buffer (20 mM Tris-HCl (pH 7.5), 100 mM KCl, 5 mM MgCl2) and incubated for 5 h at 0°C then fluorescence was measured using spectrofluorimeter (Shimadzu RF-5301PC, Japan). In parallel, 5 μl of the reaction were taken for measuring of [14C]Leu incorporation by TCA precipitation (as described above for poly(U) translation) and for analyzing protein size by SDS-PAGE with subsequent radio autography with MultiPurpose screen (MP) and CycloneTM phosphor imager (Packard Instruments, USA).
Supplementary Material
Table S1. E. coli strains and plasmids used in the work
Table S2. Primers used for PCR
Acknowledgments
We thank Vladimir Shirokov for helpful advice on cell-free translation system and Nina Bubunenko for her help in preparation of purified DNA oligonucleotides. This work was supported by the Russian Academy of Sciences, the Russian Foundation for Basic Researches and the Program for support of Outstanding Scientific Schools. The research of M.B.G. was supported by the Program for Molecular and Cellular Biology RAS and by International Research Scholar’s award from the Howard Hughes Medical Institute. This Research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and by a Trans NIH/FDA Intramural Biodefense Program Grant from NIAID (D.L.C.). This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dohme F, Nierhaus KH. Role of 5S RNA in assembly and function of the 50S subunit from Escherichia coli. Proc Natl Acad Sci USA. 1976;73:2221–2225. doi: 10.1073/pnas.73.7.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Erdmann VA, Fahnestock S, Higo K, Nomura M. Role of 5S RNA in the functions of 50S ribosomal subunits. Proc Natl Acad Sci USA. 1971;68:2932–2936. doi: 10.1073/pnas.68.12.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ammons D, Rampersad J, Fox GE. 5S rRNA gene deletions cause an unexpectedly high fitness loss in Escherichia coli. Nucl Acids Res. 1999;27:637–642. doi: 10.1093/nar/27.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dokudovskaya S, Dontsova O, Shpanchenko O, Bogdanov A, Brimacombe R. Loop IV of 5S ribosomal RNA has contacts both to domain II and to domain V of the 23S RNA. RNA. 1996;2:146–152. [PMC free article] [PubMed] [Google Scholar]
- 5.Horne JR, Erdmann VA. Isolation and characterization of 5S RNA-protein complexes from Bacillus stearothermophilus and Escherichia coli ribosomes. Mol Gen Genet. 1972;119:337–344. doi: 10.1007/BF00272091. [DOI] [PubMed] [Google Scholar]
- 6.Chen-Schmeisser U, Garrett RA. A new method for the isolation of a 5 S RNA complex with proteins L5, L18 and L25 from Escherichia coli ribosomes. FEBS Letters. 1977;74:287–291. doi: 10.1016/0014-5793(77)80866-1. [DOI] [PubMed] [Google Scholar]
- 7.Spierer P, Zimmermann RA. Stoichiometry, cooperativity, and stability of interactions between 5S RNA and proteins L5, L18, and L25 from the 50S ribosomal subunit of Escherichia coli. Biochemistry. 1978;17:2474–2479. doi: 10.1021/bi00606a002. [DOI] [PubMed] [Google Scholar]
- 8.Rohl R, Nierhaus KH. Assembly map of the large subunit (50S) of Escherichia coli ribosomes. Proc Natl Acad Sci USA. 1982;79:729–733. doi: 10.1073/pnas.79.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dabbs ER. Mutants lacking individual ribosomal proteins as a tool to investigate ribosomal properties. Biochimie. 1991;73:639–645. doi: 10.1016/0300-9084(91)90043-z. [DOI] [PubMed] [Google Scholar]
- 10.Bogdanov AA, Dontsova OA, Dokudovskaya SS, Lavrik IN. Structure and function of 5S rRNA in the ribosome. Biochem Cell Biol. 1995;73:869–876. doi: 10.1139/o95-094. [DOI] [PubMed] [Google Scholar]
- 11.Moore PB. The structure and function of 5S ribosomal RNA. In: Zimmermann RA, Dahlberg AE, editors. Ribosomal RNA: structure, evolution, processing and function in protein biosynthesis. CRC Press; Boca Raton, New York, London, Tokyo: 1996. pp. 199–236. [Google Scholar]
- 12.Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J. 5 S rRNA: structure and interactions. Biochem J. 2003;371:641–651. doi: 10.1042/BJ20020872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barciszewska MZ, Erdmann VA, Barciszewski J. Ribosomal 5S RNA: tertiary structure and interactions with proteins. Biol Rev Camb Philos Soc. 1996;71:1–25. doi: 10.1111/j.1469-185x.1996.tb00740.x. [DOI] [PubMed] [Google Scholar]
- 14.McDougall J, Wittmann-Liebold B. Comparative analysis of the protein components from 5S rRNA.protein complexes of halophilic archaebacteria. Eur J Biochem. 1994;221:779–785. doi: 10.1111/j.1432-1033.1994.tb18791.x. [DOI] [PubMed] [Google Scholar]
- 15.Tang BZ, Nazar RN. Structure of the yeast ribosomal 5 S RNA-binding protein YL3. J Biol Chem. 1991;266:6120–6123. [PubMed] [Google Scholar]
- 16.Jahn O, Hartmann RK, Boeckh T, Erdmann VA. Comparative analysis of ribosomal protein L5 sequences from bacteria of the genus Thermus. Biochimie. 1991;73:669–678. doi: 10.1016/0300-9084(91)90046-4. [DOI] [PubMed] [Google Scholar]
- 17.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucl Acids Res. 2004;32:D23–26. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gongadze GM, Tishchenko SV, Sedelnikova SE, Garber MB. Ribosomal proteins, TL4 and TL5, from Thermus thermophilus form hybrid complexes with 5 S ribosomal RNA from different microorganisms. FEBS Letters. 1993;330:46–48. doi: 10.1016/0014-5793(93)80916-i. [DOI] [PubMed] [Google Scholar]
- 19.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, et al. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001;107:679–688. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 20.Gryaznova OI, Davydova NL, Gongadze GM, Jonsson BH, Garber MB, Liljas A. A ribosomal protein from Thermus thermophilus is homologous to a general shock protein. Biochimie. 1996;78:915–919. doi: 10.1016/s0300-9084(97)86713-2. [DOI] [PubMed] [Google Scholar]
- 21.Korepanov AP, Gongadze GM, Garber MB. General stress protein CTC from Bacillus subtilis specifically binds to ribosomal 5S RNA. Biochemistry (Moscow) 2004;69:607–611. doi: 10.1023/b:biry.0000033733.60180.e3. [DOI] [PubMed] [Google Scholar]
- 22.Volker U, Engelmann S, Maul B, Riethdorf S, Volker A, Schmid R, et al. Analysis of the induction of general stress proteins of Bacillus subtilis. Microbiology. 1994;140 (Pt 4):741–752. doi: 10.1099/00221287-140-4-741. [DOI] [PubMed] [Google Scholar]
- 23.Schmalisch M, Langbein I, Stulke J. The general stress protein Ctc of Bacillus subtilis is a ribosomal protein. J Mol Microbiol Biotechnol. 2002;4:495–501. [PubMed] [Google Scholar]
- 24.Hecker M, Heim C, Volker U, Wolfel L. Induction of stress proteins by sodium chloride treatment in Bacillus subtilis. Arch Microbiol. 1988;150:564–566. doi: 10.1007/BF00408250. [DOI] [PubMed] [Google Scholar]
- 25.Frishman D, Mokrejs M, Kosykh D, Kastenmuller G, Kolesov G, Zubrzycki I, et al. The PEDANT genome database. Nucl Acids Res. 2003;31:207–211. doi: 10.1093/nar/gkg005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu Rev Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 27.Knowlton JR, Bubunenko M, Andrykovitch M, Guo W, Routzahn KM, Waugh DS, et al. A spring-loaded state of NusG in its functional cycle is suggested by X-ray crystallography and supported by site-directed mutants. Biochemistry. 2003;42:2275–2281. doi: 10.1021/bi0272508. [DOI] [PubMed] [Google Scholar]
- 28.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lupski JR, Roth JR, Weinstock GM. Chromosomal duplications in bacteria, fruit flies, and humans. Am J Hum Genet. 1996;58:21–27. [PMC free article] [PubMed] [Google Scholar]
- 30.Cerretti DP, Mattheakis LC, Kearney KR, Vu L, Nomura M. Translational regulation of the spc operon in Escherichia coli. Identification and structural analysis of the target site for S8 repressor protein. J Mol Biol. 1988;204:309–329. doi: 10.1016/0022-2836(88)90578-5. [DOI] [PubMed] [Google Scholar]
- 31.Bochner BR, Gadzinski P, Panomitros E. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 2001;11:1246–1255. doi: 10.1101/gr.186501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolb VA, Makeyev EV, Spirin AS. Co-translational folding of an eukaryotic multidomain protein in a prokaryotic translation system. J Biol Chem. 2000;275:16597–16601. doi: 10.1074/jbc.M002030200. [DOI] [PubMed] [Google Scholar]
- 33.Hirokawa G, Inokuchi H, Kaji H, Igarashi K, Kaji A. In vivo effect of inactivation of ribosome recycling factor - fate of ribosomes after unscheduled translation downstream of open reading frame. Mol Microbiol. 2004;54:1011–1021. doi: 10.1111/j.1365-2958.2004.04324.x. [DOI] [PubMed] [Google Scholar]
- 34.Janosi L, Ricker R, Kaji A. Dual functions of ribosome recycling factor in protein biosynthesis: disassembling the termination complex and preventing translational errors. Biochimie. 1996;78:959–969. doi: 10.1016/s0300-9084(97)86718-1. [DOI] [PubMed] [Google Scholar]
- 35.Hirokawa G, Demeshkina N, Iwakura N, Kaji H, Kaji A. The ribosome-recycling step: consensus or controversy? Trends Biochem Sci. 2006;31:143–149. doi: 10.1016/j.tibs.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 36.Karimi R, Pavlov MY, Buckingham RH, Ehrenberg M. Novel roles for classical factors at the interface between translation termination and initiation. Mol Cell. 1999;3:601–609. doi: 10.1016/s1097-2765(00)80353-6. [DOI] [PubMed] [Google Scholar]
- 37.Bachmann BJ. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol Rev. 1972;36:525–557. doi: 10.1128/br.36.4.525-557.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agafonov DE, Kolb VA, Spirin AS. Ribosome-associated protein that inhibits translation at the aminoacyl-tRNA binding stage. EMBO Rep. 2001;2:399–402. doi: 10.1093/embo-reports/kve091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller JH. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1972. [Google Scholar]
- 40.Thomason LC, Bubunenko M, Costantino N, Wilson H, Oppenheim A, Datta S, Court DL. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; Hoboken, N.J: 2005. Recombineering: genetic engineering in bacteria using homologous recombination; pp. 1.16.11–11.16.21. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet. 1998;20:123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 42.Staehelin T, Maglott DM, Monro RE. On the catalytic center of peptidyl transfer: a part of the 50 S ribosome structure. Cold Spring Harb Symp Quant Biol. 1969;34:39–48. doi: 10.1101/sqb.1969.034.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Hardy SJ, Kurland CG, Voynow P, Mora G. The ribosomal proteins of Escherichia coli. I. Purification of the 30S ribosomal proteins. Biochemistry. 1969;8:2897–2905. doi: 10.1021/bi00835a031. [DOI] [PubMed] [Google Scholar]
- 44.Madjar JJ, Michel S, Cozzone AJ, Reboud JP. A method to identify individual proteins in four different two-dimensional gel electrophoresis systems: application to Escherichia coli ribosomal proteins. Anal Biochem. 1979;92:174–182. doi: 10.1016/0003-2697(79)90641-9. [DOI] [PubMed] [Google Scholar]
- 45.Gavrilova LP, Smolyaninov VV. Study of the mechanism of translocation in ribosomes. 1. Polyphenylalanine synthesis in Escherichia coli ribosomes without participation of guanosine-5'-triphosphate and protein translation factors. Mol Biol (Moscow) 1971;5:710–717. [PubMed] [Google Scholar]
- 46.Bogatyreva SA, Trifonov EN, Spirin AS. Relation of polyphenylalanine synthesis in a cell-free system to the proportion of bivalent and monovalent cations. Dokl Akad Nauk SSSR (in Russian) 1970;195:213–216. [PubMed] [Google Scholar]
- 47.Crameri A, Whitehorn EA, Tate E, Stemmer WP. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 48.Gurevich VV, Pokrovskaya ID, Obukhova TA, Zozulya SA. Preparative in vitro mRNA synthesis using SP6 and T7 RNA polymerases. Anal Biochem. 1991;195:207–213. doi: 10.1016/0003-2697(91)90318-n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. E. coli strains and plasmids used in the work
Table S2. Primers used for PCR