

SUMMARY
The p53 tumor suppressor protein is a critical regulator of the cellular response to cancer initiating insults such as genotoxic stress. In this report, we demonstrate that microRNAs (miRNAs) are important components of the p53 transcriptional network. Global miRNA expression analyses identified a cohort of miRNAs that exhibit p53-dependent upregulation following DNA damage. One such miRNA, miR-34a, is commonly deleted in human cancers and, as shown here, frequently absent in pancreatic cancer cells. Characterization of the miR-34a primary transcript and promoter demonstrates that this miRNA is directly transactivated by p53. Expression of miR-34a causes dramatic reprogramming of gene expression and promotes apoptosis. Much like the known set of p53-regulated genes, miR-34a-responsive genes are highly enriched for those that regulate cell-cycle progression, apoptosis, DNA repair, and angiogenesis. Therefore, it is likely that an important function of miR-34a is the modulation and finetuning of the gene expression program initiated by p53.
INTRODUCTION
Loss of function of the p53 tumor suppressor protein is a causative event in the pathogenesis of a large fraction of human malignancies (Fridman and Lowe, 2003; Kirsch and Kastan, 1998). p53 is a transcription factor that coordinates cellular responses to stresses such as DNA damage and oncogene activation. When induced, p53 alters the expression of a large set of target genes leading to cell-cycle arrest, apoptosis, increased DNA repair, and/or inhibition of angiogenesis (Giono and Manfredi, 2006; Vogelstein et al., 2000).
A recently discovered class of small RNA molecules known as microRNAs (miRNAs) play a fundamental role in regulating gene expression in multi-cellular eukaryotes (Ambros, 2004; Bartel, 2004; He and Hannon, 2004). These 18-24 nucleotide RNAs negatively regulate the translation and stability of partially complementary target messenger RNAs (mRNAs). miRNAs are initially transcribed as long primary transcripts (pri-miRNAs) which are processed in a series of endonuclease reactions to produce the mature miRNA species (Kim, 2005). Pri-miRNAs are transcribed by RNA polymerase II and are capped, polyadenylated, and frequently spliced with the miRNA located in introns or exons (Cai et al., 2004; Kim and Kim, 2007; Lee et al., 2004; Rodriguez et al., 2004). Transcription of pri-miRNAs appears to be regulated by the same mechanisms that control mRNA expression. For instance, a number of well-studied mammalian transcription factors including c-Myc, cAMP-response element binding protein (CREB), and MyoD have been shown to control both mRNA and miRNA expression (O’Donnell et al., 2005; Rao et al., 2006; Vo et al., 2005; Zhao et al., 2005). Therefore, in order to fully elucidate the functions of a given transcription factor, miRNAs must be evaluated as potential targets.
miRNA expression is frequently dysregulated in cancer cells and specific miRNAs are known to regulate both cell-cycle progression and apoptosis (Calin and Croce, 2006; Esquela-Kerscher and Slack, 2006; Hammond, 2006). We therefore hypothesized that miRNAs might be critical downstream targets of p53 that participate in the cellular response to genotoxic stress. Here we report that miR-34a is a direct transcriptional target of p53. We demonstrate that induction of this miRNA promotes apoptosis and leads to dramatic global alterations in gene expression. Remarkably, transcripts that control the cell-cycle, apoptosis, DNA repair, and angiogenesis are highly enriched among the miR-34a-responsive genes. These data suggest that miR-34a is an important component of the p53 tumor suppressor network.
RESULTS AND DISCUSSION
Identification of candidate p53-regulated miRNAs
In order to identify miRNAs potentially regulated by p53, we utilized the p53 wild-type HCT116 colon cancer cell line (p53WT) and an isogenic cell line in which both alleles of p53 were inactivated by homologous recombination (p53-/-) (Bunz et al., 1998). Cells were treated with the DNA damaging agent adriamycin, which has previously been demonstrated to lead to induction of p53 and its downstream targets (Waldman et al., 1995), and RNA was analyzed using a custom microarray developed in our laboratory capable of monitoring the expression of 474 human miRNAs. miRNAs exhibiting a 3-fold or greater change in expression upon drug treatment in the p53WT cells and less than a 2-fold change in expression in the p53-/- cells were chosen for further study. Seven upregulated miRNAs satisfied these criteria and showed the expected expression pattern by northern blotting with adriamycin-induced accumulation specifically in p53WT cells (miR-23a, miR-26a, miR-34a, miR-30c, miR-103, miR-107, and miR-182; Figure 1A). No miRNAs that were downregulated in a p53-dependent manner following adriamycin treatment were observed. A previous study in which polysome-associated miRNAs in HCT116 p53WT and p53-/- cells were analyzed by microarray also identified miR-26a as a potential target of p53 (Xi et al., 2006).
Figure 1. miR-34a is induced by p53 following DNA damage.
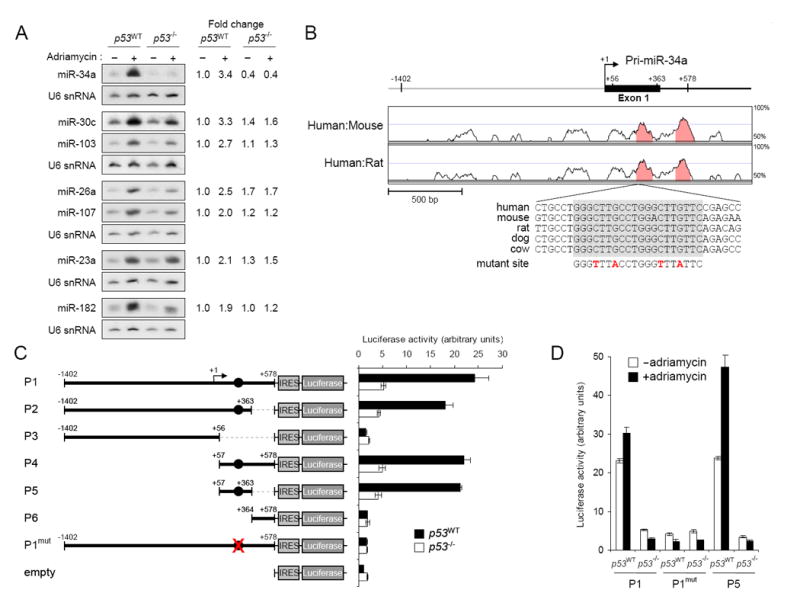
(A) Northern blot analysis of miRNA expression in p53WT and p53-/- HCT116 cells with or without adriamycin treatment. Quantification of the miRNA signals under each condition, normalized to U6 snRNA expression, is shown on the right.
(B) Phylogenetic conservation of the genomic region near the transcription start site of the miR-34a primary transcript. VISTA (http://genome.lbl.gov/vista/index.shtml) was used to generate pairwise alignments between human and mouse and human and rat. The graph is a plot of nucleotide identity for a 100 base-pair sliding window centered at a given position. Position +1 of pri-miR-34a is the most 5′ transcription start site mapped. The magnified sequence shows the location and evolutionary conservation of the p53 binding site (in gray box). Mutations introduced into the promoter reporter construct (P1mut in panel C) are shown in red.
(C) Activity of promoter constructs in p53WT and p53-/-cells. The arrow above construct P1 indicates the position of the transcription start site. Filled circles show the position of the p53 binding site. Error bars represent standard deviations from three independent transfections each measured in triplicate.
(D) Genotoxic stress activates the miR-34a promoter. The indicated promoter constructs were transfected into p53WT or p53-/- cells with or without subsequent exposure to adriamycin.
Characterization of the miR-34a primary transcript
Among the DNA-damage induced miRNAs, miR-34a exhibited the largest magnitude of upregulation. Although there are two additional human miR-34 homologues (miR-34b and miR-34c), these miRNAs were not detected by microarray analyses suggesting that they are not expressed in this cell line. In order to determine whether miR-34a is a direct transcriptional target of p53, we first elucidated the complete structure of the miR-34a primary transcript (pri-miR-34a). This miRNA is contained within the second exon of a spliced expressed sequence tag (EST) (DB286351; Supplemental Figure 1). Rapid amplification of cDNA ends (RACE) was used to define the complete 5′ and 3′ ends of this transcript. To facilitate these efforts, pri-miRNAs were stabilized by small interfering RNA (siRNA)-mediated inhibition of Drosha, the endonuclease which performs the first step in miRNA processing (Lee et al., 2003b). These experiments revealed that the 5′ end of the miR-34a pri-miRNA is heterogeneous, with all transcripts initiating in an approximately 100 base-pair (bp) region in the vicinity of the 5′ end of the DB286351 EST (Supplemental Figure 1). Although this region is within a large (>1.5 kb) CpG island, no sequences resembling a consensus TATA box (TATAAA) or initiator element (YYANTY) were present near the putative transcription start sites. 3′ RACE demonstrated that all transcripts terminate at a single position 360 bp downstream of the 3′ end of DB286351. A polyadenylation consensus site is present 22 bp upstream of this nucleotide. We also confirmed efficient splicing of the approximately 30 kb intron separating exons 1 and 2 and a lack of any intervening exons by performing reverse-transcriptase PCR (RT-PCR) with primers near the 5′ and 3′ ends of the pri-miRNA (data not shown).
p53-dependent transcriptional activity of the miR-34a promoter
We next investigated whether the miR-34a pri-miRNA is directly regulated by p53. While the region upstream of the pri-miRNA is not highly conserved between human and mouse or human and rat, a highly conserved perfect consensus p53 binding site is located just downstream of the transcription start site (Figure 1B). To test for transcriptional activity, a series of genomic fragments were cloned into a promoterless luciferase reporter plasmid (Figure 1C). Because several potential out-of-frame translation initiation codons are present between the transcription start site and the luciferase open reading frame in these reporter vectors, we also cloned an internal ribosome entry site (IRES) downstream of the putative promoter fragments. Reporter activity was then tested in HCT116 p53WT and p53-/- cells. The longest construct extended from 1.4 kb upstream to 578 bp downstream of the most 5′ transcription start site and included the putative p53 binding site (Figure 1C, P1). This fragment yielded robust p53-dependent transcriptional activity. This activity was comparable to that observed when a similar reporter vector containing the SV40 viral promoter was transfected into this cell line (data not shown). The activity of this promoter is not limited to HCT116 cells as shown by transfection experiments in HEK293T cells (Supplemental Figure 2). Removal of the p53 binding site by truncation or mutation abolished transcriptional activity (Figure 1C, P3, P6, and P1mut). Remarkably, a 307 bp fragment containing the p53 binding site located downstream of the transcription start site (Figure 1C, P5) was sufficient for full promoter activity.
To test whether genotoxic stress induces transcriptional activity of the miR-34a promoter, reporter constructs were transfected into HCT116 p53WT and p53-/- cells in the presence or absence of adriamycin. Precisely mirroring the expression of endogenous miR-34a, luciferase activity produced from the P1 and P5 reporters was induced by DNA damage in a p53-dependent manner (Figure 1D). As expected, mutation of the p53 binding site abolished this response. Providing further evidence that p53 directly regulates this miRNA, a previously published analysis of genome-wide binding sites for p53 using chromatin immunoprecipitation revealed that this protein directly binds to the genomic region defined here as the miR-34a promoter (Wei et al., 2006). This earlier study did not associate this binding site with regulation of miR-34a, likely because the miRNA is located more than 30 kb away. Taken together with the data provided here, these findings provide compelling evidence that miR-34a is a direct transcriptional target of p53.
Expression of miR-34a promotes apoptosis
In order to investigate the phenotypic consequences of miR-34a expression, we transiently transfected this miRNA into HCT116 p53WT and p53-/- cells and measured apoptotic cell death by flow cytometric measurement of DNA content (Figure 2A) and Annexin V staining (Figure 2B). Cells transfected with control oligonucleotide exhibited low levels of apoptosis [p53WT, 6.6 ± 3.4% apoptotic cells; p53-/-, 4.1 ± 1.6% apoptotic cells (means and standard deviations from 3 independent experiments reported)]. In contrast, transfection of p53WT cells with synthetic miR-34a potently induced apoptotic cell death (24.2 ± 3.8% apoptotic cells). Interestingly, apoptosis was substantially decreased but not completely abolished following transfection of miR-34a into p53-/- cells (9.3 ± 2.5% apoptotic cells), suggesting both p53-dependent and p53-independent mechanisms of miR-34a-induced cell death. These data suggest that miR-34a participates in the apoptotic program triggered by p53 activation.
Figure 2. Expression of miR-34a promotes apoptosis.
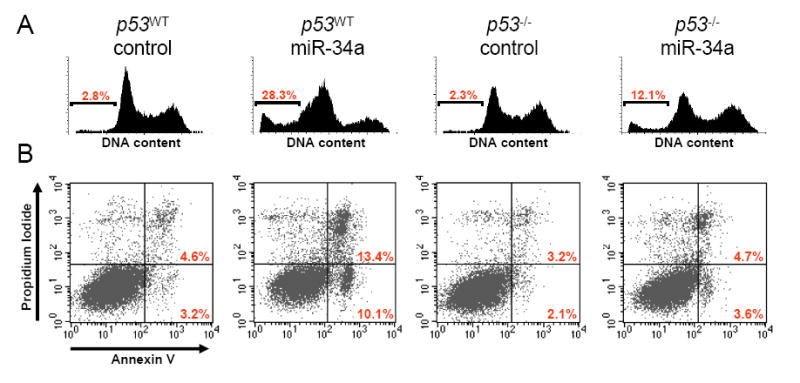
(A) p53WT and p53-/- cells were transfected with synthetic miR-34a or a control oligonucleotide and DNA content was monitored by flow cytometry. The fraction of apoptotic cells (sub-G1) is indicated.
(B) Cell death was monitored by Annexin V staining and flow cytometry. The right lower quandrant of each plot contains early apoptotic cells whereas the right upper quadrant contains late apoptotic cells. This experiment was repeated three independent times and similar results were obtained each time. Panels (A) and (B) come from two separate experiments.
Loss of miR-34a expression occurs frequently in pancreatic cancer cells
We next examined miR-34a expression in pancreatic cancer cells which frequently exhibit p53 loss-of-function. Two non-transformed pancreatic ductal epithelial cell lines (HPNE and HPDE) (Lee et al., 2003a; Ouyang et al., 2000) as well as 15 pancreatic cancer cell lines were analyzed by northern blotting (Figure 3). miR-34a was highly expressed in HPNE and HPDE cells, demonstrating that this miRNA is normally expressed in this cell type. Remarkably, all 15 pancreatic cancer cell lines showed at least a two-fold reduction in miR-34a expression as compared to expression in HPNE and HPDE. 11/15 cell lines exhibited a ten-fold reduction or complete absence of this miRNA.
Figure 3. Expression of miR-34a is frequently lost in pancreatic cancer cells.
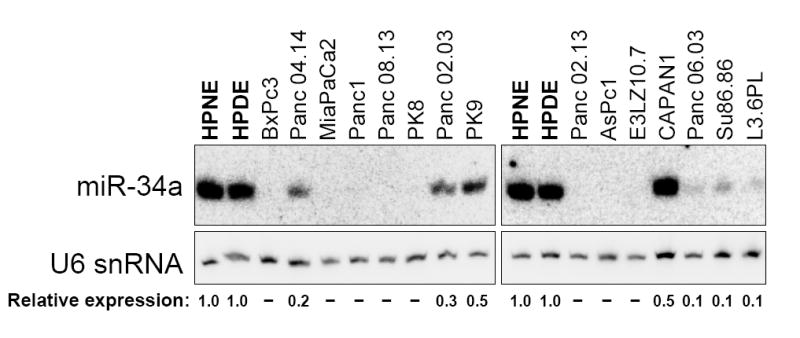
Northern blot analysis of miR-34a expression in non-transformed pancreatic ductal epithelial cell lines (HPNE and HPDE, in bold) and in pancreatic cancer cell lines. Relative expression of miR-34a in each cell line, normalized to U6 snRNA expression, is shown below the blots. Dashes indicate undetectable miR-34a expression.
Although p53 loss would be expected to reduce miR-34a expression, it is unlikely that this mechanism can account for the reduced miRNA expression in all the pancreatic cancer cell lines. There is not a direct correlation between bi-allelic loss of p53 and the magnitude of miR-34a downregulation (Figure 3 and Supplemental Table 1, compare BxPc3, PK9, CAPAN1, and Su86.86 all of which exhibit p53 loss of function). Similarly, cell lines with wild-type p53 status also exhibit low levels of miR-34a (Panc04.14 and E3LZ10.7). Thus, other mechanisms in addition to p53 inactivation likely contribute to the reduction in miR-34a abundance. In fact, deletion of the genomic interval encompassing this miRNA (1p36) is an extremely frequent event in diverse types of cancer and miR-34a is located within a recently mapped 5.4 megabase minimally-deleted region of 1p36 that commonly occurs in gliomas (Bagchi et al., 2007; Bello et al., 1995; Bieche et al., 1993; Moley et al., 1992; Mori et al., 1998; Poetsch et al., 2003). It has also very recently been reported that reduced miR-34a expression associated with loss of 1p36 is a frequent event in neuroblastoma (Welch et al., 2007). We therefore examined data from a previously published high-resolution copy number analysis of the genomes of pancreatic cancer cell lines (Calhoun et al., 2006). This study included 11/15 of the lines we studied by northern blotting experiments. Notably, hemizygous loss of the miR-34a locus was observed in 3 lines (Supplemental Table 1; Panc 02.13, Panc 04.14, and Panc 08.13). Thus, lack of transcriptional transactivation by p53, deletion, and additional unknown mechanisms likely contribute to loss of this miRNA in pancreatic cancer. Our results, together with previous studies, demonstrate that loss of miR-34a is a frequent event in diverse cancer subtypes and raise the possibility that miR-34a loss of function contributes to cancer pathogenesis.
Induction of miR-34a leads to widespread alterations in gene expression
To determine the effects of miR-34a induction on gene expression, a retroviral construct was used to generate HCT116 p53WT cells with enforced expression of this miRNA. These cells exhibited 3-4-fold higher miR-34a expression levels than cells transduced with empty virus (Figure 4A), a magnitude of induction very similar to the endogenous upregulation caused by adriamycin treatment (Figure 1A). Affymetrix gene expression profiling was then used to examine the transcriptomes of retrovirally-infected cell populations (empty virus versus miR-34a virus) in the absence of genotoxic stress. Although bona fide miR-34a targets that are not affected at the level of mRNA abundance will be missed by this approach, other groups have employed this strategy successfully to identify miRNA targets (Krutzfeldt et al., 2005; Lim et al., 2005; Voorhoeve et al., 2006). Surprisingly, cells with enforced expression of miR-34a showed a dramatically altered gene expression profile with upregulation of 532 transcripts and downregulation of 681 transcripts (Supplemental Table 2). This is a significantly greater number of transcripts showing altered expression than observed when similar experiments were performed previously with other miRNAs (Krutzfeldt et al., 2005; Lim et al., 2005). Select transcripts examined by northern blotting exhibited the expected expression changes, validating the quality of the microarray dataset (Figure 4B). To determine what fraction of the downregulated genes are likely to be direct targets of miR-34a, we examined the frequency of the hexamer complementary to the miR-34a seed sequence (ACUGCC) in the 3′ untranslated regions (UTRs) of the downregulated, unchanged, and upregulated transcripts. This motif is statistically-significantly enriched (p value = 0.023) among the top 100 downregulated transcripts (transcripts exhibiting a 2.364-fold or greater downregulation). Among transcripts showing the same fold increase in expression, this motif is statistically-significantly underrepresented (p value = 5.6 × 10-4). At this cut-off, 32/100 downregulated genes (32%), 2190/9534 unchanged genes (23%), and 18/149 upregulated genes (12%) contained the hexamer complementary to the miR-34a seed in their 3′ UTRs (Figure 4C). These findings demonstrate that the miR-34a-downregulated genes are enriched for direct targets of this miRNA and the upregulated genes are depleted of direct targets. Nevertheless, the majority of gene expression changes are likely an indirect consequence of enforced miR-34a expression. For example, only 47/185 transcripts downregulated greater than two-fold have sites complementary to the miR-34a seed sequence in their 3′ UTRs.
Figure 4. Expression of miR-34a leads to widespread alterations in gene expression.
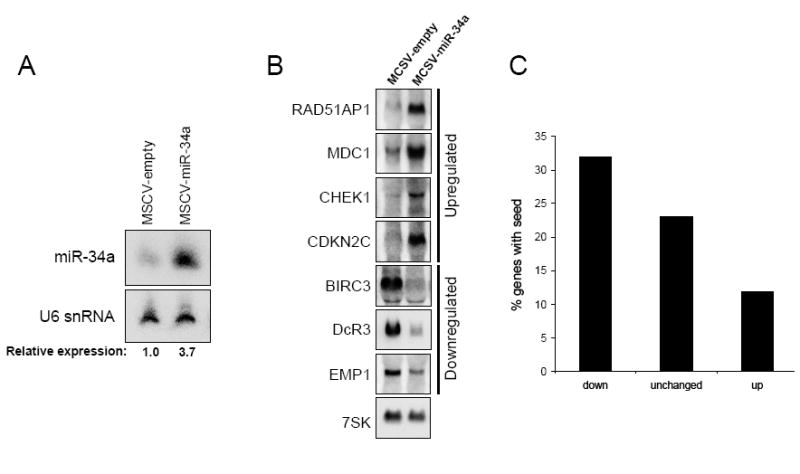
(A) Northern blot showing miR-34a expression in retrovirally-infected cell populations.
(B) Northern blot validation of selected expression changes observed by microarray analysis.
(C) Sites complementary to the miR-34a seed are enriched in the 3′ UTRs of downregulated transcripts. The graph shows the frequency of a site complementary to the miR-34a seed in the 3′ UTRs of the top 100 downregulated genes (2.364-fold or greater downregulation), the unchanged genes, and the genes upregulated by 2.364-fold or greater.
To better understand the global effects of expressing miR-34a on the transcriptome, we examined the Gene Ontology classifications of the up- and downregulated genes. Strikingly, the most highly enriched gene ontology category among the upregulated transcripts was ‘cell cycle’ (p value = 1.8 × 10-48). Genes classified as ‘DNA repair’ (p value = 6.6 × 10-23), ‘mitotic checkpoint’ (p value = 1.9 × 10-8), and ‘DNA integrity checkpoint’ (p value = 3.6 × 10-6) were also highly enriched among the upregulated transcripts. Moreover, genes assigned to the term ‘cell proliferation’ (p value = 1.9 × 10-4) and ‘angiogenesis’ (p value = 4.2 × 10-3) were significantly enriched among the downregulated genes. Similar categories of genes are known to be activated and repressed by p53 (Giono and Manfredi, 2006). Western blotting confirmed that p53 levels were unchanged in the miR-34a-expressing cells (Supplemental Figure 3). We note that while p53 and miR-34a regulate genes involved in overlapping pathways, they do not necessarily regulate the same genes. Thus, by inducing this miRNA, p53 is able to directly and indirectly regulate pathways that dictate the cellular response to genotoxic stress.
The role of miR-34a in p53-mediated apoptosis merits further discussion. Close inspection of our gene expression profiling results reveals that among the transcripts downregulated by miR-34a are several with well-documented anti-apoptotic functions including B-cell CLL/lymphoma 2 (BCL2), baculoviral IAP repeat-containing 3 (BIRC3), and decoy receptor 3 (DcR3 also known as TNFRSF6B) (Liston et al., 2003; Sheikh and Fornace, 2000; Willis et al., 2003). Indeed, the Gene Ontology term ‘programmed cell death’ is significantly enriched among the genes downregulated by miR-34a expression (p value = 7.8 × 10-5). These findings are consistent with our results indicating that miR-34a expression is sufficient to induce apoptosis. The set of miR-34a responsive genes described here will likely continue to provide mechanistic insight as additional functions of this miRNA are discovered.
Concluding Remarks
The identification of target genes of the p53 tumor suppressor protein has been the focus of intense interest. In this report, we demonstrate that miRNAs are important components of the p53 transcriptional network. Our results show that p53 directly activates expression of miR-34a and potentially other miRNAs. Expression of miR-34a is sufficient to induce apoptosis through p53-dependent and independent mechanisms. Moreover, miR-34a induction leads to dramatic reprogramming of gene expression. Much like the known set of p53-regulated genes, miR-34a-responsive genes are highly enriched for those that regulate cell-cycle progression, cellular proliferation, apoptosis, DNA repair, and angiogenesis. Interestingly, loss of 1p36, the genomic interval harboring miR-34a, is common in diverse human cancers. Consistent with this observation, we show that reduced expression of miR-34a is a very frequent feature of pancreatic cancer cells. Together, these findings suggest an important role for miR-34a in mediating p53 tumor suppressor function.
EXPERIMENTAL PROCEDURES
Cell Culture
HCT116 cells were cultured as described (Waldman et al., 1995) and treated with 0.2 μg/mL adriamycin for 24 hours to induce DNA damage.
miRNA Microarrays and Northern blotting
Custom microarrays containing oligonucleotide probes complementary to 474 human miRNAs were synthesized by Combimatrix. Probes containing 2 mismatches were included for all miRNAs. Arrays were pre-hybridized at 37° for 1 hour in 3X SSC, 0.1% SDS, 0.2% BSA. 10 μg of total RNA isolated using Trizol (Invitrogen) was labeled with Cy3 as described (Thomson et al., 2004) and hybridized to arrays at 37° overnight in 400mM Na2HPO4 (pH 7.0), 0.8% BSA, 5% SDS, and 12% formamide. Arrays were washed once at room temperature in 2X SSC, 0.25% SDS, 3 times at room temperature in 1.6X SSC, and twice in ice-cold 0.8X SSC. Hybridized arrays were then scanned using a GenePix 4000B microarray scanner (Axon) and signal intensities were extracted using the Combimatrix Microarray Imager software. The background value was determined by calculating the median signal from the mismatch probes and this value was subtracted from all perfect match probes. Signals that were less than 1.5 times background were removed and datasets were median centered prior to calculating fold-change values. Northern blotting was performed as described (Hwang et al., 2007).
RACE Mapping of Pri-miR-34a
Drosha expression was inhibited in HeLa cells as described (Hwang et al., 2007) and RACE was performed using the GeneRacer kit (Invitrogen). Primer sequences are provided in Supplemental Table 3 online.
Plasmid Construction
To generate the luciferase reporter vectors, an IRES was amplified from pMSCV-PIG (Hemann et al., 2003) and cloned into the BglII site of pGL3-basic (Promega). miR-34a promoter fragments were amplified from human genomic DNA and cloned into the XhoI site. Mutations were introduced using the Quikchange Kit (Stratagene). Primer sequences are provided in Supplemental Table 3 online.
Luciferase Assays
Twenty-four hours before transfection, 7×104 cells were plated per well in a 24-well plate. 100 ng pGL3 constructs plus 1 ng of the Renilla luciferase plasmid phRL-SV40 (Promega) were transfected using FuGENE 6 (Roche). 24 hours after transfection, luciferase assays were performed using the dual luciferase reporter assay system (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity for each transfected well. For each experimental trial, wells were transfected in triplicate and each well was assayed in triplicate. For measurement of promoter activity in cells treated with adriamycin, cells were first transfected for 24 hours prior to adding adriamycin for another 24 hour incubation. For each condition tested, the luciferase activity was normalized to the activity produced from empty vector.
Apoptosis Assays
15 nM precursor miR-34a (Pre-miR-34a) or control oligo (Pre-miR-control) (Applied Biosystems) was transfected using siPORT NeoFX (Applied Biosystems). After 60 hours, DNA content was determined by propidium iodide staining as described (Hwang et al., 2007) and Annexin V staining was performed with the Vybrant Apoptosis Assay kit (Invitrogen).
Retroviral Expression of miR-34a
miR-34a and approximately 100 bp of flanking sequence was amplified from human genomic DNA and cloned into the XhoI site of the retroviral vector pMSCV-PIG (Hemann et al., 2003). Primer sequences are provided in Supplemental Table 3 online. Following transfection of Phoenix packaging cells (G. Nolan, Stanford University, Stanford, CA), retroviral supernatants were collected, filtered, and added to recipient cells for 8 hours in the presence of 8 μg/mL polybrene. Two days after infection, puromycin was added to the media at 1 μg/mL and cell populations were selected for two weeks.
Affymetrix Gene Expression Profiling and Analysis
Hybridization of samples to Affymetrix U133 plus 2.0 microarrays was performed by the Johns Hopkins University School of Medicine Microarray Core Facility as described (Mendell et al., 2004). The quality of the microarray experiment was assessed with affyPLM and Affy, two bioconductor packages for statistical analysis of microarray data. To estimate the gene expression signals, data analysis was conducted on the chips’ CEL file probe signal values at the Affymetrix probe pair (perfect match (PM) probe and mismatch (MM) probe) level, using the statistical algorithm RMA (Robust Multi-array expression measure) (Irizarry et al., 2003) with Affy. This probe level data processing includes a normalization procedure utilizing the quantile normalization method (Bolstad et al., 2003) to reduce the obscuring variation between microarrays. Exploratory data analysis (EDA) was performed with the normalized data. Between-condition and between-replicate variation was examined with pairwise MvA plots, in which the base 2 log ratios (M) between two samples are plotted against their averaged base 2 log signals (A). After the signal intensities were averaged across technical replicates, an empirical Bayes method implemented in the bioconductor package EBarrays, was used to estimate the posterior probabilities of the differential expression of genes between the tested conditions (Kendziorski et al., 2003; Newton and Kendziorski, 2003; Newton et al., 2001). The criterion of the posterior probability > 0.5, that is to say the posterior odds favoring change, was used to produce the differentially expressed gene list (Supplemental Table 2). All Bioconductor packages are available at http://www.bioconductor.org and all computation was performed under R environment (http://www.r-project.org) (Ihaka and Gentleman, 1996).
For analysis of the frequency of the miR-34a seed hexamer, genes were first filtered for those that were called present by the Affymetrix software in both replicate control or replicate miR-34a hybridizations. Genes with annotated 3′ UTRs from the UCSC Table Browser (http://genome.ucsc.edu) were then selected (9783 total present genes with annotated 3′ UTRs). 3′ UTR sequences were downloaded from the March 2006 build of the human genome. P-values for the significance of enrichment of the seed sequence were calculated using the hypergeometric distribution. Gene ontology classification was performed using DAVID (http://david.abcc.ncifcrf.gov/) (Dennis et al., 2003).
Supplementary Material
Acknowledgments
The authors wish to thank Bert Vogelstein for HCT116 p53WT and p53-/- cells, Garry Nolan for Phoenix cells, Michael Thomson and Scott Hammond for assistance with miRNA arrays, and Francisco Martinez Murillo and Chunfa Jie for assistance with the Affymetrix microarray experiments. This work was supported by a Rita Allen Foundation Scholar Award (J.T.M.), the Lustgarten Foundation for Pancreatic Cancer Research (J.T.M), the Sol Goldman Pancreatic Cancer Research Center (A.M.), and the National Cancer Institute (R01CA120185 and R01CA113669).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimersthat apply to the journal pertain.
References
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D, Bredel M, Vogel H, Mills AA. CHD5 Is a Tumor Suppressor at Human 1p36. Cell. 2007;128:459–475. doi: 10.1016/j.cell.2006.11.052. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bello MJ, Leone PE, Vaquero J, de Campos JM, Kusak ME, Sarasa JL, Pestana A, Rey JA. Allelic loss at 1p and 19q frequently occurs in association and may represent early oncogenic events in oligodendroglial tumors. Int J Cancer. 1995;64:207–210. doi: 10.1002/ijc.2910640311. [DOI] [PubMed] [Google Scholar]
- Bieche I, Champeme MH, Matifas F, Cropp CS, Callahan R, Lidereau R. Two distinct regions involved in 1p deletion in human primary breast cancer. Cancer Res. 1993;53:1990–1994. [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ES, Hucl T, Gallmeier E, West KM, Arking DE, Maitra A, Iacobuzio-Donahue CA, Chakravarti A, Hruban RH, Kern SE. Identifying Allelic Loss and Homozygous Deletions in Pancreatic Cancer without Matched Normals Using High-Density Single-Nucleotide Polymorphism Arrays. Cancer Res. 2006;66:7920–7928. doi: 10.1158/0008-5472.CAN-06-0721. [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–9040. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- Hammond SM. MicroRNAs as oncogenes. Curr Opin Genet Dev. 2006;16:4–9. doi: 10.1016/j.gde.2005.12.005. [DOI] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Fridman JS, Zilfou JT, Hernando E, Paddison PJ, Cordon-Cardo C, Hannon GJ, Lowe SW. An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet. 2003;33:396–400. doi: 10.1038/ng1091. [DOI] [PubMed] [Google Scholar]
- Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- Ihaka J, Gentleman R. R: A language for data analysis and graphics. Journal of Computational and Graphical Statistics. 1996;5:299–314. [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Kendziorski CM, Newton MA, Lan H, Gould MN. On parametric empirical Bayes methods for comparing multiple groups using replicated gene expression profiles. Stat Med. 2003;22:3899–3914. doi: 10.1002/sim.1548. [DOI] [PubMed] [Google Scholar]
- Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch DG, Kastan MB. Tumor-suppressor p53: implications for tumor development and prognosis. J Clin Oncol. 1998;16:3158–3168. doi: 10.1200/JCO.1998.16.9.3158. [DOI] [PubMed] [Google Scholar]
- Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Lee KM, Nguyen C, Ulrich AB, Pour PM, Ouellette MM. Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem Biophys Res Commun. 2003a;301:1038–1044. doi: 10.1016/s0006-291x(03)00086-x. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003b;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Liston P, Fong WG, Korneluk RG. The inhibitors of apoptosis: there is more to life than Bcl2. Oncogene. 2003;22:8568–8580. doi: 10.1038/sj.onc.1207101. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–1078. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- Moley JF, Brother MB, Fong CT, White PS, Baylin SB, Nelkin B, Wells SA, Brodeur GM. Consistent association of 1p loss of heterozygosity with pheochromocytomas from patients with multiple endocrine neoplasia type 2 syndromes. Cancer Res. 1992;52:770–774. [PubMed] [Google Scholar]
- Mori N, Morosetti R, Spira S, Lee S, Ben-Yehuda D, Schiller G, Landolfi R, Mizoguchi H, Koeffler HP. Chromosome band 1p36 contains a putative tumor suppressor gene important in the evolution of chronic myelocytic leukemia. Blood. 1998;92:3405–3409. [PubMed] [Google Scholar]
- Newton MA, Kendziorski CM. Parametric empirical Bayes methods for microarrays. In: Parmigiani G, Garrett ES, Irizarry RA, Zeger SL, editors. The analysis of gene expression data: methods and software. New York: Springer Verlag; 2003. [Google Scholar]
- Newton MA, Kendziorski CM, Richmond CS, Blattner FR, Tsui KW. On differential variability of expression ratios: improving statistical inference about gene expression changes from microarray data. J Comput Biol. 2001;8:37–52. doi: 10.1089/106652701300099074. [DOI] [PubMed] [Google Scholar]
- O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Ouyang H, Mou L, Luk C, Liu N, Karaskova J, Squire J, Tsao MS. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol. 2000;157:1623–1631. doi: 10.1016/S0002-9440(10)64800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetsch M, Dittberner T, Woenckhaus C. Microsatellite analysis at 1p36.3 in malignant melanoma of the skin: fine mapping in search of a possible tumour suppressor gene region. Melanoma Res. 2003;13:29–33. doi: 10.1097/00008390-200302000-00006. [DOI] [PubMed] [Google Scholar]
- Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci U S A. 2006;103:8721–8726. doi: 10.1073/pnas.0602831103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh MS, Fornace AJ., Jr Death and decoy receptors and p53-mediated apoptosis. Leukemia. 2000;14:1509–1513. doi: 10.1038/sj.leu.2401865. [DOI] [PubMed] [Google Scholar]
- Thomson JM, Parker J, Perou CM, Hammond SM. A custom microarray platform for analysis of microRNA gene expression. Nat Methods. 2004;1:47–53. doi: 10.1038/nmeth704. [DOI] [PubMed] [Google Scholar]
- Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A. 2005;102:16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, Liu YP, van Duijse J, Drost J, Griekspoor A, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007 doi: 10.1038/sj.onc.1210293. Advance Online Publication. [DOI] [PubMed] [Google Scholar]
- Willis S, Day CL, Hinds MG, Huang DC. The Bcl-2-regulated apoptotic pathway. J Cell Sci. 2003;116:4053–4056. doi: 10.1242/jcs.00754. [DOI] [PubMed] [Google Scholar]
- Xi Y, Shalgi R, Fodstad O, Pilpel Y, Ju J. Differentially regulated micro-RNAs and actively translated messenger RNA transcripts by tumor suppressor p53 in colon cancer. Clin Cancer Res. 2006;12:2014–2024. doi: 10.1158/1078-0432.CCR-05-1853. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.