

Abstract
The synthesis of N-acyl 3,4-disubstituted pyrroles can be accomplished directly from hydrazine and an aldehyde via a Piloty-Robinson pyrrole synthesis. The use of microwave radiation for the cyclization and pyrrole formation greatly reduces the time necessary for this process and facilitates moderate to good yields from hydrazine for the corresponding 3,4-disubstituted products (5–12). By simple hydrolysis, the free N–H pyrroles can be accessed after the Piloty-Robinson reaction and then used directly in the synthesis of octaethylporphyrin (H2OEP, 14) and octaethyltetraphenylporphyrin (H2OETPP, 15).
Pyrroles with substituents at C3 and C4 are important compounds for the synthesis of pharmaceuticals, natural products1–5 and porphyrins.6–10 Consequently, there are numerous general methods to access this important aromatic heterocycle with various substitution patterns.11,12 Many established approaches for the synthesis of pyrroles are based on the venerable Paal-Knorr13–16 and Hantzsch17 reactions which were developed in the late 19th century. Even with the substantial work in this area spanning the last hundred years, new reports that provide efficient and versatile access to pyrroles continue to appear, underscoring the importance of this heterocycle in various areas of science.18–20 For example, contemporary strategies for the construction of pyrroles include transition metal-mediated and multi-component coupling routes.16,21–25 The symmetric 3,4-disubstitued pyrrole core has special interest since the combination of these monomers with an aldehyde results in highly substituted porphyrins. These resulting four-fold symmetric macrocycles are key molecules that are the basis for a large variety of synthetic and physiochemical studies.6,26,27 Over the last three decades, two compounds have emerged as useful model systems in the investigations of general properties of metalloporphyrinate derivatives: octaethylporphyrin (H2OEP)28–33 and tetraphenylporphyrin (H2TPP).34 For example, iron complexes of octaethylporphyrin have received significant attention due to its homology to heme b of the heme proteins.26,35,36 Although H2OEP is a more appropriate model compound due to its substitution pattern and homology to the heme cofactor, H2TPP is more widely used due in part to its convenient synthesis from pyrrole and benzaldehyde.37 An inexpensive and reliable synthesis of H2OEP and related porphyrin derivatives remains a challenging goal since it relies on an efficient route to the requisite 3,4-disubstituted pyrroles. In our continuing studies on metalloporphrynate compounds (W.R.S.), we typically synthesize H2OEP by way of 3,4-diethyl pyrrole. The H2OEP synthesis procedures reported independently by Inhoffen,29 Whitlock,30 Dolphin31 and Sessler33 all provide access to H2OEP, but the synthesis of the monomer precursor 3,4-diethyl pyrrole remains challenging due to the number of synthetic steps, the cost of the starting materials, or both. In all these approaches, 2-carboxy or 2,5-bis-carboxy pyrroles are converted into the key 3,4-diethyl pyrrole, which is then employed in the subsequent H2OEP syntheses.
With our interests in the efficient syntheses of nitrogen heterocycles (K.A.S.),23,38–40 we recognized that a straightforward and reliable strategy to 3,4-diethylpyrrole would provide for a streamlined H2OEP synthesis with a minimal number of purification steps. A facile route to the desired 3,4-disubstituted heterocycles is the Piloty-Robinson pyrrole synthesis that involves the conversion of ketone-derived azines into pyrroles.41–43 A seminal study by Baldwin in 1982 expanded this useful reaction sequence to include azines derived from aldehydes.44 The reported two-step process delivers symmetric N-benzoyl pyrroles, but suffers from the elevated temperatures (140 °C) and prolonged reaction times (3 days) required for low to moderate yields of products (<35%).
With the emergence of microwave irradiation as a useful tool in organic synthesis, we sought to apply this technique to this reaction.45–53 In this Note, we report the use of microwave radiation in the Piloty-Robinson pyrrole synthesis to afford disubstituted N-acyl pyrroles (Scheme 1). The overall sequence involves the synthesis of a symmetric azine (2) followed by exposure of the unpurified material to two equivalents of an aroyl chloride and pyridine. With the aid of significant thermal energy, the acylation of the azine nitrogen atoms under these conditions promotes tautomerization to intermediate I and then subsequent [3,3]-sigmatropic rearrangement delivers a 1,4-bis imine (II). This acyclic intermediate then undergoes cyclization and aromatization to generate the desired N-acylated pyrrole core with substitution at C3 and C4 (3) and benzamide (4).
Scheme 1.
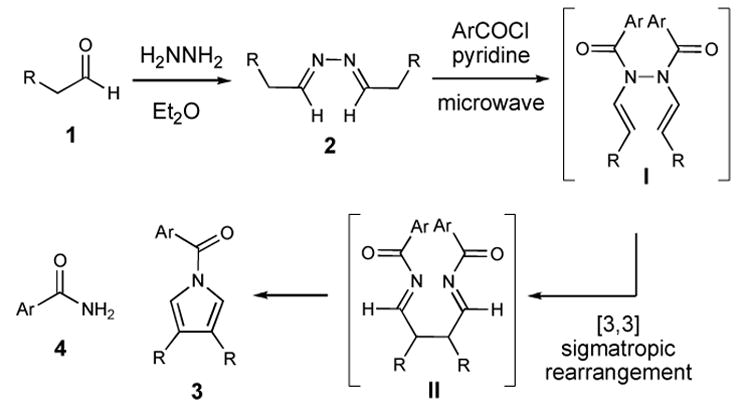
A key goal in streamlining our approach was to determine the optimal set of reaction conditions that would minimize the number of purification steps while still providing pure N-aroyl pyrroles from simple and inexpensive starting materials. Given the well-known propensity for pyrroles to undergo oxidative processes over prolonged reaction times and temperatures, we sought to employ a rapid heating step to induce the Piloty-Robinson sequence which could potentially increase the yield and ease purification. Accessing symmetrical azines (2) is easily accomplished by the slow addition of aqueous hydrazine (1 equiv) to a 0 °C ethereal solution of an aldehyde (1, 2 equiv). After removing the water (anhydrous potassium carbonate), the reaction is filtered and the solvent is removed under reduced pressure to afford the unpurified azines in >90% yield.
With a quick route to the symmetrical precursors, we initially examined the reaction of the azine derived from butyraldehyde with benzoyl chloride using microwave conditions in order to optimize the thermal parameters. We chose to omit additional solvent since Baldwin had reported earlier that pyridine was used to promote the acylation/rearrangement step of the Piloty sequence and we anticipated that the pyridine alone would facilitate adequate mixing and heat transfer in the microwave-assisted reaction. Accordingly, the heating of the azine, benzoyl chloride (2 equiv.) and pyridine (3.7 equiv.) in a sealed microwave reaction vessel provides a dark suspension that can be passed through a course fritted funnel with the aid of ethyl acetate. The filtrate was concentrated in vacuo, passed through a short pad of silica gel using chloroform and then initially purified by distillation to afford the symmetric N-benzoyl pyrrole (3, Ar = Bz, R = Et). After surveying various times and thermal parameters, we determined that heating reaction mixtures containing 3–5 mmol of azine for 30–60 min at 180 °C provided the best yields for the desired pyrroles. Larger scale reactions (25–30 mmol of azine) required longer reaction times to convert all of the bis-acylated materials (II) to products.
With the microwave parameters optimized, we surveyed the reaction scope as a function of different acid chlorides and different starting aldehydes (Table 1). In all cases, the unpurified azine is carried directly into the Piloty-Robinson reaction. While the reaction is limited to aromatic acid chlorides in the cases we have examined, the use of butyraldehyde produces the corresponding 3,4-diethylpyrroles in moderate yields based on the amount of aldehyde used (entries 1–4). A short survey of the aldehyde structure of indicates that changes in the length or substitution does not greatly impact the overall yield of the process. Currently, the pyridinium chloride produced in the reaction and high temperatures rule out the use of highly sensitive protecting groups on either reaction component, but the overall sequence should be amenable to a variety of different functional groups. It should be noted that while the maximum reaction flask size for the microwave reactor used in these studies is a 20 mL vessel (Biotage Initiator), the process can be performed multiple times in a single day to afford significant amounts of unpurified pyrrole (>20 g).
Table 1.
Scope of N-Aroyl Pyrrole Synthesisa
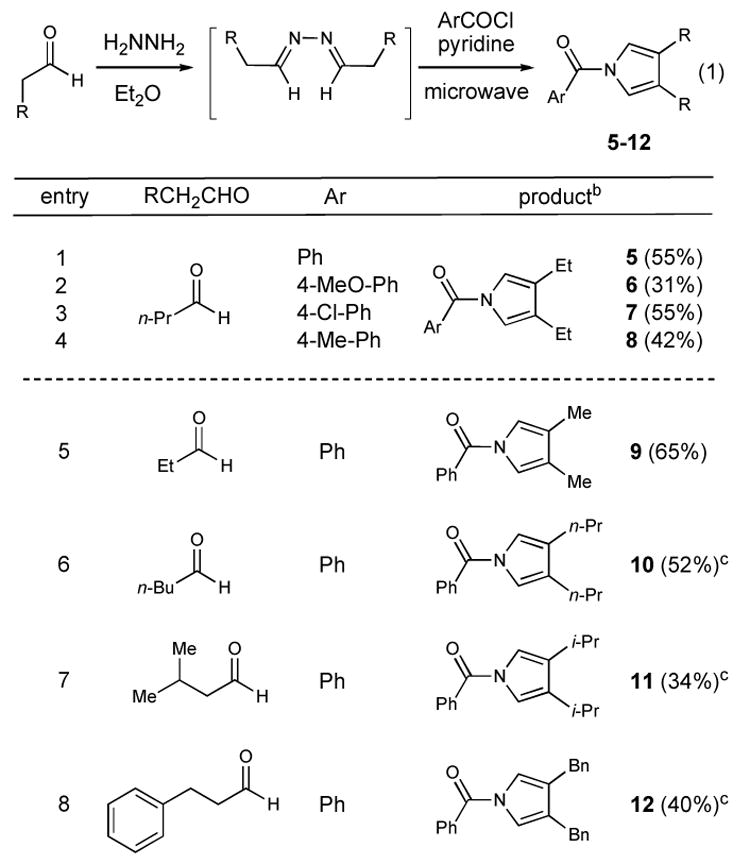
1 equiv azine, 2.1 equiv. ArCOCl, 3.7 equiv. pyridine. Biotage Initiator microwave reactor. 180 °C for 30 min h.
Isolated yields calculated from azine after purification.
180 °C for 1 h.
An attractive feature of this reaction sequence is that it delivers the N-acylated pyrroles. Importantly, the placement of an electron withdrawing substituent on the pyrrole nitrogen deactivates the pyrrole core towards unwanted side reactions and can induce crystallinity to ease purification. In this manner, the N-benzoylated pyrroles from the Piloty-Robinson synthesis can be stored indefinitely with only minimal precautions to exclude air or moisture. The removal of the benzoyl group can be cleanly accomplished in high yield by exposure of the N-benzoylated pyrroles to aqueous KOH in ethanol (eq 2). After 15–20 minutes, the reaction is diluted with water and extracted with hexanes. A wash of the organic layer with aqueous sodium bicarbonate removes any remaining benzoic acid, thereby affording >95% pure N-H pyrrole without purification.
This material can be taken directly into the synthesis of various porphyrins, including octaethylporphyrin (H2OEP, 14) and octaethyltetraphenylporphyrin (H2OETPP, 15). After the hydrolysis of 5, the exposure of the unpurified N-H pyrrole (13) to aqueous formaldehyde and oxygen with benzene as solvent generates the desired H2OEP macrocycle in 51% yield after recrystallization.54–56 By integrating our approach with the porphyrin synthesis described by Smith,57 we can also access the more substituted H2OETPP from the initial porphyrinogen formation with BF3·OEt (76%) and subsequent oxidation with DDQ (68%, 51% overall yield from 5). The combination of our Piloty-Robinson protocol with these hydrolysis and cyclization steps affords a streamlined, four-step porphyrin synthesis starting from hydrazine, benzoyl chloride and a saturated aldehyde (e.g. butyraldehyde). Notably, the process uses inexpensive starting materials and requires only two purification steps- one for the N-aroyl pyrrole and the second for the final porphyrin product.
Due to the potential issues with hydrazine and related nitrogenous compounds, we examined the safety and thermodynamic parameters of the Piloty-Robinson reaction using calorimetry. Based on our initial differential scanning calorimetry, the only intermediate in the overall process that has an exothermic event below 300 °C is the unpurified azine (see Supporting Information for details). For the azine derived from butyraldehyde and hydrazine hydrate, an exothermic event is observed at 240 °C. We have also used reaction calorimetry (Mettler-ASI RC1) to study the azine formation and Piloty-Robinson sequence. Not surprisingly, the addition of hydrazine hydrate to the butyraldehyde in the azine formation is exotherminc, with a heat of reaction of −692 J/g butyraldehyde. The heat generation for this process can be controlled by cooling the reaction to 0 °C and the addition rate of hydrazine. With the use of a chemical reactivity calorimeter (Omincal SuperCRC), the thermal parameters of the benzoyl chloride addition were determined. The combination of the acid chloride and azine in the presence of pyridine is exothermic with the heat of reaction being −1383 J/g azine. The estimated maximum adiabatic temperature rise is 130 °C, i.e. if the energy of the reaction were released at 25 °C and the heat were not removed, the reaction mass could self-heat to 115 °C and vaporize approximately 18% of the pyridine. This heat is solely due to events that occur at room temperature. Overall, the formation of the azine and subsequent Piloty-Robinson sequence can be performed safely at lab scale with current commercial microwave reactors equipped with appropriate temperature control and safety precautions associated with operating at elevated pressures. If traditional heating protocols are employed (i.e. non-microwave), then metal contact should be avoided since adiabatic testing of the Piloty-Robinson sequence in titanium test cells (accelerating rate calorimetry) led to a significant increase in exothermic activity and potential runaway reaction as indicated by the much higher self-heating rates shown in Figure 1.
Figure 1.
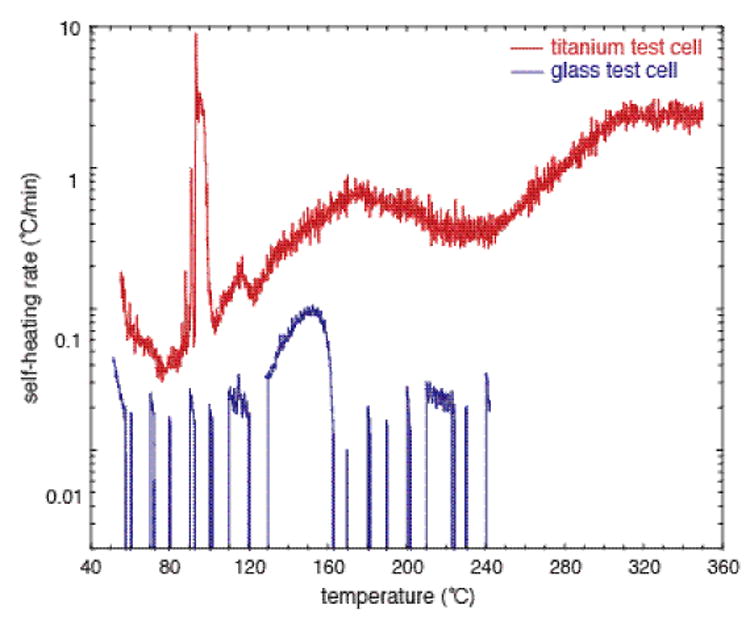
Adiabatic Calorimetry Self-Heating Data for Piloty-Robinson Reaction Mixturea
In summary, we have developed a concise and cost-effective synthesis of symmetric 3,4-disubstituted pyrroles based on the Piloty-Robinson synthesis. The use of microwave radiation allows for short reaction times that deliver N-benzoylated pyrroles from hydrazine and a saturated aldehyde in moderate yields with a single purification. The products from this acylation/rearrangement/cyclization can be converted to the free N-H pyrroles using basic conditions and the unpurified pyrroles have been used directly in the synthesis of octaethylporphyrin (H2OEP) and octaethyltetraphenylporphyrin (H2OETPP). The methodology detailed in this study should facilitate the synthesis of highly substituted porphyrin derivatives for use in bioinorganic chemistry and materials research.
Experimental Section
(3,4-diethyl-1H-pyrrol-1-yl)(phenyl)methanone (5)
To a round bottom flask equipped with a magnetic stir bar was added diethyl ether (90 mL) and butyraldehyde (64.3 mL, 713 mmol). The solution was cooled to 0 °C and hydrazine hydrate (17.3 mL, 357 mmol) was added dropwise over 30 min. The reaction was warmed to 23 °C and stirred for 30 minutes. The water produced in the reaction was then removed by pipette and anhydrous potassium carbonate was added to dry the reaction further. The mixture was filtered and the resulting solution was concentrated carefully under reduced pressure (400 mbar) to afford the 1,2-dibutylidenehydrazine (41.9 g, 299 mmol) of which a portion (18.8 g, 134 mmol) is used directly in the subsequent Piloty-Robinson reaction.
To a dry 20 mL microwave vial equipped with a magnetic stir bar was added unpurified azine (3.76 g, 27 mmol) and pyridine (8.1 mL, 100 mmol). Benzoyl chloride (6.7 mL, 58 mmol) was then added slowly and the vial capped with a rubber septum. The vial was shaken vigorously and then heated in the microwave at 170 °C for 60–75 min (Biotage Initiator). After heating, the vessel was cooled, diluted with ethyl acetate (1 L) and filtered through a course frit funnel. The above microwave procedure was repeated five times and the resulting combined solution was concentrated under reduced pressure. The remaining material was dissolved in chloroform and filtered through a 400 mL silica gel pad to remove the benzamide. The eluent (2 L) was collected and removed under reduced pressure to afford a brown residue. This material was purified by vacuum distillation (145–155 °C @ 200 mT), yielding 15.9 g (52%) of (3,4-diethyl-1H-pyrrol-1-yl)(phenyl)methanone (5) as a tan oil that crystallized upon standing: Rf = 0.71 (20:80 ethyl acetate/hexanes); mp = 43 °C; IR (film) 2965, 1688, 1377, 1317, 1274, 1246, 1136, 884, 807, 717, 668 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 7.3 Hz, 2H), 7.57 (dd, J = 7.3 Hz, 1H), 7.50 (dd, J = 7.3 Hz, 2H), 7.00 (s, 2H), 2.43 (q, J = 7.3 Hz, 4H), 1.21 (t, J = 7.3 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ164.7, 133.8, 131.7, 130.8 129.2 128.3 117.4 18.4 13.4; LRMS (ESI): Mass calculated for C15H17NO [M]+, 227.3. Found [M+H]+, 228.5.
Supplementary Material
Scheme 2.
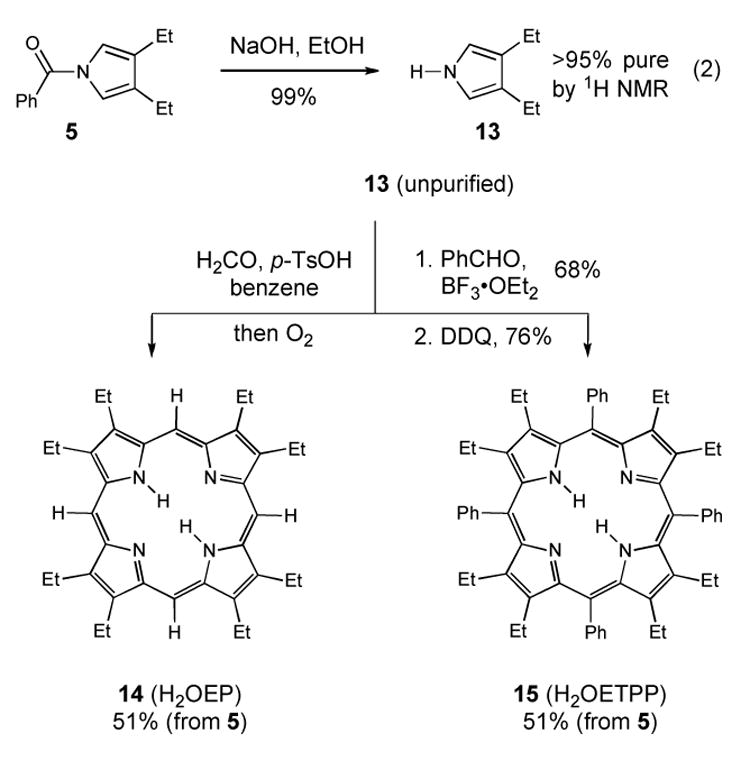
Synthesis of H2OEP and H2OETPP from N-Benzoyl-3,4-diethylpyrrole
Acknowledgments
Support for this research has been provided by the NSF (CAREER CHE-0348979 to K.A.S.), and the NIH (GM 38401 to W.R.S.). K.A.S. thanks Abbott Laboratories, Amgen, Boehringer-Ingelheim and 3M for generous unrestricted research support. B.C.M. thanks Northwestern University and Sigma-Aldrich for summer research fellowships. We thank Steve Paeschke (Biotage) for early technical assistance and initially providing microwave equipment. The funding for the NU Analytical Services Laboratory has been furnished in part by the NSF (CHE-9871268).
Footnotes
Accelerating rate calorimetry (ARC) performed with TIAX Accelerating Rate Calorimeter in glass test cell (in blue) and titanium test cell (in red). See Supporting Information for details.
Supporting Information Available: Experimental procedures and spectral data for all new compounds, (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.LeQuesne PW, Dong Y, Blythe TA. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. Vol. 13 Elsevier; Amsterdam, The Netherlands: 1999. [Google Scholar]
- 2.O'Hagan D. Nat Prod Rep. 2000;17:435–446. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez C, Zhu LL, Brana AF, Salas AP, Rohr J, Mendez C, Salas JA. Proc Nat Acad Sci USA. 2005;102:461–466. doi: 10.1073/pnas.0407809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishizawa T, Gruschow S, Jayamaha DHE, Nishizawa-Harada C, Sherman DH. J Am Chem Soc. 2006;128:724–725. doi: 10.1021/ja056749x. [DOI] [PubMed] [Google Scholar]
- 5.Hinze C, Kreipl A, Terpin A, Steglich W. Synthesis. 2007:608–612. [Google Scholar]
- 6.Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Academic Press; San Diego, CA: 2000. [Google Scholar]
- 7.Smith KM. In: Comprehensive Heterocyclic Chemistry. Katritzky AR, Rees WE, editors. Vol. 4. Pergamon Press; Oxford: 1984. p. 377. [Google Scholar]
- 8.Ono N, Kawamura H, Bougauchi M, Maruyama K. Tetrahedron. 1990;46:7483–7496. [Google Scholar]
- 9.Ono N, Miyagawa H, Ueta T, Ogawa T, Tani H. J Chem Soc, Perkins Trans 1. 1998:1595–1601. [Google Scholar]
- 10.Merz A, Schwarz R, Schropp R. Adv Mater. 1992;4:409–411. [Google Scholar]
- 11.Jones RA, editor. Pyrroles Part I: The Synthesis and the Physical and Chemical Aspects of the Pyrrole Ring. Part 1 Vol. 48. Wiley; New York: 1990. [Google Scholar]
- 12.Jones RA, editor. Pyrroles Part 2 The Synthesis, Reactivity, and Physical Properties of Substituted Pyrroles. Part 2 Vol. 48. Wiley; New York: 1992. [Google Scholar]
- 13.Paal C. Chem Ber. 1885:367–371. [Google Scholar]
- 14.Knorr L. Chem Ber. 1885;18:299–311. [Google Scholar]
- 15.Amarnath V, Anthony DC, Amarnath K, Valentine WM, Wetterau LA, Graham DG. J Org Chem. 1991;56:6924–6931. [Google Scholar]
- 16.Braun RU, Zeitler K, Muller TJJ. Org Lett. 2001;3:3297–3300. doi: 10.1021/ol0165185. [DOI] [PubMed] [Google Scholar]
- 17.Hantzsch A. Chem Ber. 1890;23:1474–1483. [Google Scholar]
- 18.Tang JS, Verkade JG. J Org Chem. 1994;59:7793–7802. [Google Scholar]
- 19.Lash TD, Bellettini JR, Bastian JA, Couch KB. Synthesis. 1994:170–172. [Google Scholar]
- 20.Yu M, Pagenkopf BL. Org Lett. 2003;5:5099–5101. doi: 10.1021/ol036180+. [DOI] [PubMed] [Google Scholar]
- 21.Buchwald SL, Wannamaker MW, Watson BT. J Am Chem Soc. 1989;111:776–777. [Google Scholar]
- 22.Kel'in AV, Sromek AW, Gevorgyan V. J Am Chem Soc. 2001;123:2074–2075. doi: 10.1021/ja0058684. [DOI] [PubMed] [Google Scholar]
- 23.Bharadwaj AR, Scheidt KA. Org Lett. 2004;6:2465–2468. doi: 10.1021/ol049044t. [DOI] [PubMed] [Google Scholar]
- 24.Dhawan R, Arndtsen BA. J Am Chem Soc. 2004;126:468–469. doi: 10.1021/ja039152v. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal S, Knolker HJ. Org Biomol Chem. 2004;2:3060–3062. doi: 10.1039/B412206B. [DOI] [PubMed] [Google Scholar]
- 26.Buchler JW. Angew Chem Int Ed. 1978;17:407–423. doi: 10.1002/anie.197804071. [DOI] [PubMed] [Google Scholar]
- 27.Scheidt WR, Reed CA. Chem Rev. 1981;81:543–555. [Google Scholar]
- 28.Eisner U, Lichtarowicz A, Linstead RP. J Chem Soc. 1957:733–739. [Google Scholar]
- 29.Inhoffen HH, Fuhrhop JH, Voigt H, Brockman H. Liebigs Ann Chem. 1966;695:133–139. [Google Scholar]
- 30.Whitlock HW, Hanauer R. J Org Chem. 1968;33:2169–2171. [Google Scholar]
- 31.Paine JB, Kirshner WB, Moskowitz DW, Dolphin D. J Org Chem. 1976;41:3857–3860. doi: 10.1021/jo00886a018. [DOI] [PubMed] [Google Scholar]
- 32.Paine JB, Dolphin D. J Org Chem. 1988;53:2787–2795. [Google Scholar]
- 33.Sessler JL, Mozaffari A, Johnson M, Fischer J, Winterfeldt E. Organic Synthesis. 1992;70:68–74. [Google Scholar]
- 34.Adler AD, Longo FR, Finarelli JD, Goldmach J, Assour J, Korsakoff L. J Org Chem. 1967;32:476. [Google Scholar]
- 35.Ellison MK, Schulz CE, Scheidt WR. J Am Chem Soc. 2002;124:13833–13841. doi: 10.1021/ja0207145. [DOI] [PubMed] [Google Scholar]
- 36.Hu CJ, Roth A, Ellison MK, An J, Ellis CM, Schulz CE, Scheidt WR. J Am Chem Soc. 2005;127:5675–5688. doi: 10.1021/ja044077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The cost of tetraphenylporphyrin (Sigma-Aldrich) is $21 per mmol. The cost of 2,3,4,7,8,12,13,17,18-octaethylpophyrin (Sigma-Aldirch) is $245 per 1 mmol.
- 38.Galliford CV, Beenen MA, Nguyen ST, Scheidt KA. Org Lett. 2003;5:3487–3490. doi: 10.1021/ol035292y. [DOI] [PubMed] [Google Scholar]
- 39.Galliford CV, Scheidt KA. J Org Chem. 2007;72:1811–1813. doi: 10.1021/jo0624086. [DOI] [PubMed] [Google Scholar]
- 40.Galliford CV, Scheidt KA. Chem Comm. 2007:631–633. doi: 10.1039/b609155e. [DOI] [PubMed] [Google Scholar]
- 41.Piloty O. Chem Ber. 1910;43:489. [Google Scholar]
- 42.Robinson R, Robinson GM. J Chem Soc. 1918;43:639–644. [Google Scholar]
- 43.Posvic H, Dombro R, Ito H, Telinski T. J Org Chem. 1974;39:2575–2580. [Google Scholar]
- 44.Baldwin JE, Bottaro JC. J Chem Soc, Chem Commun. 1982:624–625. [Google Scholar]
- 45.Kuhnert N. Angew Chem Int Ed. 2002;41:1863–1866. doi: 10.1002/1521-3773(20020603)41:11<1863::aid-anie1863>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 46.Larhed M, Moberg C, Hallberg A. Acc Chem Res. 2002;35:717–727. doi: 10.1021/ar010074v. [DOI] [PubMed] [Google Scholar]
- 47.Lew A, Krutzik PO, Hart ME, Chamberlin AR. J Comb Chem. 2002;4:95–105. doi: 10.1021/cc010048o. [DOI] [PubMed] [Google Scholar]
- 48.Hayes BL. Aldrichimica Acta. 2004;37:66–77. [Google Scholar]
- 49.Xu Y, Guo QX. Heterocycles. 2004;63:903–974. [Google Scholar]
- 50.Kappe CO. Angew Chem Int Ed. 2004;43:6250–6284. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- 51.de la Hoz A, Diaz-Ortiz A, Moreno A. Chem Soc Rev. 2005;34:164–178. doi: 10.1039/b411438h. [DOI] [PubMed] [Google Scholar]
- 52.Molteni V, Ellis DA. Curr Org Synth. 2005;2:333–375. [Google Scholar]
- 53.Roberts BA, Strauss CR. Acc Chem Res. 2005;38:653–661. doi: 10.1021/ar040278m. [DOI] [PubMed] [Google Scholar]
- 54.Rothemund P. J Am Chem Soc. 1939;61:2912–2915. [Google Scholar]
- 55.Rothemund P. J Am Chem Soc. 1936;58:625–627. [Google Scholar]
- 56.Sessler JL, Mozaffari A, Johnson MR. Org Syn. 1992;70:242–249. [Google Scholar]
- 57.Barkigia KM, Berber MD, Fajer J, Medforth CJ, Renner MW, Smith KM. J Am Chem Soc. 1990;112:8851–8857. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.