

Abstract
Epidemiological evidence suggests that epilepsy and depression are comorbid diseases. In fact, depression is the most common neuropsychiatric disorder associated with epilepsy, particularly temporal lobe epilepsy, and individuals with a history of depression are at a higher risk for developing epilepsy than the general population. Despite the epidemiological evidence for this link, there has been little experimental evidence to support the connection or elucidate possible underlying mechanisms. In an effort to address this problem and develop an animal model of epilepsy and depression comorbidity, we assessed seizure susceptibility and severity parameters in rats selectively bred for either susceptibility (the SwLo, SUS, and HYPER lines) or resistance (the SwHi, RES, and MON RES lines) to depression-like phenotypes. We found that rats bred for susceptibility to depression-like phenotypes experienced higher mortality following kainic acid-induced seizures than their resistant counterparts. In contrast, most line differences were not recapitulated when flurothyl was used to elicit seizures. Stress reduced kainic acid-induced mortality rates in all lines except the HYPER rats, supporting previously established indications that the stress response of HYPER rats is abnormal. These combined results support a neurobiological link between epilepsy and depression, advancing us towards an animal model of their comorbidity.
Keywords: epilepsy, depression, comorbidity, rats, seizure, stress, animal model, kainic acid, flurothyl
1. Introduction
Epidemiological evidence has demonstrated that depression is the most common comorbid psychiatric condition associated with epilepsy, and the risk that an epileptic will develop major depression is approximately five-fold higher than average (Kanner and Nieto, 1999; Wiegartz et al., 1999; Hermann et al., 2000; Harden, 2002; Kanner and Balabanov, 2002; Gaitatzis et al., 2004; Swinkels et al., 2005). Individuals with temporal lobe epilepsy (TLE), in particular, are especially prone to develop depression (Kanner and Nieto, 1999; Paradiso et al., 2001; Harden, 2002). Depression remains underdiagnosed in the epileptic population, however, at least in part because both patients and clinicians often assume that low spirits must be a “normal” response to living with a debilitating neurological disease. Yet that explanation alone cannot fully account for the comorbidity of epilepsy and depression, because it is bidirectional (Kanner and Balabanov, 2002). Epilepsy occurs with approximately five-fold greater frequency among individuals with a history of depression (or even a family history of depression) than among the general population, indicating that the bidirectional relationship is more than a psychosocial phenomenon and that the two disorders likely share common pathogenic mechanisms (Fosgren and Nystrom, 1990; Kanner and Nieto, 1999; Hesdorffer et al., 2000; Kanner and Balabanov, 2002; Thapar et al., 2005; Kanner, 2005). In fact, major depressive episodes and suicide attempts independently increase the risk for developing unprovoked seizures and epilepsy (Hesdorffer et al., 2006), and depression scores and seizure frequency are significant predictors of each other both within and across time (Thapar et al., 2005).
Understanding the biological link between epilepsy and depression is desirable for a host of reasons. Depression has a more profound impact on an epileptic patient’s quality of life than seizure severity or frequency (Johnson et al., 2004; Meldolesi et al., 2006). Furthermore, treating these comorbid individuals presents problems, because some antidepressant drugs increase seizure susceptibility, and, conversely, some anticonvulsant drugs precipitate depressive episodes (Brendt et al., 1987; Trimble, 1996; Alldredge, 1999; Kanner and Nieto, 1999; Pisani et al., 1999). Despite a clear epidemiological link between the two diseases, there is little experimental evidence to support a shared pathology, and there are few animal models that might reveal the potential underlying mechanisms, a deficiency much lamented in recent reviews (eg Kanner, 2003).
In an attempt to experimentally confirm the striking comorbidity between TLE and depression and to simultaneously create an animal model, we tested whether rats selectively bred for depression-like phenotypes are more susceptible to kainic acid-induced seizures.
2. Experimental Procedures
2.1 Selectively bred rats
With the idea of expanding upon the rodent models of depression then available, Weiss and colleagues have selectively bred normal Sprague-Dawley rats for a number of phenotypes relevant to human depression (Scott et al., 1996; Weiss et al., 1998; Weiss et al., 2000; Weiss et al., 2005; West and Weiss, 2005; our unpublished data). These lines include: Swim-Lo active (SwLo), with low motor activity in a Porsolt forced swim test (FST); Swim-Hi active (SwHi), with high motor activity in the FST; Swim-test susceptible (SUS), which are highly susceptible to having their swim-test behavior disrupted by mild stress; Swim-test resistant (RES), which are highly resistant to having their swim-test behavior disrupted by mild stress; Monitor Hyperactive (HYPER), which experience long-lasting hyperactivity following stress; and Monitor Resistant (MON RES), which are resistant to changes in activity following stress. Randomly bred (nonselected; NS) rats served as controls. The following generations were used: SwLo (39–41), SwHi (36, 41), SUS (33, 34, 36), RES (32, 33, 35), HYPER (30, 32, 33), MON RES (28, 32, 33, 35), and NS (36, 38, 39).
Male rats were used for all experiments, and each rat was used in a seizure experiment only once. Animals were housed in ventilated racks (2–3 rats per cage) at the Emory University Briarcliff vivarium. The colony room was maintained on a 12-hour light-dark cycle (lights on from 7 am to 7 pm), and the rats received standard laboratory chow and water ad libitum. Animals were treated in accordance with the Guidelines for Animal Care and Use of the National Institutes of Health, and all experiments were approved by the Emory University Institutional Animal Care and Use Committee.
2.2 Seizure induction
Kainic Acid
Kainic acid (opika-1, Ocean Produce International, Shelburne, Nova Scotia, Canada) was administered to rats as previously described (Ben-Ari et al., 1980; McKhann et al., 2003), but with slight modifications. Rats were injected intraperitoneally (ip) with kainic acid (10 mg/kg), then placed in a clear container and observed closely for behavioral seizures. Latency to various seizure behaviors was scored using a modified version of the Racine scale (Racine, 1972): 0 = no seizure, 1 = staring, 2 = wet-dog shakes, 3 = forelimb clonus, 4 = rearing and falling, and 5 = clonic-tonic (CT) seizure. If a CT seizure was not observed in the first hour, rats were given booster injections of kainic acid (5 mg/kg) every hour until they experienced a CT seizure. Maximal seizure severity was assessed by mortality 24 hours after the CT seizure.
Flurothyl
Rats were placed in an airtight Plexiglas chamber (15 cm × 19.5 cm × 35.5 cm), and the volatile convulsant 2,2,2-trifluroethylether (flurothyl, Sigma-Aldrich, St Louis, MO) was infused at a rate of 20 μL/min onto filter paper, from which it evaporated. Flurothyl was infused until the occurrence of a CT seizure, after which the rat was removed from the chamber. Seizure susceptibility was assessed by measuring latency to CT seizure and by maximal seizure severity (whether or not the animals progressed to tonic extension and death). Tonic extension was defined as when the hind limbs pass through a 90 degree angle with the body and then reach maximum extension pointing straight down, accompanied by full body rigidity.
2.3 Stress induction
Some of the selectively bred rat lines exhibited their depressive-like behaviors only following specific stressors. Therefore, seizure susceptibility was also assessed following exposure to the conditions that elicited those phenotypes. SUS and RES rats were exposed to 30 minutes of 90–95 dB white noise in a novel environment, which is used to elicit their depression-relevant phenotypes, and immediately thereafter were tested for seizure susceptibility. Similarly, HYPER and MON RES rats received 3 hours of tail shock, as described (Scott et al., 1996), then they were tested for seizure susceptibility two days later.
2.4 Statistics
For analysis of seizure threshold (the dose of chemoconvulsant required to produce a CT seizure) and latency (the time from the initial chemoconvulsant administration that a particular stage of seizure occurred), Student’s t-tests were used for comparison between two groups with equal variance, and Mann-Whitney tests were used for comparison between two groups with unequal variance. Fisher’s exact test was used for a comparison of seizure mortality incidence between two groups. Logistic regression was used for analysis of correlation between seizure threshold and mortality rate. GraphPad InStat (version 3.0) and Prism (version 4.0) software for Macintosh were used for all statistical analyses.
3. Results
3.1 Increased kainic acid-induced seizure mortality in rats selectively bred for depression
To determine whether a genetic susceptibility to depression-related behavioral characteristics correlates with an increased susceptibility to limbic seizures, we assessed the response to kainic acid in rats selectively bred for susceptibility or resistance to depression-like phenotypes. Rats of all strains followed a typical behavioral progression following kainic acid administration (10 mg/kg, followed by 5 mg/kg/hr booster doses until CT seizure occurred). All rats tested from required at least one booster dose to produce a CT seizure, with the exception of one SwLo rat and three NS rats.
Within 30 minutes of injection, the rats began to stare, followed by increasingly severe seizure behaviors, including wet-dog shakes, forelimb clonus, rearing and falling, and CT seizures. We found no significant differences in latency to staring, wet-dog shakes, forelimb clonus, rearing and falling, or CT seizures (data not shown). We also did not observe any differences in number of seizures of any stage per unit time. However, following the elicitation of seizures, there were striking differences in mortality among the lines. In general, all lines bred for susceptibility to depression-like phenotypes had higher mortality rates than their depression-resistant counterparts (significant difference for SwLo vs SwHi and SUS vs RES, trend for HYPER vs MON RES) (Fig. 1A). The mortality rate for randomly bred control (NS) rats fell between the susceptible and resistant lines. Of the rats that died, many expired during the first CT seizure, while others continued to have sporadic seizures and died either later that day or were found dead the next morning. The dose of kainic acid required to produce a CT seizure was equivalent between each pair of strains, except for the SUS rats, for which the dose was slightly but significantly increased (Fig. 1B). However, the higher dose probably cannot account for the increased mortality of the SUS rats, because dose and mortality were not significantly correlated across all lines (p > 0.05). For example, some NS control rats that received a cumulative dose of 20 mg/kg survived, while others that received 15 mg/kg or even as little as 10 mg/kg died, and similar patterns were also observed among the other strains.
Fig. 1.
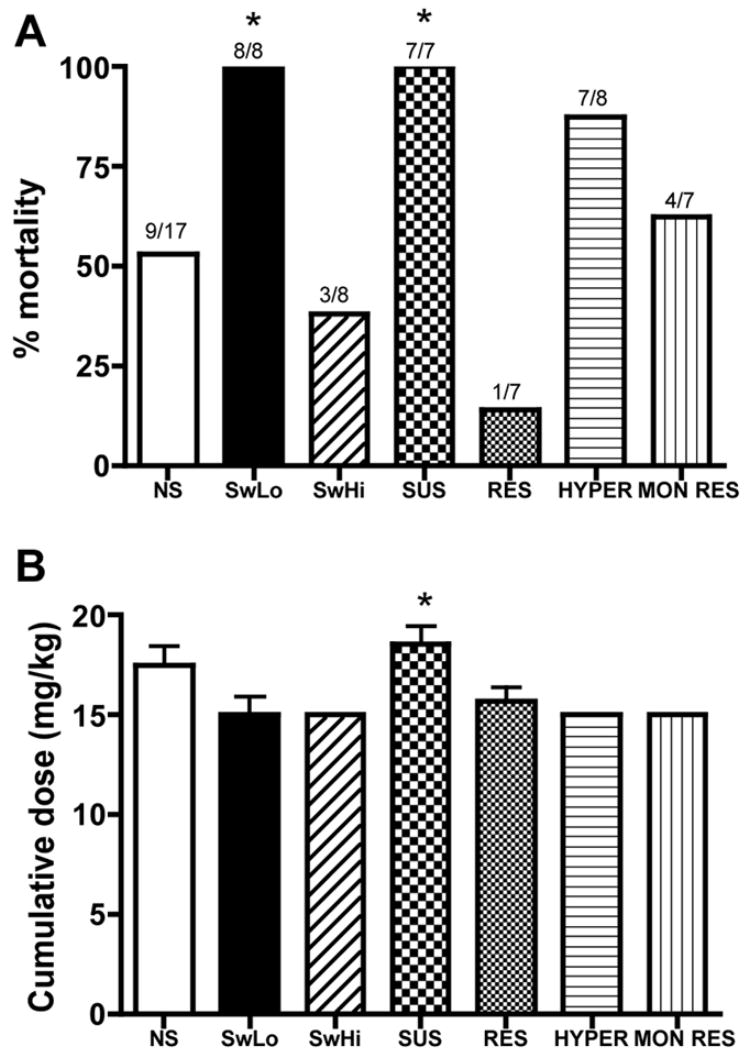
Kainic acid-induced seizures. Kainic acid was administered to rats at a dose of 10 mg/kg, followed by 5 mg/kg booster injections until a clonic-tonic seizure was produced. Shown is (A) mortality, with the number of animals that died over the total tested above each bar, and (B) the cumulative kainic acid dose (mean±SEM) required to produce a CT seizure. * P <0.05 for the depression-susceptible line compared to its depression-resistant counterpart (SwLo vs SwHi, SUS vs RES, HYPER vs MON RES, respectively).
3.2 Rats selectively bred for depression are not more susceptible to flurothyl-induced seizures
To determine whether the SwLo, SUS, and HYPER rats were particularly susceptible to mortality following kainic acid-induced seizures, or whether they were also more susceptible to seizures produced by other compounds, we assessed their response to flurothyl. There were no differences in latency to CT seizure between depression-susceptible lines and their depression-resistant counterparts, with the exception of MON RES rats, which were more resistant (ie, longer latency) than HYPER rats (Fig. 2). None of the animals we tested progressed to tonic extension, nor did any die. These findings suggest that the increased seizure severity leading to mortality observed in the depression-sensitive lines unlikely the result of a general increase in neuronal excitability.
Fig. 2.
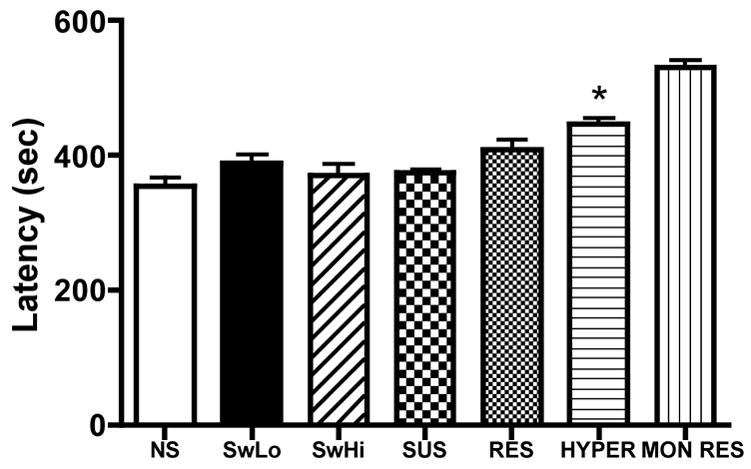
Flurothyl-induced seizures. Flurothyl was administered to rats at a rate of 20 μL/min, and latency to CT seizure was observed. Shown is latency to CT seizure (mean±SEM) for each line. N = 6–16 per group. * P <0.05 for the depression-susceptible line compared to its depression-resistant counterpart (SwLo vs SwHi, SUS vs RES, HYPER vs MON RES, respectively).
3.3 The effects of stress on kanic acid-induced seizures
The selectively bred phenotypes for some lines are observed only following stress. For example, the SUS rats have normal basal activity in the forced swim test, but they display increased immobility following stress (Scott et al., 1996; West and Weiss, 2005). Further, the hyperactive phenotype of the HYPER rats is greatly increased by stress (Weiss et al., 2000; Weiss et al., 2005; our unpublished data). We wished to assess whether the stress paradigms used to elicit depression-like phenotypes also alter seizure susceptibility for each line. We found that stress (ie, 30-minute exposure to 90–95 dB white noise in a novel environment for SUS and RES rats, 3-hour tail shock for HYPER, MON RES, and NS control rats) decreased kainic acid-induced seizure mortality in all lines, except the HYPER (Fig. 3A). Stress did not consistently alter the dose of kainic acid required to produce a CT seizure (ie, no effect in NS or HYPER, significantly decreased dose in SUS and RES, trend towards increased dose in MON RES) (Fig. 3B). Again, the effects of stress on the dose were unrelated to the effects on mortality, because the two measures were not significantly correlated. For example, stress did not alter the kainic acid dose in control rats, but prevented lethality, and most of the SUS rats that received 15 mg/kg kainic acid following stress survived, while all the SUS rats that received 15 mg/kg kainic acid in the absence of stress died. For the flurothyl-induced seizures, stress did not alter any parameters, except for increasing latency to CT seizure in MON RES rats (Fig. 4 and data not shown).
Fig. 3.
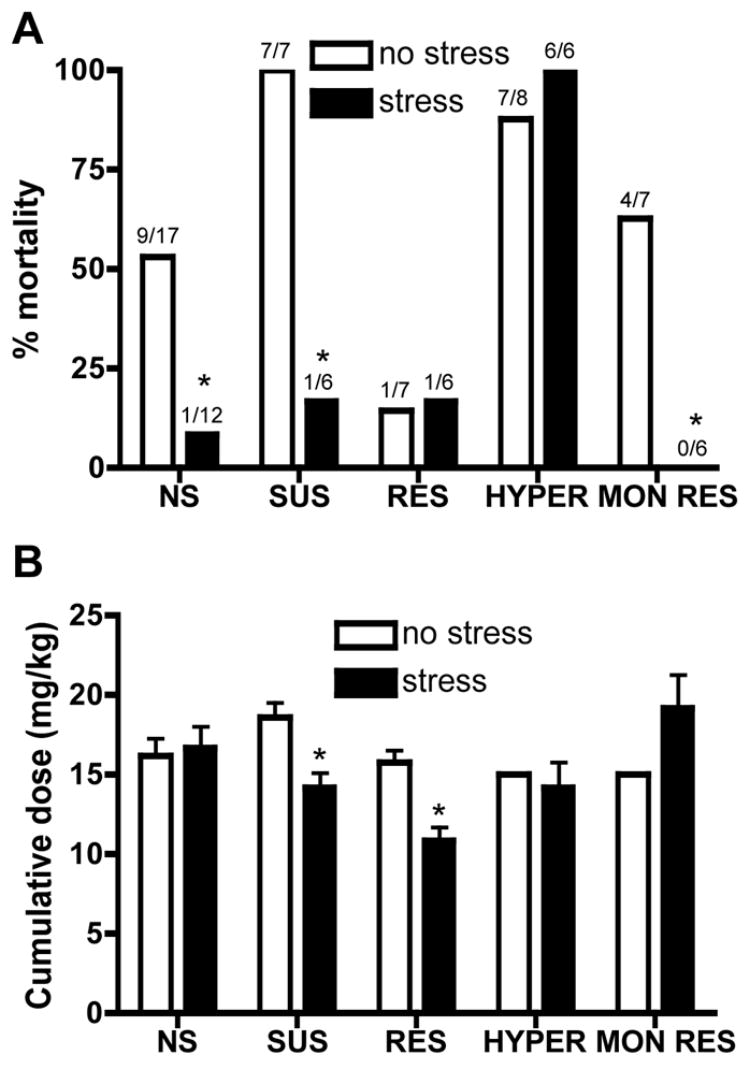
The effects of stress on kainic acid-induced seizures. Kainic acid-induced seizures were assessed following exposure of the rats to stress paradigms used to produce the depression-like phenotype in each line. SUS and RES rats were exposed to 30 minutes of 90–95 dB white noise in a novel environment, while HYPER, MON RES, and NS control rats received 3 hours of tail shock. Shown is (A) mortality, with the number of animals that died over the total tested above each bar, and (B) the cumulative kainic acid dose (mean±SEM) required to produce a CT seizure. * P <0.05 compared to the no-stress condition for each line.
Fig. 4.
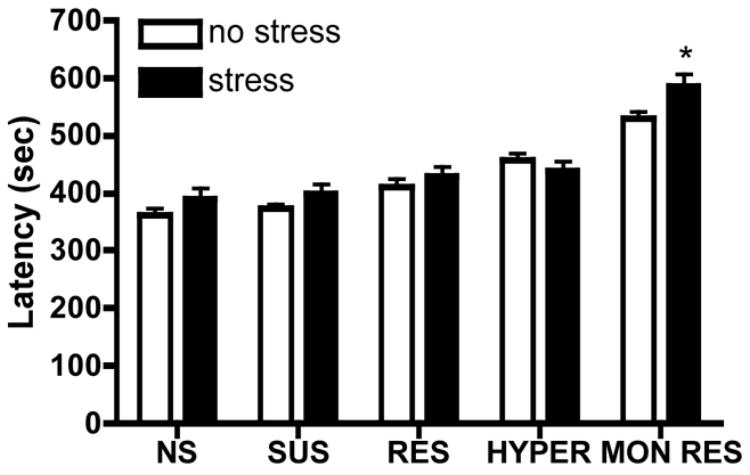
The effects of stress on flurothyl-induced seizures. Flurothyl-induced seizures were assessed following exposure of the rats to stress paradigms used to produce the depression-like phenotype in each line. SUS and RES rats were exposed to 30 minutes of 90–95 dB white noise in a novel environment, while HYPER, MON RES, and NS control rats received 3 hours of tail shock. NS control rats also received 3 hours of tail shock. Shown is latency (mean±SEM) to CT seizure following flurothyl administration (20 μL/min). N = 6–18 per group. * P <0.05 compared to the no-stress condition for each strain.
4. Discussion
Epilepsy and depression appear to be comorbid disorders, and depression in epilepsy is clearly much more than a mere “psychosocial” phenomenon (Kanner, 2005). Despite the wealth of epidemiological evidence linking these diseases, there is a paucity of experimental evidence to support the link, and few animal models have been developed for studying the possible underlying mechanisms. Contó et al (2005) reported that Wistar rats that were susceptible to convulsions induced by methyl-6,7-dimethoxy-4-ethyl-beta-carboline-3-carboxylate (DMCM), a benzodiazepine inverse agonist, were more anxious than DMCM-resistant rats when tested on the elevated plus-maze, but they demonstrated normal depression-like behavior in the forced swim test. Sarkisova and Kulikov (2006) likewise found that rats susceptible to audiogenic seizures displayed increased levels of anxiety but, again, no consistent differences in the forced swim test. Perhaps the best characterized model of epilepsy and depression co-morbidity is the genetically epilepsy-prone rat (GEPR), developed by Jobe and colleagues (reviewed by Daily et al., 1989). These rats were selectively bred for susceptibility to audiogenic seizures, but they have also been reported to display behavioral responses related to depression, such as increased immobility in the forced swim test and decreased saccharin consumption that is possibly indicative of anhedonia (Jobe et al., 1999; Jobe and Weber, 2005).
Relative to the studies described above, we have approached the question of comorbidity from the opposite direction. That is, instead of asking whether seizure susceptibility leads to depression-like behaviors, we asked whether the predisposition to depression-like behaviors increases seizure susceptibility. Our model possesses two other salient features that are particularly relevant to the topic at hand. First, genetic load is a critical component of both epilepsy and depression, and a family history of depression is itself a risk factor for developing epilepsy (Robertson et al., 1987; Kanner and Nieto, 1999; Harden, 2002). The selective breeding approach embodies a strong genetic component, because we used rats selectively bred for more than 30 generations for susceptibility or resistance to behaviors associated with human depression. Of particular note, all three lines bred for depression-relevant behavioral phenotypes were also susceptible to kainic acid-induced seizure mortality, compared to their counterparts that are resistant to these phenotypes, which suggests the lines may share genetic changes that contribute to the phenotypes. Furthermore, both the depression-like and seizure phenotypes were stable over multiple generations, indicating coheritability. Second, the incidence of major depression is particularly high in patients with TLE, ranging from ~20–70% (Kanner and Balabanov, 2002; Kuhn et al., 2003). We found that our rat lines differed in response to kainic acid, which is commonly used to model TLE, but not to any seizure phenotype related to flurothyl, which produces generalized seizures. While it is extremely difficult to tell exactly why an animal dies using any seizure model, it is generally thought that very severe seizures propagate to and impair regions of the brain essential for life, such as those controlling respiration and cardiovascular function (ie brainstem). It is important to note that while the origin of kainic acid-induced seizures is the limbic system, our rats experienced tonic-clonic seizures, indicating seizure generalization. It is unclear why mortality resulted from “secondarily generalized” (kainic acid) seizures, but not “primarily generalized” seizures (flurothyl), but they may involve different brain regions.
Alternatively, sustained severe seizure activity following kainic acid administration may be more devestating than the single tonic-clonic seizure induced by our flurothyl paradigm. Another possibility is that the depression-sensitive animals we used have cardiovascular impairment that is responsible for their deaths. We find it unlikely that all three depression-sensitive lines that we have studied have compromised cardiovascular or other organ systems, and simply died for this reason, while the depression-resistant lines happen to have particularly robust heart function. Also, as part of the selective breeding paradigm, these lines commonly undergo procedures that put significant stress on the cardiovascular system, such as forced swim or a prolonged session (i.e. three hours) of tail shock, and the rats never die during such manipulations. Thus, we attribute the higher kainic acid-induced mortality of the depression-sensitive lines to an increase in seizure propagation and severity. However, a study of heart physiology in the rat lines will be required to rule out the possibility of cardiovascular involvement.
Some of the rat lines were bred for phenotypes that only emerge following stress. While chronic stress can certainly exacerbate seizures, acute stress is typically anticonvulsant, at least in animals (reviewed by Reddy, 2006). We found that stress reduced mortality following kainic acid-induced seizures in all lines except for HYPER rats. This would indicate that the HYPER rats have an abnormal response to stress, as previously shown using other paradigms (Weiss et al., 2000, 2005; our unpublished data).
While we are encouraged by the promise of the new epilepsy and depression co-morbidity models presented in this paper, they have some important limitations. First and foremost, none of the selectively bred rats, even those from the “depression-susceptible” lines, are epileptic. The seizure phenotypes we observed were induced by kainic acid, and we have never observed an unprovoked, spontaneous seizure in these lines. It will be necessary to test these rats in models of epileptogenesis, such as kindling- or pilocarpine-induced epilepsy. Second, it is doubtful that a rodent can experience anything truly analogous to full human depression. The general consensus in the field has become that modeling individual behaviors relevant to human depression will likely yield better results than simultaneously modeling the host of complex cognitive and behavioral features encompassed by depression itself. That said, the forced swim test is still the most widely used animal model of depression and antidepressant drug efficacy, and the SwLo/SwHi and SUS/RES rats have significantly improved on previous incarnations of this model (Weiss et al., 1996, 1998; West and Weiss, 1998; 2005). While psychomotor hyperactivity is a feature of agitated/psychotic depression, the phenotype of the HYPER rats more closely resembles mania or bipolar disorder (Weiss et al., 2000; 2005). Intriguingly, some anticonvulsant drugs, such as valproic acid and carbamazepine, are also first-line treatments for bipolar disorder (Spina and Perugi, 2004; Nasrallah et al., 2006). Finally, we presently know little about the genetics or the neurobiology of these rats. Although a genetic strategy (selective breeding) was used to generate these lines, we have only begun the process of thorough genetic analysis to identify those genes that contribute to the observed phenotypes (Weinshenker et al., 2005). Insofar as the depression-related behaviors and kainic acid-induced seizure differences are coinherited, we would predict that at least some genetic factors underlie both phenotypes.
The greatest value of any animal model of disease is as a tool for studying the disease mechanism(s); in this case, monoamine dysfunction in the hippocampus (and other limbic structures) is an appealing candidate, as originally proposed by Jobe and colleagues (1999; Jobe and Weber, 2005). The hippocampus is heavily implicated in both epilepsy and depression (Hecimovic et al., 2003; Campbell and Macqueen, 2004; Kanner, 2005; Warner-Schmidt and Duman, 2006) and receives dense noradrenergic input from the locus coeruleus and serotonergic input from the dorsal raphe nucleus. Furthermore, norepinephrine and serotonin have both antidepressant and anticonvulsant properties, and an abnormality in either neurotransmitter system could produce both low mood and seizure susceptibility (Jobe et al., 1999; Kanner and Balabanov, 2002). Drugs that block norepinephrine and/or serotonin reuptake are the most common antidepressant drugs used clinically, and these same drugs can also reverse depressive-like behavior in SwLo and SUS rats in the forced swim test (Weiss et al., 1998; West and Weiss, 1998, 2005). Interestingly, both the GEPR-3 rats and our selectively bred rats have monoamine abnormalities (Jobe et al., 1999; Scott et al., 1996; our unpublished data). Thorough characterization of monoamine and other systems in these rats, as well as their neuronal and synaptic organization, may yield clues about the underlying mechanisms of epilepsy and depression comorbidity, leading eventually to improved therapy for individuals who suffer from both disorders.
Acknowledgments
We thank J Moore for technical assistance and C Strauss for critical reading of the manuscript and helpful suggestions. This work was funded by grants from the National Alliance for Research on Schizophrenia and Depression (NARSAD Young Investigator’s Award) and NIH/NINDS (NS053444).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alldredge BK. Seizure risk associated with psychotropic drugs: clinical and pharmacokinetic considerations. Neurology. 1999;53(Suppl 2):S68–S75. [PubMed] [Google Scholar]
- Ben-Ari Y, Tremblay E, Ottersen OP, Meldrum BS. The role of epileptic activity in hippocampal and “remote” cerebral lesions induced by kainic acid. Brain Res. 1980;191:79–97. doi: 10.1016/0006-8993(80)90316-9. [DOI] [PubMed] [Google Scholar]
- Brent DA, Crumrine PK, Varma RR, Allan M, Allman C. Phenobarbital treatment and major depressive disorder in children with epilepsy. Pediatrics. 1987;80:909–917. [PubMed] [Google Scholar]
- Campbell S, Macqueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci. 2004;29:417–426. [PMC free article] [PubMed] [Google Scholar]
- Contó MB, de Carvalho JG, Benedito MA. Behavioral differences between subgroups of rats with high and low threshold to clonic convulsions induced by DMCM, a benzodiazepine inverse agonist. Pharmacol Biochem Behav. 2005;82:417–426. doi: 10.1016/j.pbb.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Dailey JW, Reigel CE, Mishra PK, Jobe PC. Neurobiology of seizure predisposition in the genetically epilepsy-prone rat. Epilepsy Res. 1989;3:3–17. doi: 10.1016/0920-1211(89)90063-6. [DOI] [PubMed] [Google Scholar]
- Forsgren L, Nystrom L. An incident case-referent study of epileptic seizures in adults. Epilepsy Res. 1990;6:66–81. doi: 10.1016/0920-1211(90)90010-s. [DOI] [PubMed] [Google Scholar]
- Gaitatzis A, Carroll K, Majeed AW, Sander J. The epidemiology of the comorbidity of epilepsy in the general population. Epilepsia. 2004;45:1613–1622. doi: 10.1111/j.0013-9580.2004.17504.x. [DOI] [PubMed] [Google Scholar]
- Harden CL. The co-morbidity of depression and epilepsy: epidemiology, etiology, and treatment. Neurology. 2002;59(Suppl 4):S48–S55. doi: 10.1212/wnl.59.6_suppl_4.s48. [DOI] [PubMed] [Google Scholar]
- Hecimovic H, Goldstein JD, Sheline YI, Gilliam FG. Mechanisms of depression in epilepsy from a clinical perspective. Epilepsy Behav. 2003;4 (Suppl 3):S25–S30. doi: 10.1016/j.yebeh.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Hermann BP, Seidenberg M, Bell B. Psychiatric comorbidity in chronic epilepsy: identification, consequences, and treatment of major depression. Epilepsia. 2000;41 (Suppl 2):S31–S41. doi: 10.1111/j.1528-1157.2000.tb01522.x. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Hauser WA, Annegers JF, Cascino G. Major depression is a risk factor for seizures in older adults. Ann Neurol. 2000;47:246–249. [PubMed] [Google Scholar]
- Hesdorffer DC, Hauser WA, Olafsson E, Ludvigsson P, Kjartansson O. Depression and suicide attempt as risk factors for incident unprovoked seizures. Ann Neurol. 2006;59:35–41. doi: 10.1002/ana.20685. [DOI] [PubMed] [Google Scholar]
- Jobe PC, Dailey JW, Wernicke JF. A noradrenergic and serotonergic hypothesis of the linkage between epilepsy and affective disorders. Crit Rev Neurobiol. 1999;13:317–356. doi: 10.1615/critrevneurobiol.v13.i4.10. [DOI] [PubMed] [Google Scholar]
- Jobe PC, Weber RW. Affective disorder and epilepsy comorbidity in the genetically epilepsy prone-rat (GEPR) In: Gilliam F, Kanner AM, Sheline YI, editors. Depression and Brain Dysfunction. Taylor & Francis Medical Books; London: 2005. pp. 121–157. [Google Scholar]
- Johnson EK, Jones JE, Seidenberg M, Hermann BP. The relative impact of anxiety, depression, and clinical seizure features on health-related quality of life in epilepsy. Epilepsia. 2004;45:544–550. doi: 10.1111/j.0013-9580.2004.47003.x. [DOI] [PubMed] [Google Scholar]
- Kanner AM. Depression in epilepsy: prevalence, clinical semiology, pathogenic mechanisms, and treatment. Biol Psychiatry. 2003;54:388–398. doi: 10.1016/s0006-3223(03)00469-4. [DOI] [PubMed] [Google Scholar]
- Kanner AM. Depression in epilepsy: a neurobiologic perspective. Epilepsy Curr. 2005;5:21–27. doi: 10.1111/j.1535-7597.2005.05106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner AM, Balabanov A. Depression and epilepsy: how closely related are they? Neurology. 2002;58(Suppl 5):S27–S39. doi: 10.1212/wnl.58.8_suppl_5.s27. [DOI] [PubMed] [Google Scholar]
- Kanner AM, Nieto JC. Depressive disorders in epilepsy. Neurology. 1999;53(Suppl 2):S26–S32. [PubMed] [Google Scholar]
- Kuhn KU, Quednow BB, Thiel M, Falkai P, Maier W, Elger CE. Antidepressive treatment in patients with temporal lobe epilepsy and major depression: a prospective study with three different antidepressants. Epilepsy Behav. 2003;4:674–679. doi: 10.1016/j.yebeh.2003.08.009. [DOI] [PubMed] [Google Scholar]
- McKhann GM, 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122:551–561. doi: 10.1016/s0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- Meldolesi GN, Picardi A, Quarato PP, Grammaldo LG, Esposito V, Mascia A, Sparano A, Morosini P, Di Gennaro G. Factors associated with generic and disease-specific quality of life in temporal lobe epilepsy. Epilepsy Res. 2006;69:135–146. doi: 10.1016/j.eplepsyres.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Nasrallah HA, Ketter TA, Kalali AH. Carbamazepine and valproate for the treatment of bipolar disorder: a review of the literature. J Affect Disord. 2006;95:69–78. doi: 10.1016/j.jad.2006.04.030. [DOI] [PubMed] [Google Scholar]
- Paradiso S, Hermann BP, Blumer D, Davies K, Robinson RG. Impact of depressed mood on neuropsychological status in temporal lobe epilepsy. J Neurol Neurosurg Psychiatry. 2001;70:180–185. doi: 10.1136/jnnp.70.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani F, Spina E, Oteri G. Antidepressant drugs and seizure susceptibility: from in vitro data to clinical practice. Epilepsia. 1999;40 (Suppl 10):S48–S56. doi: 10.1111/j.1528-1157.1999.tb00885.x. [DOI] [PubMed] [Google Scholar]
- Reddy DS. Physiological role of adrenal deoxycorticosterone-derived neuroactive steroids in stress-sensitive conditions. Neuroscience. 2006;138:911–920. doi: 10.1016/j.neuroscience.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Robertson MM, Trimble MR, Townsend HR. Phenomenology of depression in epilepsy. Epilepsia. 1987;28:364–372. doi: 10.1111/j.1528-1157.1987.tb03659.x. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure Electroencephalogr. Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Sarkisova KY, Kulikov MA. Behavioral characteristics of WAG/Rij rats susceptible and non-susceptible to audiogenic seizures. Behav Brain Res. 2006;166:9–18. doi: 10.1016/j.bbr.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Scott PA, Cierpial MA, Kilts CD, Weiss JM. Susceptibility and resistance of rats to stress-induced decreases in swim-test activity: a selective breeding study. Brain Res. 1996;725:217–230. doi: 10.1016/0006-8993(96)00093-5. [DOI] [PubMed] [Google Scholar]
- Spina E, Perugi G. Antiepileptic drugs: indications other than epilepsy. Epileptic Disord. 2004;6:57–75. [PubMed] [Google Scholar]
- Swinkels WA, Kuyk J, van Dyck R, Spinhoven P. Psychiatric comorbidity in epilepsy. Epilepsy Behav. 2005;7:37–50. doi: 10.1016/j.yebeh.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Thapar A, Roland M, Harold G. Do depression symptoms predict seizure frequency--or vice versa? J Psychosom Res. 2005;59:269–274. doi: 10.1016/j.jpsychores.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Trimble MR. Anticonvulsant-induced psychiatric disorders. The role of forced normalisation. Drug Saf. 1996;15:159–166. doi: 10.2165/00002018-199615030-00001. [DOI] [PubMed] [Google Scholar]
- Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16:239–249. doi: 10.1002/hipo.20156. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, Wilson MM, Williams KM, Weiss JM, Lamb NE, Twigger SN. A new method for identifying informative genetic markers in selectively bred rats. Mamm Genome. 2005;16:784–791. doi: 10.1007/s00335-005-0047-6. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Boss-Williams KA, Moore JP, Demetrikopoulos MK, Ritchie JC, West CH. Testing the hypothesis that locus coeruleus hyperactivity produces depression-related changes via galanin. Neuropeptides. 2005;39:281–287. doi: 10.1016/j.npep.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Cierpial MA, West CH. Selective breeding of rats for high and low motor activity in a swim test: toward a new animal model of depression. Pharmacol Biochem Behav. 1998;61:49–66. doi: 10.1016/s0091-3057(98)00075-6. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Demetrikopoulos MK, McCurdy PM, West CHK, Bonsall RW. Depression seen through an animal model: an expanded hypothesis of pathophysiology and improved models. In: Davidson RJ, editor. Anxiety, Depression, and Emotion. Oxford University Press; Oxford: 2000. pp. 3–35. [Google Scholar]
- West CH, Weiss JM. Effects of antidepressant drugs on rats bred for low activity in the swim test. Pharmacol Biochem Behav. 1998;61:67–79. doi: 10.1016/s0091-3057(98)00076-8. [DOI] [PubMed] [Google Scholar]
- West CH, Weiss JM. A selective test for antidepressant treatments using rats bred for stress-induced reduction of motor activity in the swim test. Psychopharmacology (Berl) 2005;182:9–23. doi: 10.1007/s00213-005-0048-x. [DOI] [PubMed] [Google Scholar]
- Wiegartz P, Seidenberg M, Woodard A, Gidal B, Hermann B. Co-morbid psychiatric disorder in chronic epilepsy: recognition and etiology of depression. Neurology. 1999;53(Suppl 2):S3–S8. [PubMed] [Google Scholar]