

Summary
Steroid receptors (SR), which are ligand activated transcription factors, and their coactivators are phosphoproteins whose activities are regulated by cell signaling pathways. Many of the identified phosphorylation sites in these proteins contain Ser/Thr-Pro motifs suggesting that they are substrates for cyclin dependent kinases and/or for mitogen activated protein kinases. An analysis of the roles of cyclins and their kinases in regulating receptor action has revealed that there are both stimulatory and inhibitory actions of cyclins, that some of the actions are independent of the partner kinases and that these activities are receptor specific. Consistent with this finding, the limited analyses of receptor activity as a function of cell cycle reveal distinct patterns of activation. SR often regulate cell proliferation. Thus, the cross talk between cyclins and their kinases and the SR provides a means for integrating the actions of the SR with the cell cycle status of cells.
Keywords: steroid receptor, cyclin, progesterone receptor, androgen receptor, cell cycle
Introduction
SR belong to a family of ligand activated transcription factors (Evans, R.M., 1988) that contain carboxyl terminal ligand binding domains, centrally located DNA binding domains, and poorly conserved amino terminal domains that vary greatly in length and in their overall contribution to the transcriptional activity of the SR. In the best characterized mode of SR action, upon hormone binding the SR dissociates from a heat shock protein complex, forms a homodimer and binds to a sequence specific DNA element recruiting a series of coactivator complexes that remodel chromatin and facilitate transcription of target genes. In an alternative mode of action, SR can induce or inhibit transcription through protein/protein interactions with other transcription factors without binding directly to DNA. Finally, there is evidence that these same SR residing at the cell membrane or in the cytoplasm activate a variety of cell signaling pathways through interactions with Src, G proteins, and other signaling molecules. Activation of these pathways can impact the genomic actions of the SR (Edwards, D.P., et al., 2003; Watson, C.S. and Lange, C.A., 2005). The SR and their coactivators are phosphorylated (Weigel, N.L., 1996) and these phosphorylations regulate activity. (Rowan, B.G., et al., 2000; Wu, R.-C., et al., 2002). Studies of the p160 coactivator SRC-3 (AIB1) show that different patterns of phosphorylation allow this coactivator to preferentially activate subsets of transcription factors (Wu, R.-C., et al., 2002; Wu, R.-C., et al., 2004). The finding that coactivators and SR contain many phosphorylation sites with Ser/Thr-Pro motifs, a minimal requirement for phosphorylation by cyclin dependent kinases (Cdk) and mitogen activated kinases (MAPK) suggests that the phosphorylation status and activities of SR might be regulated as a function of cell cycle and that cyclins and their partner kinases play a role in regulating activity. Studies to date reveal a surprising diversity in the roles of cyclins in regulating SR actions. In some cases the actions of the cyclins are independent of their partner kinases and, in contrast to many of the well characterized coactivators and corepressors that are shared by all of the SR, these actions are SR specific.
Regulation of SR by cyclins and cyclin dependent kinases
There is increasing evidence that cyclins and cyclin dependent kinases play important roles in regulating SR function. In contrast to the well characterized SR coactivators, the actions of the cyclins differ substantially depending on the target SR. Both kinase dependent and kinase independent actions have been identified. Cyclins that stimulate SR activity often increase the agonist activity of partial antagonists. Levels of cyclins are often elevated in cancers potentially decreasing the utility of antagonists as therapeutic agents.
Cyclin D1
The first cyclin identified as having a role in regulating SR action was cyclin D1. D type cyclins are expressed in G1 and act as partners for Cdk4 and Cdk6. Both hormone dependent and hormone independent transcriptional activities of estrogen receptor (ER) are stimulated by cyclin D1 independent of its partner kinase, whose co-expression reduces the potentiation of ER activity by cyclin D1 (Zwijsen, R.M.L., et al., 1997). Tamoxifen, an ER antagonist in breast, does not block cyclin D1 dependent induction of ER activity. Cyclin D1 binds p160 coactivator complexes; it interacts with the ER LBD (ligand binding domain) (Zwijsen, R.M.L., et al., 1997) and recruits coactivators to the ER complex in the absence of agonist. In cells that express high levels of endogenous cyclin D1, co-transfection of a mutant cyclin D1 lacking the ability to bind to coactivators but capable of binding ER reduces ER transcriptional activity (Zwijsen, R.M., et al., 1998). Thus, endogenous ER activity is potentiated by cyclin D1 in cells that overexpress cyclin D1.
In contrast, multiple investigators have shown that cyclin D1 inhibits androgen receptor (AR) activity (Knudsen, K.E., et al., 1999). Mutant cyclin D1 unable to bind its partner kinase, Cdk4, is equally effective in inhibiting AR action. Multiple AR interaction sites and mechanisms of action have been suggested. Pestell’s group demonstrated that an isolated fragment containing amino acids 633–668 in the hinge region interacts with cyclin D1 (Reutens, A.T., et al., 2001). Cyclin D1 also competed with AR for interaction with P/CAF providing an additional means for inhibition independent of cyclin D1 binding directly to AR. Knudsen’s group found that cyclin D1 interacted with AR through the FXXLF motif blocking the amino terminal AR interaction with the carboxyl terminal of AR that is important for full transcriptional activation (Burd, C.J., et al., 2005). Cyclin D1 is expressed as splice variants. The cyclin D1a splice variant of cyclin D1 inhibits AR activity and AR dependent cell growth (Burd, C.J., et al., 2006). In contrast, a second variant, cyclin D1b, is less effective in inhibiting AR action although the differences are target gene specific. Strikingly, the cyclin D1b splice variant does not inhibit AR dependent cell growth (Burd, C.J., et al., 2006). Consistent with a role for cyclin D1a in inhibiting AR action, the transcriptional activity of AR measured as a function of cell cycle is lowest at G1/S when cyclin D1a is expressed (Martinez, E.D. and Danielsen, M., 2002).
Cyclin E
Cyclin E is expressed in late G1 and is a partner for Cdk2. The activities of AR, PR, and GR are all influenced by Cyclin E although the modes of action differ. Cyclin E stimulates AR activity independent of its partner kinase, Cdk2 (Yamamoto, A., et al., 2000). Overexpression of cyclin E also stimulates AR activity in the presence of the AR antagonist, hydroxyflutamide, suggesting that aberrant overexpression of cyclin E has the potential to induce growth of androgen dependent prostate cancer cells despite the presence of an anti-androgen.
The actions of cyclin E on other SR are less well characterized. Overexpression of cyclin E alone had no effect on the activity of ER or PR (Yamamoto, A., et al., 2000). Interestingly, overexpression of cyclin E reduced PR expression in HeLa cells stably expressing PR-B and mutation of one of the PR phosphorylation sites, S400, blocked the reduction in expression (Pierson-Mullany, L.K. and Lange, C.A., 2004). Moreover, a Cdk2 inhibitor enhanced PR expression although inhibition of Cdk2 activity/expression inhibits PR activity. Ser400 has been shown previously to be a candidate Cdk2 site in in vitro phosphorylation studies and a Cdk2 inhibitor blocked EGF dependent phosphorylation of this site implicating Cdk2 in its phosphorylation. The glucocorticoid receptor (GR) is a substrate for cyclin E/Cdk2 in vitro; Ser224, one of the authentic GR phosphorylation sites is phosphorylated by this kinase. When GR is expressed in yeast, elimination of CDC28 (the cyclin dependent kinase that complexes with both G1 and G2 cyclins), reduces the transcriptional activity of GR as does elimination of subsets of G1 or G2 cyclins (Krstic, M.D., et al., 1997). Thus, cyclin E/Cdk2 may potentiate GR action in vivo.
Cyclin A
Cyclin A is an S phase kinase that can partner with either Cdk2 or Cdk1. Cyclin A potentiates the activity of several SR although the mechanisms of potentiation differ. AR activity is increased by the overexpression of cyclin A2 (Narayanan, R., et al., 2005a), the cyclin A form expressed in adult tissues. Cyclin A1, which is more highly expressed during development, is re-expressed in prostate cancer and in prostate cancer cell lines. The combination of cyclin A1, AR, and Rb (retinoblastoma protein) enhances expression of VEGF (vascular endothelial cell growth factor) (Wegiel, B., et al., 2005). Although AR is required for this induction, the mechanism of potentiation by cyclin A1 has not been addressed. In vitro, cyclin A2/Cdk2 phosphorylates GR on two amino-terminal sites, Ser224 and Ser232 (Krstic, M.D., et al., 1997), both of which are phosphorylated in vivo. The kinase and cyclin studies in yeast described above are also consistent with a role for cyclin A/Cdk2 in positively modulating GR activity.
The activity of ER is also stimulated by overexpression of cyclin A and Cdk2 is required (Rogatsky, I., et al., 1999). Cyclin A potentiates ligand independent activity of ER as well as enhancing tamoxifen induced activity. In vitro phosphorylation studies revealed that serines 104 and 106, two previously identified ER phosphorylation sites, are phosphorylated by cyclin A/Cdk2. The potentiation of ER activity by cyclin A is dependent upon these phosphorylation sites (Rogatsky, I., et al., 1999).
The transcriptional activity of PR is also enhanced by cyclin A, but the mechanism of potentiation differs from that of ER. The activity also is dependent upon the partner kinase, Cdk2 (Narayanan, R., et al., 2005a). The most striking difference in mechanism is that potentiation of PR activity is independent of Cdk2 dependent phosphorylation of PR. PR is expressed as two isoforms, PR-B and the shorter PR-A form that lacks the first 164 amino acids of PR-B; the activities of both are potentiated by cyclin A (Narayanan, R., et al., 2005a). Although a number of the in vivo phosphorylation sites identified in PR can be phosphorylated by cyclin A/Cdk2 in vitro (Knotts, T.A., et al., 2001), mutation of every candidate Ser/Thr-Pro site in PR-A has no effect on the ability of cyclin A to potentiate PR activity (Narayanan, R., et al., 2005a). This finding led to the hypothesis that cyclin A/Cdk2 might be acting as a PR coactivator. Subsequent studies demonstrated that PR interacts with cyclin A in vitro as well as in cells. Hormone dependent recruitment of cyclin A to a stably integrated MMTV promoter was detected (Narayanan, R., et al., 2005a).
Inhibition of Cdk activity with roscovitine blocks PR dependent induction of a PR responsive luciferase reporter as did transfection with a Cdk2 siRNA (Narayanan, R., et al., 2005a). Roscovitine also blocked hormone dependent induction of an endogenous PR responsive gene, metallothionein IIA, in T47D cells but had no effect on cadmium induction of metallothionein IIA. Thus, the inhibition was PR specific. Chromatin immunoprecipitation (ChIP) studies of a stably transfected MMTV promoter revealed that roscovitine treatment had no effect on PR binding to the promoter or on recruitment of cyclin A. However, recruitment of the p160 coactivator, SRC-1, was greatly reduced (Narayanan, R., et al., 2005a). Subsequent in vitro studies showed that phosphatase treatment of SRC-1 reduced its ability to bind to PR and that rephosphorylation with cyclin A/Cdk2 restored binding. This suggests that the ability of PR to bind cyclin A/Cdk2 creates a local high concentration of kinase facilitating phosphorylation of SRC-1 on a site(s) that enhances affinity for PR (Narayanan, R., et al., 2005a). Previous studies had shown that the LXXLL motifs in SRC-1 are required for the interaction with PR. These studies show that there is an additional requirement for SRC-1 phosphorylation.
Cyclin H and cyclin T
In addition to the cell cycle regulated cyclins, there are other cyclins that associate with kinases whose best characterized function is the phosphorylation of the C-terminal tail of RNA polymerase II. Cyclin H/Cdk7, components of Cdk-activating kinase (CAK), enhances the activity of SR and, in some cases, directly phosphorylates the SR. Overexpression of CAK stimulates the transcriptional activity of AR (Lee, D.K., et al., 2000). CAK interacts with AR through its amino-terminal region and in vitro binding studies show that both Cdk7 and cyclin H interact well with the amino-terminus and that the third component of CAK, Mat1, interacts more weakly. ER activity is also increased by overexpression of Cdk7 (Chen, D., et al., 2000). Mutation of one of the ER phosphorylation sites, Ser118, eliminated potentiation by Cdk7. Subsequent in vivo and in vitro studies showed that the kinase does phosphorylate Ser118 (Chen, D., et al., 2000). Whether CAK phosphorylates AR as well as ER remains to be determined.
P-TEFb (cyclin T/Cdk9) also phosphorylates the CTD of Pol II. Dominant negative Cdk9 reduces AR activity and AR interacts with the kinase subunit both in vivo and in vitro through the amino terminal domain of AR (Lee, D.K., et al., 2001). Cyclin T1 interacts with ER through the ligand binding domain of ER and is recruited in a hormone dependent manner to the promoter of the pS2 gene (Wittmann, B.M., et al., 2005). Overexpression of cyclin T1 enhances ER activity measured with an ER responsive reporter. Thus, this kinase potentiates the activity of both ER and AR, but similar to some of the other cyclin/kinases, the sites of interaction are distinct.
Cell cycle dependence of SR function
The distinct responses of SR to overexpression of cyclins and/or enhanced kinase activity leads to the prediction that SR activity should vary as a function of the cell cycle and that cell cycle dependence of activity should be SR specific (Bodwell, J.E., et al., 1998). Early studies of GR activity suggested that GR activity was inhibited in G2. In a hepatoma cell line expressing rat GR, G2 synchronization in the presence of Hoechst 33342 reduced the ability of GR to induce a stably transfected MMTV reporter, but not to inhibit dexamethasone dependent repression of the proliferin promoter (Hsu, S.-C. and DeFranco, D.B., 1995). However, subsequent studies in mouse neuronal HT-22 cells and in CHO cells suggested that GR is capable of transactivation in G2 and that the observed inhibition may be a direct effect of Hoechst 33342 on GR activity (Abel, G.A., et al., 2002).
Cell cycle analysis of AR action in murine L929 fibroblasts showed that AR activity was highest in G0, strongly reduced at the G1/S interface and partially restored in S phase (Martinez, E.D. and Danielsen, M., 2002). AR levels were also greatly reduced in G1/S, but activity was reduced more than can be accounted for by the differences in protein expression. The inhibition of activity was AR specific as GR activity in the same cells was only modestly reduced in G1/S. Treatment with the histone deacetylase inhibitor, trichostatin A (TSA), strongly increased the activity of AR in G1/S suggesting that the defect is due at least in part to inadequate histone acetyl transferase (HAT) activity.
The activity of PR has also been measured as a function of cell cycle using a stably transfected MMTVCAT reporter in T47D breast cancer cells. In contrast to GR and AR, PR activity was highest in S phase where cyclin A is most highly expressed with reduced activity in G1 and in G2/M (Narayanan, R., et al., 2005b). However, some of the reduction in G2/M can be attributed to the reduction in PR expression. Consistent with the Cdk2 inhibitor studies, the ability of PR to recruit the p160 coactivators, SRC-1 and SRC-3, was reduced in G1 relative to S phase. Treatment with TSA restored transcriptional activity in G1 and in G2/M (Narayanan, R., et al., 2005b).
Summary
The studies summarized above demonstrate that cyclins have unexpected functions independent of their partner kinases. The studies of the regulation of SR activity show that the influence of cyclins and their partner kinases are SR specific. In most cases, only the classical transcriptional activation through a steroid response element has been studied. Whether cyclins and their partner kinases differentially regulate SR dependent repression of transcription or activation of transcription that is dependent upon protein/protein interactions remains to be determined. The difference in response to cyclins and their partner kinases is supported by the unique cell cycle dependent patterns of SR activity although this, too, is an area that requires more investigation.
Figure 1.
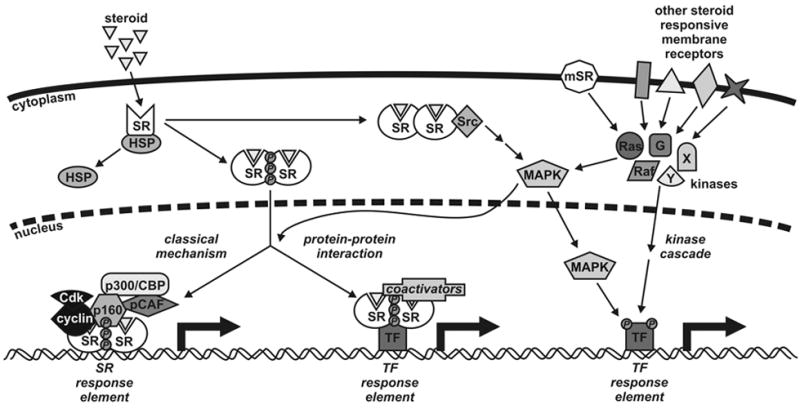
Model for steroid receptor (SR) action. Steroid hormones difuse across the cell membrane where they bind to their cognate receptor in the cytoplasm of target cells. Ligand binding induces conformational changes in the receptor, dissociation of heat shock proteins (HSPs), dimerization and nuclear translocation. In the classical mechanism for SR action, the SR dimer binds to specific DNA response elements situated in the regulatory regions of target genes. This is followed by recruitment of coactivators, such as p160s, cyclins, p300/CBP and pCAF, and other components of the general transcription machinery enabling RNA synthesis. SRs also influence transcription regulated by other transcription factors (TFs) through protein-protein interactions and coactivator recruitment rather than DNA binding. These mechanisms are in turn influenced by phosphorylation of the receptor by multiple kinase pathways such as Src, MAPK, Ras, Raf and G-proteins. Both cytoplasmic and membrane bound receptors (mSRs) influence kinase signaling cascades leading to the phosphorylation of transcription factors. Other steroid responsive receptors with no homology to nuclear receptors are also capable of altering cellular signaling.
Acknowledgments
These studies were supported in part by NIH grants, CA57539 (NLW and NLM) and DK65252 (NLW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel GA, Wochnik GM, Rüegg J, Rouyer A, Holsboer F, Rein T. Activity of the GR in G2 and mitosis. Mol Endocrinol. 2002;16:1352–1366. doi: 10.1210/mend.16.6.0842. [DOI] [PubMed] [Google Scholar]
- Bodwell JE, Webster JC, Jewell CM, Cidlowski JA, Hu JM, Munck A. Glucocorticoid receptor phosphorylation: overview, function and cell cycle-dependence. J Steroid Biochem Mol Biol. 1998;65:91–99. doi: 10.1016/s0960-0760(97)00185-4. [DOI] [PubMed] [Google Scholar]
- Burd CJ, Petre CE, Moghadam H, Wilson EM, Knudsen KE. Cyclin D1 binding to the androgen receptor (AR) NH2-terminal domain inhibits activation function 2 association and reveals dual roles for AR corepression. Mol Endocrinol. 2005;19:607–620. doi: 10.1210/me.2004-0266. [DOI] [PubMed] [Google Scholar]
- Burd CJ, Petre CE, Morey LM, Wang Y, Revelo MP, Haiman CA, Lu S, Fenoglio-Preiser CM, Li J, Knudsen ES, Wong J, Knudsen KE. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci USA. 2006;103:2190–2195. doi: 10.1073/pnas.0506281103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Riedl T, Washbrook E, Pace PE, Coombes RC, Egly JM, Ali S. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol Cell. 2000;6:127–137. [PubMed] [Google Scholar]
- Edwards DP, Wardell SE, Boonyaratanakornkit V. Progesterone receptor interacting proteins and cross-talk with cell signaling pathways. J Steroid Biochem Mol Biol. 2003;83:173–186. doi: 10.1016/s0960-0760(02)00265-0. [DOI] [PubMed] [Google Scholar]
- Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SC, DeFranco DB. Selectivity of cell cycle regulation of glucocorticoid receptor function. J Biol Chem. 1995;270:3359–3364. doi: 10.1074/jbc.270.7.3359. [DOI] [PubMed] [Google Scholar]
- Knotts TA, Orkiszewski RS, Cook RG, Edward DP, Weigel NL. Identification of a phosphorylation site in the hinge region of the human progesterone receptor and additional amino-terminal phosphorylation sites. J Biol Chem. 2001;276:8475–8483. doi: 10.1074/jbc.M009805200. [DOI] [PubMed] [Google Scholar]
- Knudsen KE, Cavenee WK, Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res. 1999;59:2297–2301. [PubMed] [Google Scholar]
- Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol. 1997;17:3947–3954. doi: 10.1128/mcb.17.7.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DK, Duan HO, Chang C. Androgen receptor interacts with the positive elongation factor P-TEFb and enhances the efficiency of transcriptional elongation. J Biol Chem. 2001;276:9978–9984. doi: 10.1074/jbc.M002285200. [DOI] [PubMed] [Google Scholar]
- Lee DK, Duan HO, Chang C. From androgen receptor to the general transcription factor TFIIH. J Biol Chem. 2000;275:9308–9313. doi: 10.1074/jbc.275.13.9308. [DOI] [PubMed] [Google Scholar]
- Martinez ED, Danielsen M. Loss of androgen receptor transcriptional activity at the G(1)/S transition. J Biol Chem. 2002;277:29719–29729. doi: 10.1074/jbc.M112134200. [DOI] [PubMed] [Google Scholar]
- Narayanan R, Adigun AA, Edwards DP, Weigel NL. Cyclin-dependent kinase activity is required for progesterone receptor function: novel role for cyclin A/Cdk2 as a progesterone receptor coactivator. Mol Cell Biol. 2005a;25:264–277. doi: 10.1128/MCB.25.1.264-277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Edwards DP, Weigel NL. Human progesterone receptor displays cell cycle-dependent changes in transcriptional activity. Mol Cell Biol. 2005b;25:2885–2898. doi: 10.1128/MCB.25.8.2885-2898.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson-Mullany LK, Lange CA. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol. 2004;24:10542–10557. doi: 10.1128/MCB.24.24.10542-10557.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reutens AT, Fu M, Wang C, Albanese C, McPhaul MJ, Sun Z, Balk SP, Janne OA, Palvimo JJ, Pestell RG. Cyclin D1 binds the androgen receptor and regulates hormone-dependent signaling in a p300/CBP-associated factor (P/CAF)-dependent manner. Mol Endocrinol. 2001;15:797–811. doi: 10.1210/mend.15.5.0641. [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Trowbridge JM, Garabedian MJ. Potentiation of human estrogen receptor α transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J Biol Chem. 1999;274:22296–22302. doi: 10.1074/jbc.274.32.22296. [DOI] [PubMed] [Google Scholar]
- Rowan BG, Weigel NL, O’Malley BW. Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation sites and phosphorylation through the mitogen-activated protein kinase pathway. J Biol Chem. 2000;275:4475–4483. doi: 10.1074/jbc.275.6.4475. [DOI] [PubMed] [Google Scholar]
- Watson CS, Lange CA. Steadying the boat: integrating mechanisms of membrane and nuclear-steroid-receptor signalling. EMBO Rep. 2005;6:116–119. doi: 10.1038/sj.embor.7400336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel B, Bjartell A, Ekberg J, Gadaleanu V, Brunhoff C, Persson JL. A role for cyclin A1 in mediating the autocrine expression of vascular endothelial growth factor in prostate cancer. Oncogene. 2005;24:6385–6393. doi: 10.1038/sj.onc.1208795. [DOI] [PubMed] [Google Scholar]
- Weigel NL. Steroid hormone receptors and their regulation by phosphorylation. Biochem J. 1996;319:657–667. doi: 10.1042/bj3190657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann BM, Fujinaga K, Deng H, Ogba N, Montano MM. The breast cell growth inhibitor, estrogen down regulated gene 1, modulates a novel functional interaction between estrogen receptor alpha and transcriptional elongation factor cyclin T1. Oncogene. 2005;24:5576–5588. doi: 10.1038/sj.onc.1208728. [DOI] [PubMed] [Google Scholar]
- Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, Tsai MJ, O’Malley BW. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) coactivator activity by IκB kinase. Mol Cell Biol. 2002;22:3549–3561. doi: 10.1128/MCB.22.10.3549-3561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O’Malley BW. Selective phosphorylations of the SRC-3/AIBI coactivator integrate genomic responses to multiple cellular signaling pathways. Mol Cell. 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Hashimoto Y, Kohri K, Ogata E, Kato S, Ikeda K, Nakanishi M. Cyclin E as a coactivator of the androgen receptor. J Cell Biol. 2000;150:873–880. doi: 10.1083/jcb.150.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwijsen RM, Buckle RS, Hijmans EM, Loomans CJ, Bernards R. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Dev. 1998;12:3488–3498. doi: 10.1101/gad.12.22.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwijsen RML, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJAM. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–415. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]