

Abstract
Exposure to environmental toxins increases the risk of neurodegenerative diseases including Parkinson’s disease (PD). Rotenone is a neurotoxin that has been used to induce experimental parkinsonism in rats. We used the rotenone model of experimental parkinsonism to explore a novel aspect of extra-nigral degeneration, the neurodegeneration of spinal cord (SC), in PD. Rotenone administration to male Lewis rats caused significant neuronal cell death in cervical and lumbar SC as compared to control animals. Dying neurons were motoneurons as identified by double immunofluorescent labeling for TUNEL+ cells and ChAT-immunoreactivity. Neuronal death was accompanied by abundant astrogliosis and microgliosis as evidenced from GFAP-immunoreactivity and OX-42-immunoreactivity, respectively, implicating an inflammatory component during neurodegeneration in SC. However, the integrity of the white matter in SC was not affected by rotenone administration as evidenced from the non co-localization of any TUNEL+ cells with GFAP-immunoreactivity and MBP-immunoreactivity, the selective markers for astrocytes and oligodendrocytes, respectively. Increased activities of 76 kD active m-calpain and 17/19 kD active caspase-3 further demonstrated involvement of these enzymes in cell death in SC. The finding of ChAT+ cell death also suggested degeneration of SC motoneurons in rotenone-induced experimental parkinsonism. Thus, this is the first report of its kind in which the selective vulnerability of a putative parkinsonian target outside of nigrostriatal system has been tested using an environmental toxin to understand the pathophysiology of PD. Moreover, rotenone-induced degeneration of SC motoneuron in this model of experimental parkinsonism progressed with upregulation of calpain and caspase-3.
Keywords: spinal motoneuron, neurodegeneration, Parkinson’s disease, calpain, caspase-3
Environmental toxins, especially pesticides and herbicides, attack the central nervous system (CNS), often latently, leading to development of a range of neurodegenerative diseases (Uversky, 2004; Carvey et al., 2006). Epidemiological studies have linked exposure to such toxins with the etiology of Parkinson’s disease (PD) in genetically predisposed animals (Gorell et al., 1998; Priyadarshi et al., 2000; Uversky et al., 2002; Uversky, 2004). PD is a complex devastating neurodegenerative disease that progressively impairs the control of movement. It affects 1% of the population above the age of 60 worldwide. Neuropathology of PD is characterized by loss of dopaminergic neurons in the midbrain substantia nigra pars compacta (SNpc) with diminishing dopaminergic neurotransmission in the terminal region (striatum), which is further documented by response of all types of PD to L-DOPA treatment in early stages (Dauer and Przedborski, 2003). The affected neurons show marked presence of proteinaceous intraneuronal or intraglial inclusions, called Lewy bodies and Lewy neurites, the histopathological hallmark of PD (Spillantini et al., 1997; Goedert, 2001). Despite substantial evidence of nigrostriatal degeneration in PD, neurodegeneration in other CNS areas are also suggested in progression of PD (Ray et al., 2000b; Chera et al., 2002; Chera et al., 2004; Yanagisawa, 2006).
Spinal cord (SC), the final coordinator of movement, is connected to brain by various descending and ascending projections and metabolic inter-neuronal circuits. Several independent investigators have suggested that SC is involved in progression of PD. For example, one common correlation among these findings is the marked presence of α-synuclein immunoreactive lesions containing Lewy bodies (Wakabayashi and Takahashi, 1997; Bloch et al., 2006; Klos et al., 2006). Further support for neuropathological changes outside the brain comes from an animal study in which significant neuronal loss and Lewy body-like inclusion has been reported in SC of A53T mutant mice that express mutant α-synuclein gene, which also occurs in autosomal dominant familial PD (Martin et al., 2006). Further, previous studies from our laboratory have demonstrated the degeneration of SC neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in mice (Ray et al., 2000b; Chera et al., 2002; Chera et al., 2004).
Molecular mechanisms underlying SC degeneration in PD becomes relevant because all symptoms of PD, in particular early symptoms of the disease, can not be adequately explained by damage only to SNpc, thus, suggesting that degeneration in other CNS regions may have some role preceding the degeneration in SNpc. Selective vulnerability of nigrostriatal system to environmental toxins is well documented in PD; however, knowledge regarding the susceptibility of other extra-nigral targets such as SC to environmental toxins is meager. Therefore, the investigation of a role for environmental toxins in SC degeneration would reveal further contribution of these toxins to progression of parkinsonian disorder.
Rotenone, an environmental toxin, is a naturally occurring insecticide and a high-affinity specific inhibitor of mitochondrial complex I (NADH dehydrogenase). Systemic administration of rotenone in low doses (1.5–2.5 mg/kg) has been used to create a chronic progressive animal model of PD (Betarbet et al., 2000; Alam and Schmidt, 2002). This model mimics the mild systemic inhibition of mitochondrial complex I as found in human PD (Mizuno et al., 1989; Schapira et al., 1989). Neurodegeneration in rotenone-induced parkinsonism is not purely a bio-energetic defect due to complex I inhibition (Sherer et al., 2003). Rotenone causes neurodegeneration also via multiple mechanisms that include moderate inhibition of complex I, enhanced production of reactive oxygen species (ROS), oxidative damage (Sherer et al., 2002; Tada-Oikawa et al., 2003), induction of apoptosis (Fiskum et al., 2003), activation of microglia (Gao et al., 2002), and acceleration of α-synuclein aggregation and fibrillation (Uversky et al., 2002; Diaz-Corrales et al., 2005).
Cysteine proteases such as calpains (Ray and Banik, 2003) and caspases (Nunez et al., 1998) are crucial players in mediation of apoptosis. Sustained mitochondrial dysfunction could release Ca2+ contributing to aberrant Ca2+ homeostasis in the cytosol leading to activation of Ca2+-dependent proteolytic enzymes such as calpain. Activation of caspase-3 has been reported in apoptosis of ventral mesencephalic dopaminergic neurons (Ahmadi et al., 2003) and human neuroblastoma cells (Sherer et al., 2002) following rotenone exposure.
Increased expression of calpain occuring in the mesencephalon of PD patients and primate PD models but not in other neurodegenerative disorders has been demonstrated (Mouatt-Prigent et al., 1996). Importantly, inhibition of calpain prevents neuronal and behavioral deficits in MPTP-induced PD (Crocker et al., 2003). Calpain involvement in rotenone-induced apoptosis has recently been shown in cultured neocortical neurons (Chen et al., 2006). Calpain and caspases may cross-talk for mediation of apoptosis independently or synergistically.
In the present study, we have explored the rotenone-toxicity in degeneration in SC and studied the activation of calpain and caspase-3, as probable mechanisms of contributing to motoneurons degeneration in SC. Data generated from these studies support the possiblilty of an involvement of increased calpain and caspase-3 activitities in rotenone-induced neurotoxicity in SC of Lewis rats.
EXPERIMENTAL PROCEDURES
Animals
Adult male Lewis rats (weight ≈ 425 g) from Charles River Laboratories (Wilmington, MA, USA) were used in this study. Rats were housed under standard conditions (12 h light-dark cycles, 23°C, and 55% relative humidity). Water and food pellets were given ad libitum. Rotenone administration and animal care were conducted in accordance with the guidelines outlined in the “Guide for the Care and Use of Laboratory Animals” of the U.S. Department of Health and Human Services (National Institute of Health, Bethesda, MD, USA) and approved by the Institutional Animal Care and Use Committee (IACUC) of the Medical University of South Carolina (Charleston, SC, USA).
Rotenone administration to Lewis rats
Rotenone (Sigma, St. Louis, MO, USA), dissolved in 1:1 (v/v) dimethylsulfoxide (DMSO; Sigma) and polyethylene glycol (PEG-300; Sigma), was injected in a low dose of 2.5 mg/kg subcutaneously (s.c.) once daily on days 1–4 and then on days 6, 9, 12, 15, 18, and 21 (total 25 mg/kg over 21 days). Control animals received s.c. injection of vehicle (1:1, DMSO/PEG-300) on corresponding days. Animals were weighed and monitored for general health conditions daily. Rats were anesthetized by injection of ketamine (80 mg/kg, i.p.) and sacrificed by decapitation. Brain and SC (cervical and lumbar) tissues were dissected on ice. Samples were either immediately immersed into frozen tissue embedding media (Histo Prep; Fisher Scientific, Fair Lawn, NJ, USA) or freshly frozen for biochemical analysis. All tissues were stored at −70°C until analysis.
Tissue processing for immunofluorescent and biochemical analysis
Brain and SC samples were warmed to −18°C before slicing. Thin (10 μm for SN and NCP, 5 μm for SC) sections were cut at −18°C using Leica CM1850 cryostat (Germany). Sections were fixed in 95% ethanol for 10 min, rinsed 3×5 min in phosphate-buffered saline (PBS, containing 137 mM NaCl, 2.7 mM KCl, 11.9 mM phosphates, pH 7.4) and stored in the same buffer at 4°C for further studies within one week.
For Western blotting, freshly frozen SC tissues (from the cervical and lumbar SC) were homogenized in an ice-cold homogenizing buffer (50 mM Tris-HCl, pH 7.4, 5 mM EGTA, and 1 mM PMSF). Protein concentrations were determined using Coomassie Plus™ Protein Assay Reagent (Pierce, Rockford, IL, USA) and spectrophotometric measurements at 595 nm (Spectronic, Rochester, NY, USA). Samples were diluted (1:1) in sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, and 5 mM β-mercaptoethanol), boiled for 5 min, and stored at −20°C. Bromophenol blue (0.025%) was added to the samples before loading on to the gels.
In situ TUNEL assay and double immunofluorescent labeling
Our previously described method (Ray et al., 2000a) employing terminal deoxynucleotidyl transferase recombinant (TdT)-mediated dUTP nick-end labeling (TUNEL) assay coupled with double immunofluorescent labeling was used to analyze tissue samples for cell death in different neuronal subtypes in SC. Briefly, sections prefixed in 95% ethanol were further fixed in 4% methanol-free formaldehyde (Polysciences, Warrington, PA, USA) and washed in PBS. After equilibration in TdT buffer (Apoptosis Detection System, Promega, Madison, WI, USA), samples were saturated with digoxigenin-labeled nucleotides (Roche, Indianapolis, IN, USA) and recombinant TdT (Promega) and incubated at 37°C for 1 h in a humidified Omnislide thermal Cycler (Hybaid Ltd, UK). TUNEL reaction was stopped by immersing in 2x NaCl/Na-citrate (SSC) at room temperature (RT) for 15 min. Unincorporated nucleotides were removed with washing in PBS three times, 5 min each.
In situ single and double immunofluorescent labelings
Brain and SC slices were blocked for 1 h in PBS containing 2% serum (horse, goat or sheep, Sigma), incubated with respective primary IgG antibodies prepared in the same diluent for 1 h at RT or in the case of antibody for motoneuron detection (ChAT) and oligodendrocyte detection (MBP), for 1 to 2 h at RT followed by 18 h at 4°C. Unbound primary antibodies were removed by rinsing in PBS three times, 5 min each. Slices were incubated with either FITC- or Texas red-conjugated appropriate secondary IgG antibodies for 1 h at RT, rinsed 3×5 min in PBS, once in distilled water, and mounted with anti-fade medium Vectashield™ (Vector Laboratories, Burlingame, CA, USA).
The images were viewed using an Olympus BH-2 microscope at 200X magnification at a pixel resolution of 1280×1024. Images were captured with a Dage CCD100 integrating camera (DAGE-MTI, Michigan City, IN, USA) and a Flashpoint 128 capture board (Integral Technologies, Indianapolis, IN, USA). Image capture was performed on a Pentium IV Imaging workstation (Dell Computers, Round Rock, TX, USA), using Magna Fire Image Pro Plus software (Media Cybernetics, Silver Spring, MD, USA). Image formatting was done using Photoshop software (Adobe, San Jose, CA, USA).
Antibodies and dilutions for immunofluorescent labelings
Antibodies used in the study were: mouse monoclonal anti-neuronal nuclei (NeuN, 1:100; Chemicon International, Temecula, CA, USA); mouse monoclonal anti-choline acetyltransferase (ChAT, 1:100; Chemicon); mouse monoclonal anti-glial fibrillary acidic protein (GFAP, 1:400; Chemicon); mouse monoclonal anti-rat CD11b/c (OX-42, 1:100; Invitrogen, BioSource™, Camarillo, CA, USA); rabbit polyclonal anti-m-calpain (domain IV) C-terminal, that recognizes 76 kD active m-calpain {1:100, raised in our laboratory, (Banik et al., 1983)}, rabbit polyclonal cleaved caspase-3 (1:100; Cell Signaling Technology, Boston, MA, USA), rabbit polyclonal anti-myelin basic protein {MBP, 1:500, raised and characterized in our laboratory, (Li and Banik, 1995) and mouse monoclonal anti-tyrosine hydroxylase (TH, 1:100; Chemicon).
The following fluorescent secondary antibodies were used: fluorescein-conjugated horse anti-mouse IgG for NeuN, ChAT, GFAP and OX-42 (1:100; Vector Laboratories, Burlingame, CA, USA); anti-digoxigenin-rhodamine-coupled, Fab fragments (1:100; Roche, Germany) for TUNEL; Texas red-conjugated goat anti-rabbit IgG (1:100; Vector Laboratories) for m-calpain, cleaved caspase-3 and MBP.
Semi-quantitative analysis of Mean Fluorescence Intensities (MFIs) of TUNEL-immunofluorescence was performed using NIH image J software (Kalkonde et al., 2007). Four images of sections stained for NeuN and TUNEL or ChAT and TUNEL were obtained per SC (cervical and lumbar) tissue derived from the control and rotenone-treated rats respectively. Collaged images of TUNEL and NeuN or ChAT staining were converted into an 8-bit format and the background was subtracted. An intensity threshold was set and was kept constant for all images analyzed. NeuN or ChAT-positive neurons were outlined using an outlining tool in any neuronal or motoneuronal-staining panel of the collage and the outline was moved over to the respective TUNEL staining panel of the collage to delineate the neurons or specifically the motoneurons. MFIs of TUNEL were measured in these outlined areas. MFI per unit area (MFI/area) was calculated by dividing the MFI units by the area of outlined neurons or motoneurons, and the data were represented as arbitrary units ± standard error of mean (S.E.M.).
Western blot analysis
SC samples were subjected to Western blot analysis to determine the levels of inactive 80 kD and active 76 kD calpain. Calpain and caspase-3 activities were determined by measuring the 145 kD calpain specific breakdown product (SBDP) and 120 kD caspase-3 specific SBDP following the previously reported protocol from our laboratory (Karmakar et al., 2006; Sribnick et al., 2006).
Protein samples (25 μg) were loaded onto 4–20% gradient of precast sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE), which were electrophoresed at 200 V for 30 min for resolution of proteins (Bio-Rad Laboratories, Hercules, CA, USA). For resolution of SBDP bands, 5% precast SDS-PAGE gels were used and electrophoresis was carried out at 100 V for 2 h (Bio-Rad). The resolved proteins were transferred from gels to Immobilon™-P polyvinylidene fluoride micro-porous membranes (Millipore, Bedford, MA, USA) in a transfer buffer [25 mM Tris-HCl, 192 mM glycine, 20% (v/v) methanol], using Genie apparatus (Idea Scientific, Minneapolis, MN, USA) Membranes were blocked in 5% non-fat milk in Tris-buffer (20 mM Tris-HCl, pH 7.6), containing 0.1% Tween-20 (T-TBS), incubated overnight at 4°C with a primary antibody, [anti-m-calpain (1:500) or mouse monoclonbal anti-α-spectrin (1:10,000; Biomol International, Butler Pike, Plymouth Meeting, PA, USA)] followed by incubation with horseradish peroxidase-conjugated secondary IgG antibodies. Goat anti-rabbit secondary IgG antibody (1:2,000; MP Biomedicals, LLC, Solon, Ohio, USA) was used to recognize m-calpain; goat anti-mouse secondary IgG antibody (1:2,000; MP Biomedicals) was used to recognize SBDP.
Between exposures to each antibody, membranes were washed with T-TBS. Membranes were shortly incubated with enhanced chemiluminescence reagents (ECL or ECLplus, Amersham Biosciences, Piscataway, NJ, USA), and exposed to X-ray film (Kodak X-OMAT XAR-2 (Kodak, Rochester, NY, USA). Autoradiograms were scanned on a UMAX PowerLook Scanner (UMAX Technologies, Fremont, CA, USA) using SilverFast Scanner software (LaserSoft Imaging, Kiel, Germany); optical density (OD) of each band was determined using Quantity One software (Bio-Rad).
Calpain blots were reprobed using mouse monoclonal anti-β-actin primary IgG antibody (1:15,000, Sigma) followed by HRP-conjugated goat anti-mouse secondary IgG antibody (1:2,000, Biomedicals) to monitor β-actin expression, which served as loading controls. For SBDP, separate 4–20% SDS-PAGE gels were run and probed for β-actin.
Statistical analysis
Quantitative data of immunofluorescent images were represented through pixel analyses using the public domain NIH Image 1.63 software. Semi-quantitative analysis of Mean Fluorescence Intensities (MFIs) of TUNEL imunofluorescence was performed using NIH image J software. The data were presented as arbitrary units ± standard error of mean (SEM). The results obtained from Western blot experiments were analyzed using the StatView software (Abacus Concepts, Berkley, CA, USA). Data were expressed as mean ± SEM of separate experiments (n≥4). Differences were considered significant at P≤0.05.
RESULTS
Rotenone reduced body weight in Lewis rats
We measured body weight of animals daily to assess the effect of rotenone on general metabolism of experimental animals (Fig. 1). Because of rotenone administration, rats lost body weight steadily during the first week, and any further weight gain of the experimental animals was arrested. Control animals gained body weight throughout the three-week time course of the experiment (Fig. 1). We recorded 33% mortality in rotenone treated group.
Fig. 1.
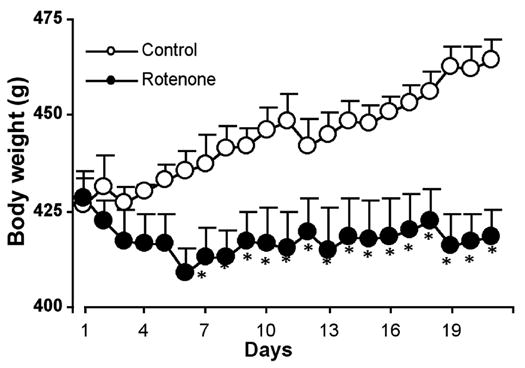
Effect of rotenone on body weight. Rats were injected with rotenone (2.5 mg/kg/day) subcutaneously (s.c.) once daily during the first 4 days, followed by single injection of the same dose on day 6, 9, 12, 15, 18, and 21 (total 25 mg/kg over 21 days). Daily records of body weight of rats injected with rotenone (closed circles) or vehicle (open circles) are represented in g ± SEM. *P<0.05 (n=9).
Rotenone induced cell death in the cervical and lumbar regions of SC
To test whether SC (the final coordinator of movement in the body) was affected in rotenone-administered rats, we examined neuronal death in sections from upper (cervical) and lower (lumbar) segments of SC from rotenone-injected animals and compared with those of vehicle-treated animals. Combined TUNEL and double immunofluorescent labeling was used for detecting TUNEL (as a DNA fragmentation marker)-positive NeuN (as a neuronal marker) cells. Microscopic evaluation of control samples from both cervical and lumbar SC regions revealed different types of neurons, which could be easily distinguished due to the distinct morphology (Fig. 2). The small nuclei of sensory neurons in the dorsal horn (Fig. 2 A, left panels) and the large nuclei of the motoneurons in ventral horn (Fig. 2 A, right panels) were strongly immunostained with NeuN. We did not detect TUNEL-positive neurons in any of the control samples (Fig. 2 A: Merge controls). However, microscopic examination revealed degeneration of a number of neurons in SC sections from the rotenone-injected animals. Cell death, as evaluated by TUNEL-positive NeuN-IR was observed throughout the dorsal horn as well as ventral horn of both the cervical and lumbar SC segments in rats treated with rotenone. Merged images demonstrated pronounced co-localization of TUNEL and NeuN (Fig. 2 A), showing neuronal death in SC in rotenone-injected animals. Semi-quantitative analysis of MFIs of TUNEL immunofluorescence per unit area of neurons showed that Compared controls, the levels of TUNEL-positive neurons in the rotenone-injected animals were significantly (P<0.05) increased by 90% and 87% in cervical and lumbar SC sections, respectively, of the dorsal horn, and also significantly (P<0.05) increased by 97% and 89% in cervical and lumbar SC sections, respectively, of the ventral horn (Fig. 2 B), These results clearly indicated susceptibility of neuronal populations of the SC to rotenone-induced neurotoxicity.
Fig. 2.
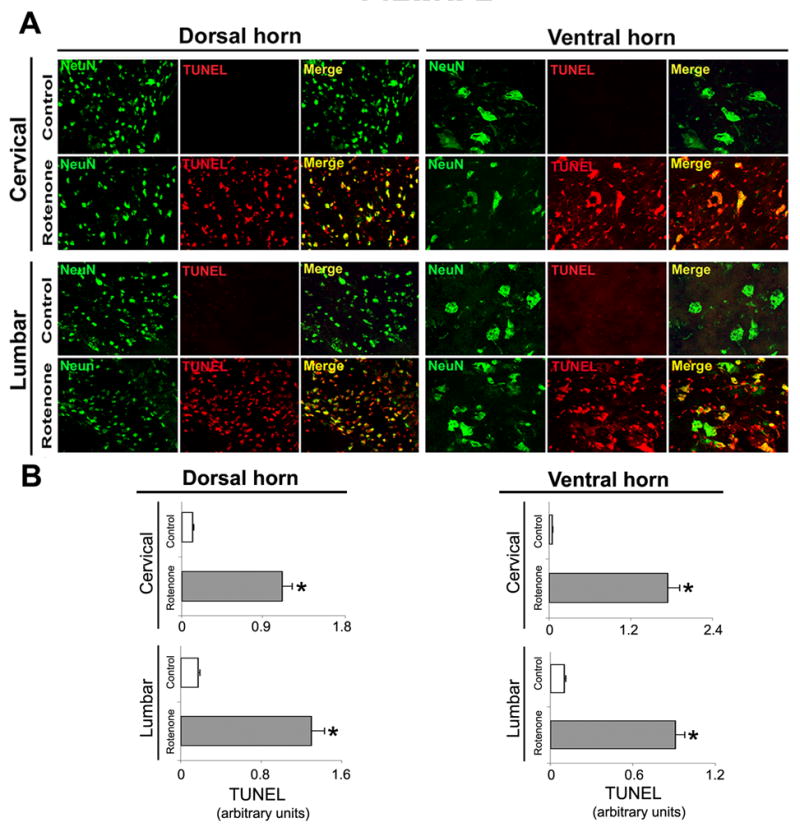
Effect of rotenone on neuronal viability in cervical and lumbar SC. (A) Representative photomicrographs of double immunofluorescent staining for NeuN (green) and TUNEL (red) performed in cervical and lumbar SC coronal sections (5 μm). No TUNEL+ neurons were identified in the sections from control animals (see Merge); however, cell death as evaluated by TUNEL-positive IR in many dorsal and ventral neurons was found in sections from cervical and lumbar SC segments in rotenone-injected animals (yellow, merge). Images taken at 200X magnification (n≥4). (B) The lower panels show semi-quantitative analysis of TUNEL mean fluorescence intensities (MFI) per unit area of neurons. Data show significant differences in the arbitrary units between samples from control animals and after rotenone exposure. *P≤0.05 (n≥ 4).
Rotenone caused motoneuron death in SC
Because significant amounts of neuronal death occurred in the SC of rats following rotenone administration, next we performed experiments to identify the types of the dying neurons. We identified the death of motoneurons by TUNEL staining in combination with the cholinergic motoneuron marker ChAT (Fig. 3). Microscopic examination of the SC sections from the control animals showed strong ChAT-IR healthy motoneurons in both cervical and lumbar regions in control rats. No TUNEL-positive motoneurons were seen in control slices from the cervical (Fig. 3, panels A, B, and C) or the lumbar (Fig. 3, panels G, H, and I) segments of SC. However, we found co-labeling of cells with TUNEL and ChAT-IR in slices obtained of the cervical (Fig. 3, panels D, E, and F) and the lumbar (Fig. 3, panels J, K, and L) segments of SC from rotenone-injected rats. Semi-quantitative analysis of MFIs of TUNEL immunofluorescence per unit area of ventral motoneurons showed that compared to controls, the levels of TUNEL staining were significantly (P<0.05) increased by 92% and 96% in the cervical and lumbar SC sections, respectively, of the ventral horn in rotenone-injected animals (Figure 3). These data specifically showed the vulnerability of ventral motoneurons of the SC to rotenone toxicity.
Fig. 3.
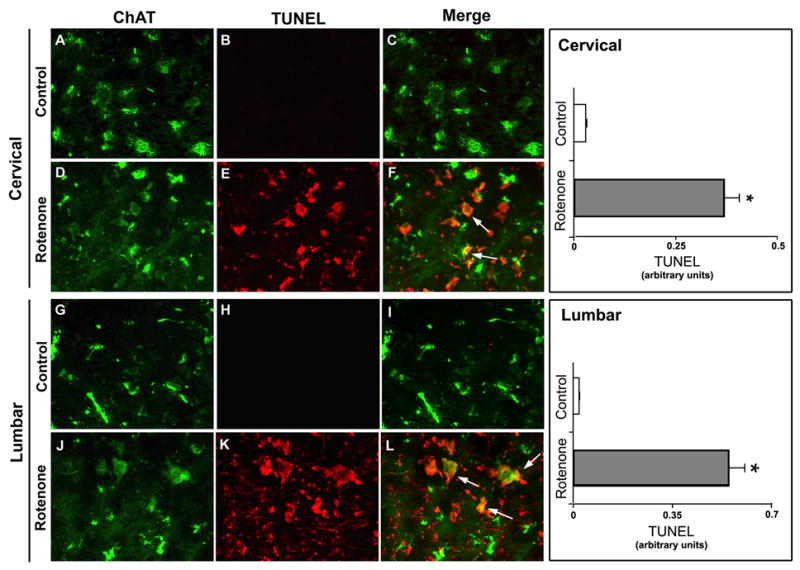
Effect of rotenone on motoneurons in cervical and lumbar SC. Representative photomicrographs of double immunofluorescent staining for TUNEL (red) and ChAT (green) a marker of motoneuron, performed in cervical and lumbar SC coronal slices (5 μm). In cervical and lumbar slices from control rat SC, ChAT-positive motoneurons did not show TUNEL-positive IR (A, B, C and G, H, I). TUNEL-positive IR in ventral motoneurons appeared after rotenone exposure, depicting motoneuronal cell death, evaluated by ChAT and TUNEL co-localization sites (D, E, F and J, K, L, respectively, arrows indicate co-staining). Images taken at 200x magnification (n≥4). Semi-quantitative analysis of TUNEL mean fluorescence intensities (MFI) per unit area of ventral motoneurons has been represented in the respective right panels. Data showed significant differences in the arbitrary units between samples from control animals and after rotenone exposure. *P≤0.05 (n≥4).
Rotenone evoked astrogliosis as well as microgliosis in SC
Rotenone-induced inflammatory responses were examined in SC sections (Fig. 4). The responses of both astroglial and microglial cells to rotenone neurotoxicity were tested in SC slices from control and rotenone-treated rats by immunofluorescent staining using the cell marker antibodies GFAP (for astrocytes) and OX-42 (for activated microglia and macrophages).
Fig. 4.
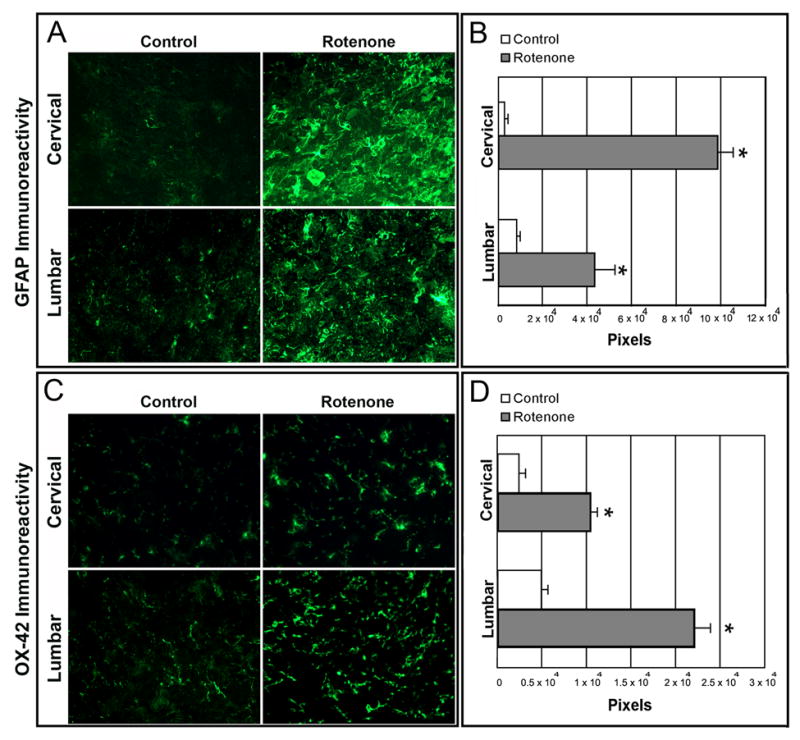
Effect of rotenone on GFAP and OX-42 immunoreactivity in cervical and lumbar SC. In control rat SC sections, there were a few GFAP and OX-42 positive cells. Intense astrogliosis, detected by increased GFAP-IR (A) and microgliosis, detected by OX-42-IR (C) seen in both cervical and lumbar SC slices from rotenone-injected animals. Data analyzed with NIH image 1.63 showed significant differences in the number of pixels in sections from control and rotenone-injected animals (B and D). *P≤0.05 (n≥ 4).
Microscopic observations revealed only a few GFAP immunoreactive astroglial or OX-42 immunoreactive microglial cells in slices from cervical and lumbar SC segments of control rats, indicating lack of astrogliosis (Fig. 4A, control panels) or microgliosis (Fig. 4C, control panels). In contrast, examination of samples from rotenone-treated animals showed profound GFAP-IR and OX-42-IR in both cervical and lumbar SC sections. Quantitative evaluation of the captured images showed that compared to controls, the levels of GFAP-IR were significantly (P<0.05) increased by 97% and 81% in cervical and lumbar SC sections, respectively, and also the level of OX-42-IR were significantly (P<0.05) increased by 77% and 77% (*P<0.05), respectively, in cervical and lumbar SC segments of the rotenone-injected animals. These results indicated that rotenone caused intense astrogliosis and microgliosis in both the cervical and lumbar SC regions in experimental animals.
Rotenone did not induce astroglial death in SC
Enhanced astrogliosis in the ventral SC led us to examine the integrity of SC white matter. Combination of TUNEL and GFAP stainings was used to identify any astrocytic death (Fig. 5). Microscopic examination of the SC sections showed healthy astrocytes in both cervical and lumbar regions in control rats. Co-localization of TUNEL and GFAP stainings did not occur in cervical and lumbar SC segments from the control rats or the rotenone-injected animals, indicating absence of death of the astrocytes. Notably, the intensity of GFAP-IR in SC sections was more in rotenone-injected rats than in control animals (Fig. 5).
Fig. 5.
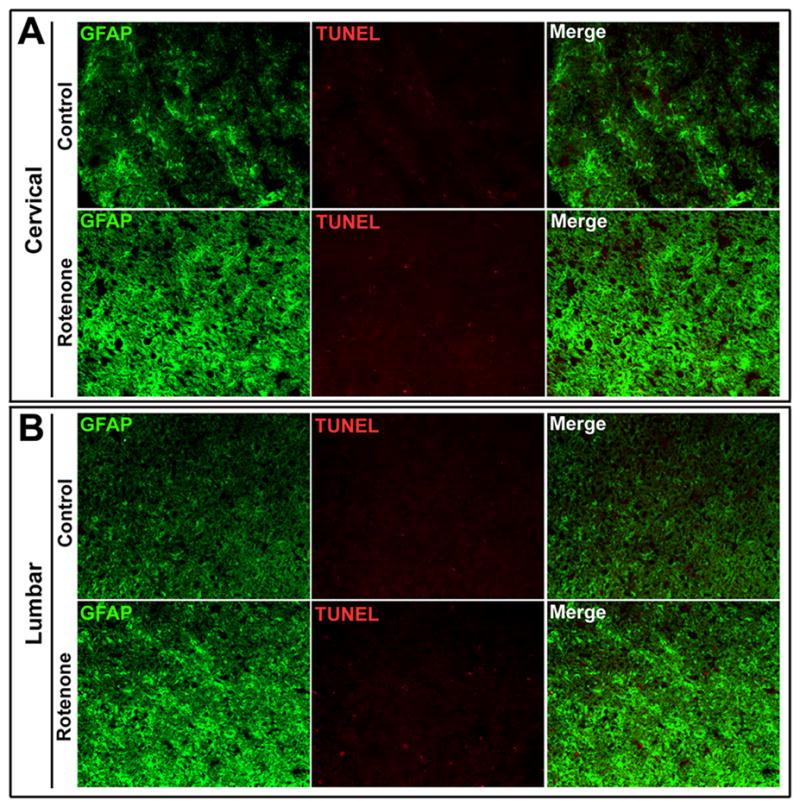
Effect of rotenone on astrocytes in cervical and lumbar SC. Representative photomicrographs of double immunofluorescent staining for TUNEL (red) and GFAP (green) a marker for astrocytes, performed in cervical and lumbar SC coronal slices (5 μm). In cervical and lumbar slices from SC, GFAP-positive astrocytes did not show TUNEL-positive IR in control or rotenone administered rats. GFAP-immunostaining was enhanced in experimental rat SC. Images taken at 200x magnification (n≥4).
Rotenone did not induce death in oligodendrocytes in SC
White matter integrity of the SC was further tested in the rotenone-treated rats by combination of TUNEL assay with MBP-IR, the selective marker for oligodendrocytes (Fig. 6). Microscopic examination showed MBP-positive oligodendrocytes in both the cervical and lumbar SC sections in control rats. Co-locatization of TUNEL and MBP-IR was not seen in cervical or lumbar SC segments in control and rotenone-injected rats (Fig. 6), indicating that white matter containing oligodendrocytes was spared in course of rotenone toxicity.
Fig. 6.
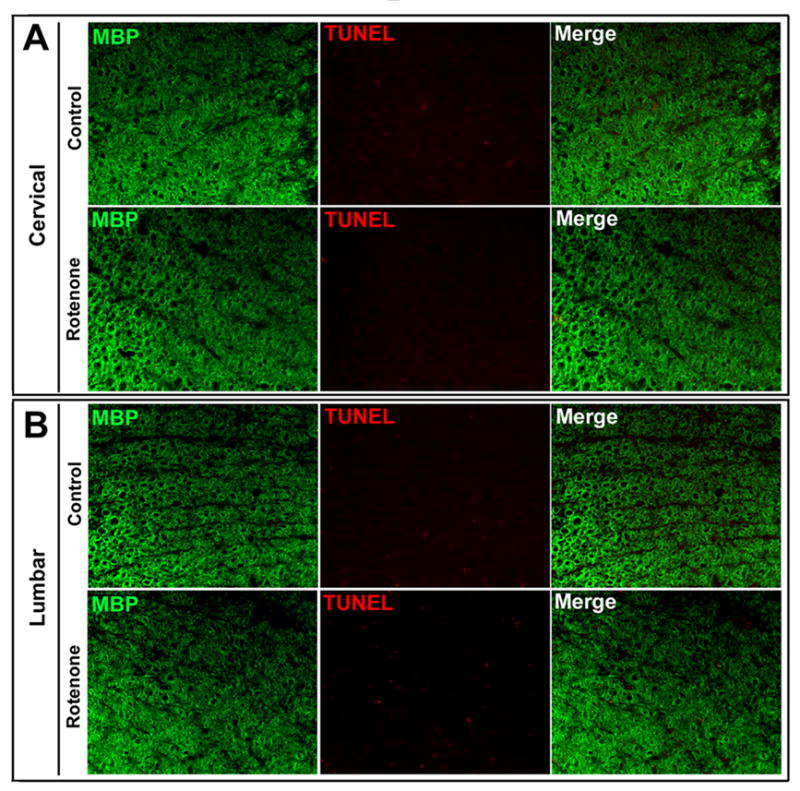
Effect of rotenone on oligodendrocytes in cervical and lumbar SC. Representative photomicrographs of double immunofluorescent staining for TUNEL (red) and MBP (green) a marker of oligodendrocytes, performed in cervical and lumbar SC coronal slices (5 μm). MBP-positive oligodendrocytes did not show any TUNEL-positive IR in cervical and lumbar slices from control or the experimental rat SC. Images taken at 200x magnification (n≥4).
Rotenone caused activation of calpain in SC
We examined activation of calpain and co-localization of active calpain and the neuronal maker NeuN in SC sections (Fig. 7). Changes in calpain content following rotenone injection were monitored by Western blotting of cervical and lumbar SC samples (Fig. 7A). We used an antibody to monitor the levels of two molecular forms, 80 kD (inactive) and 76 kD (active), of m-calpain in SC samples from the control and rotenone-treated rats. Uniform levels of 42 kD β-actin expression showed equal loading of samples in all lanes. Although the 80 kD (inactive) calpain bands on representative Western blots apparently showed different thicknesses between the control and rotenone groups, quantitative analysis showed no discernible statistical difference (Fig. 7A). However, administration of rotenone significantly elevated the levels of 76 kD (active) calpain by 86% and 90% in cervical and lumbar SC tissues, respectively, from rotenone-treated rats as compared to control animals.
Fig. 7.
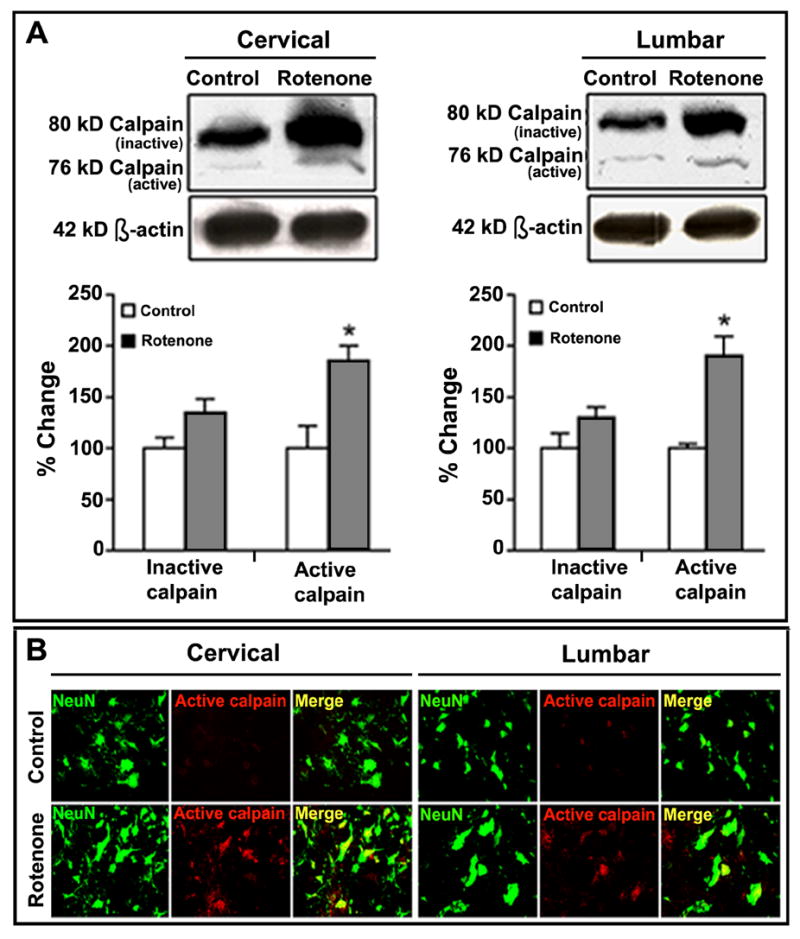
Effect of rotenone on calpain content in cervical and lumbar SC. Determination of calpain content by Western blotting (A). Representative blot pictures (upper panels) showing bands of 80 kD (inactive) and 76 kD (active) calpain, as well as 42 kD β-actin (loading control), detected in cervical and lumbar SC tissues from control and rotenone-injected animals respectively. Densitometric analysis showed significant percent changes in 76 kD active calpain bands in both cervical and lumbar SC segments in rotenone-injected rats. *P≤0.05 (n≥4). Representative photomicrographs (B) of double immunofluorescent staining for 76 kD m-calpain (red) and NeuN (green), performed in cervical and lumbar SC coronal slices (5 μm). No active calpain-IR was found in the sections from control animals. Significant content of active calpain was observed in many ventral motoneurons in cervical and lumbar SC segments of rotenone-injected animals, evaluated by co-localization of 76 kD calpain-IR and NeuN-IR (Merge indicate co-staining in rotenone panel). Images are taken at 200x magnification (n≥4).
Our findings from the Western blot analysis were further confirmed by double immunofluorescent labeling of cervical and lumbar SC sections using active calpain and NeuN antibodies (Fig. 7B). Microscopic observation of the cervical and lumbar SC sections from control rats hardly showed presence of active calpain-IR (Fig. 7B, control panels). The SC sections from rotenone-treated rats showed a pronounced labeling for active calpain and co-localization active calpain with NeuN-IR in both the cervical and lumbar segments (Fig. 7B, rotenone panels). Thus, rotenone treatment raised the content of active calpain in neurons.
Rotenone induced activation of caspase-3 in SC
Cleavage of pro-caspase-3 to its active form was examined in SC sections from the control and rotenone-treated rats (Fig. 8). Neither cervical nor lumbar SC sections of control rats had any detectable amount of active caspase-3-IR; however, SC sections of experimental animals showed prominent co-localization of active caspase-3-IR and NeuN-IR (Fig. 8), indicating a role for active caspase-3 in neuronal death.
Fig. 8.
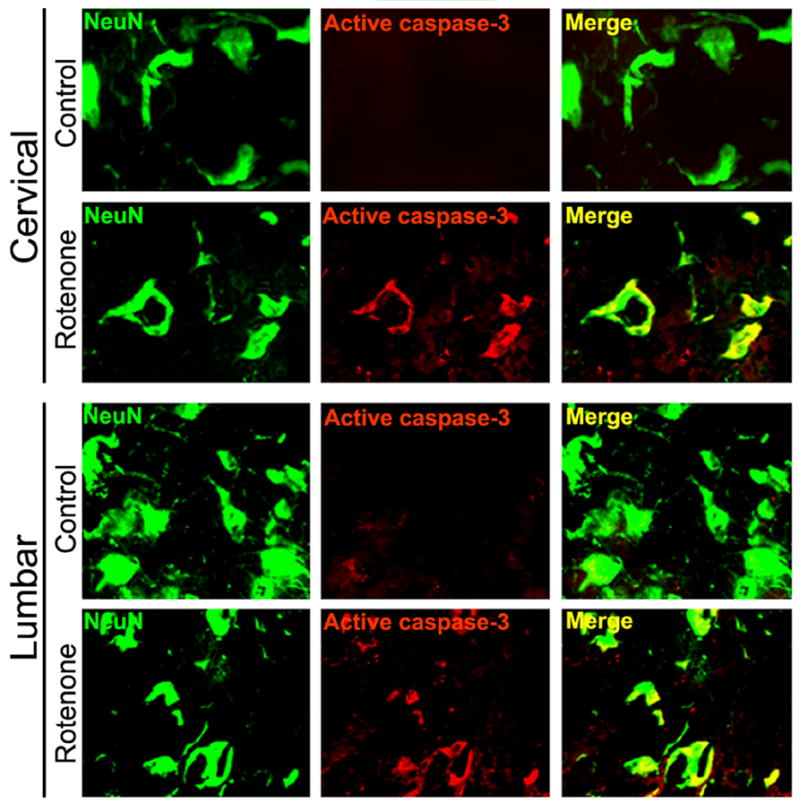
Effect of rotenone on activation of caspase-3 in cervical and lumbar SC. Representative photomicrographs of double immunofluorescent staining for active caspase-3 (red) and NeuN (green), performed in cervical and lumbar SC coronal slices (5 μm). No active caspase-3-IR was found in the sections from control animals. Significant amount of active caspase-3 was observed in many ventral motoneurons in cervical and lumbar SC segments of rotenone-injected animals, evaluated by increased co-localization of active caspase-3-IR and NeuN-IR (Merge indicate co-staining in rotenone panel). Images are taken at 200x magnification (n≥4).
Rotenone increased activities of calpain and caspase-3 in SC
We performed Western blotting to monitor the increased activities of calpain and caspase-3 in the degradation of 270 kD α-spectrin to the calpain-specific 145 kD SBDP and caspase-3-specific 120 kD SBDP (Fig. 9). Rotenone administration caused increases in the calpain and caspase-3 activities in both the cervical and lumbar SC segments. Levels of 42 kD β-actin expression, used as a loading control, were uniform in all samples. Densitometric analysis showed that compared to control rats, the levels of the calpain-specific 145 kD SBDP were increased by 55% and 35% in the cervical and lumbar SC segments, respectively, and the levels of caspase-3-specific 120 kD SBDP were increased by 60% and 30% in the cervical and lumbar SC segments, respectively, in rotenone-treated rats (Fig. 9, bar graphs).
Fig. 9.
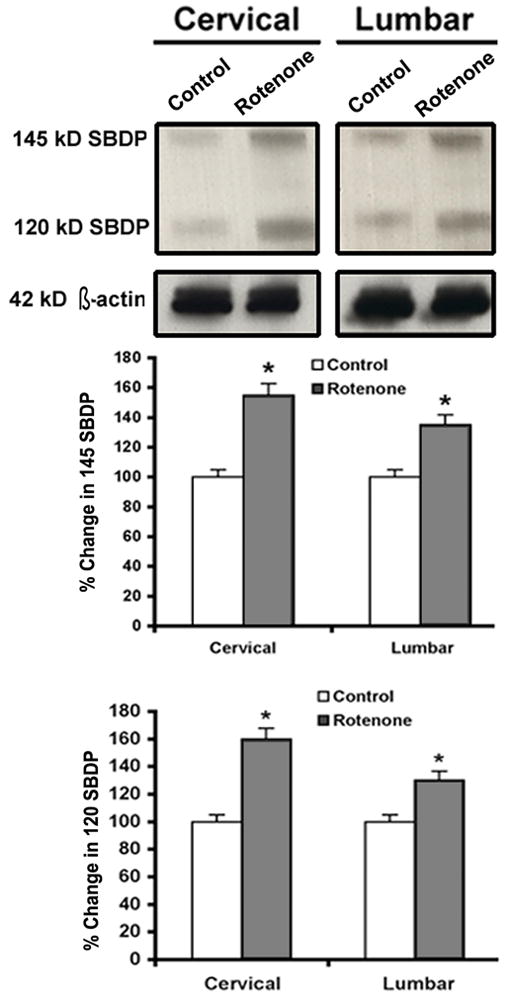
Effect of rotenone on calpain and caspase-3 activities in cervical and lumbar SC. Representative blot pictures, showing bands of calpain-specific 145 kD SBDP and caspase-3-specific 120 kD SBDP, as well as 42 kD β-actin (loading control) in cervical and lumbar SC tissues from control and rotenone-injected animals respectively. Densitometrc analysis shows significant percent changes in calpain and caspase-3 SBDPs in both cervical and lumbar SC segments in rotenone-injected rats. *P<0.05 (n≥4).
Rotenone caused nigrostriatal degeneration along with SC neurodegeneration
We assessed concomitant neurodegeneration in SC and nigrostriata in the rotenone-administered rats (Fig. 10). The TH-IR was used as the phenotypic marker of the dopaminergic neurons and fibres. We examined the coronal slices for the nigral cell body region and the terminal striatal region in control and rotenone-injected rats. There was marked reduction in dopaminergic TH-IR in SNpc (Fig. 10, panels A vs B). In contrast, there was no apparent loss of dopaminergic TH-IR in ventral tegmental area in both the control and experimental animals. Further, data showed dramatic decrease in TH-IR in dopaminergic terminals in striatum in rotenone-injected rats as compared with control animals (Fig. 10, panels C vs D). These data demonstrated concomitant nigrostriatal degeneration in all experimental animals after 21 days following rotenone administration.
Fig. 10. Effect of rotenone on TH-IR.
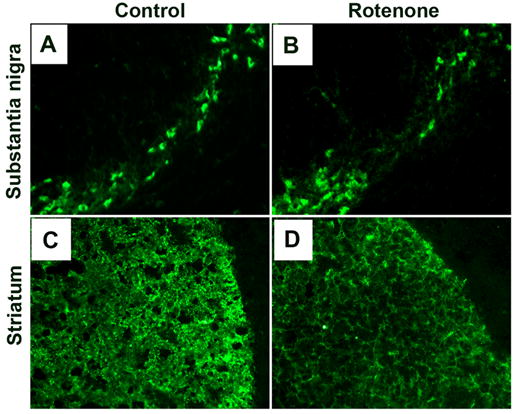
Representative photomicrographs of immunofluorescent staining for TH-IR in coronal brain sections (10 μm) passing through SN and striatal region from control rats (A and C) and rotenone-injected rats (B and D). In control animals, numerous TH-positive cells and intense TH-IR are seen in SNpc (A) and striatum (C), respectively. Rotenone administration caused substantial decrease in TH-IR in both SN and striatum. Images were taken at 200X magnification. (n≥4).
DISCUSSION
Spinal cord degeneration upon exposure to an environmental toxin has not been reported earlier, even though we have previously demonstrated neuronal death in SC in the pathophysiology of MPTP-induced PD mice (Ray et al., 2000b; Chera et al., 2002; Chera et al., 2004). PD is a movement disorder in which motor control is progressively lost due to degeneration of the motor control unit in midbrain. Although less appreciated, it is clear that multiple brain nuclei are affected in PD and the complex pathology is probably responsible for the other non-motor symptoms that respond poorly to L-dopa (Braak et al., 2003).
SC, where locomotion centers are located, is known to play a pivotal physiological role in reflex responses, either by promoting movements (phasic muscle contractions promote reposition of limbs) or maintaining posture (tonic muscle contractions counteract gravity and maintain the body in an upright position). However, it is unclear whether SC motoneurons are affected in PD. Obviously, SC with damaged neurons and motoneurons, may seriously affect reflex activities as well as impair motor functions, thus contributing to the progression of motor dysfunctions, which are also related to PD characteristics.
Investigations from our present study indicate significant degeneration of SC neurons of Lewis rats in rotenone-induced experimental parkinsonism. We detected neuronal degeneration in dorsal and ventral horn and motoneuron death in cervical and lumbar SC. In the present investigation, we found that integrity of the white matter of SC was not impaired by rotenone administration, suggesting some semblance of selectivity in rotenone-induced ventral motoneuron degeneration in our model. In addition, there was enhanced astrogliosis and microgliosis in ventral, cervical and lumbar SC with upregulation of proteolytic enzymes calpain and caspase-3, suggesting participation of inflammation and proteolytic system in damage.
The involvement of SC in the onset, neuropathology, or progression of PD symptoms remains debatable due to lack of sufficient clinical and experimental data to support the hypothesis. To date, well-documented clinical PD symptoms such as tremor, rigidity, bradykinesia/akinesia, and postural instability (Wichmann and DeLong, 1993) are mainly attributed to dysfunctions of the striatum, basal ganglia, pedunculopontine nucleus, thalamus, limbic system and frontal lobe (Yanagisawa, 2006). Only autonomic disturbances, which appear prior to changes in motor symptoms, are attributed to lesions in SC (Yanagisawa, 2006). However, the presence of Lewy bodies in SC of elderly subjects with incidental Lewy Body disease (ILBD) has been recently confirmed independently by at least two different groups (Bloch et al., 2006; Klos et al., 2006). If ILBD represents pre-symptomatic PD, then it is reasonable to suggest that SC degeneration may play a role in the pathophysiology of PD and precede nigrostriatal degeneration like that of the early degeneration in the lower brain stem nuclei (Braak et al., 2001; Braak et al., 2002). Damage to SC may even be as a consequence of diminished axonal input from the degeneration of higher brain nuclei.
Here. we investigated SC damage in a different experimental PD model induced by the dopamine transporter-independent agent rotenone to further confirm our earlier findings of SC neuron damage in MPTP-induced parkinsonism in mice (Ray et al., 2000b; Chera et al., 2002; Chera et al., 2004). Although, rotenone is variously administered to model PD, we selected subcutaneous delivery of rotenone in low doses to Lewis rats in the present study to minimize the peripheral toxic affects of systemic administration. Since there is enhanced sensitivity of dopaminergic neurons to rotenone-induced toxicity with aging, we focused on the usefulness of aged rats for inducing PD in our study (Phinney et al., 2006). However, in doing so we had encountered 33% mortality by day 4, which prompted us to inject rotenone on every third day thereafter. Several other investigators have also faced a similar rate of mortality in rotenone-induced PD model (Biehlmaier et al., 2006; Huang et al., 2006). Cumulative rotenone dosage administered was 25 mg/kg, which was 27% of the total rotenone (90 mg/kg) that was attempted for delivery by microsphere (Huang et al., 2006). In our model, there was moderate (30–40%) nigrostriatal dopaminergic degeneration as evidenced by TH-IR, consistent amongst the experimental rats.
We did not see any distinct behavioral symptoms in the rotenone-treated rats as compared to controls. This may be due to cumulative dosage of rotenone, being lesser than the threshold required for the manifestation of symptoms. In other words, we may consider these animals to be in pre-symptomatic phase of parkinsonian degeneration. However, all the surviving animals treated with rotenone showed consistent arrest in body weight gain, which implied occurrence of moderate metabolic compromise in these animals and gave a valid platform to assess the biochemical and molecular mechanisms of SC degeneration concomitant to nigrostriatal degeneration.
Accessibility of rotenone to SC is highly probable as rotenone is extremely lipophilic and thus membrane permeable. Passage of rotenone through the membrane is independent of transporters (unlike 1-methyl-4-phenylpyridinum, MPP+) and it diffuses into the brain very rapidly (Betarbet et al., 2000). Rotenone may rapidly enter into the SC as well, where it accumulates in mitochondria and inhibits activity of NADH-ubiquinone oxidoreductase (complex I) of the mitochondrial electron transport chain (Degli Esposti, 1998; Talpade et al., 2000) inducing multiple disruptions in the chain of interrelated metabolic reactions within the cells. Eventual cell death is attributed to moderate mitochondrial complex I deficiency followed by a multitude of overlapping and cross-linked factors such as reduced ATP production (Greenamyre et al., 1999), generation of reactive oxygen species and oxidative stress (Sherer et al., 2003; Tada-Oikawa et al., 2003), altered Ca2+ homeostasis (Sousa et al., 2003), protein misfolding (Mytilineou et al., 2004), microtubule depolymerization (Ren et al., 2005), apoptosis and/or necrosis of affected cells and tissues (Fiskum et al., 2003, Li et al., 2003).
In our study, we adopted a dorsal versus ventral bias while assessing cell death in SC sections to evaluate the degree of susceptibility of dorsal sensory neurons or the ventral motoneurons to rotenone-toxicity. While rotenone affected both types of neurons, at this juncture we concentrated selectively on the motoneurons as our primary goal was to evaluate the fate of motoneurons in an experimental model of PD. Rotenone induces cell death in SC mixed cultures, attacking the neurons preferably more than the astrocytes (Litsky et al., 1999). Rotenone toxicity has been evaluated in retinal sensory system (Biehlmaier et al., 2006; Zhang et al., 2006) and in DRG neurons (Dedov et al., 2001); however, there are no reports on rotenone-induced impairment on the SC sensory neurons especially in an experimental model of PD where the rotenone administration is usually low and over an extended period of time.
Rotenone-induced selective vulnerability of motoneurons as compared to non-motor neurons has been shown in rat spinal motoneurons which is mediated by ionotropic glutamate receptor-mediated excitotoxicity (Kanki et al., 2004) and also in a cellular model of familial amyotrophic lateral sclerosis (Rizzardini et al., 2005; Rizzardini et al., 2006). Thus, it is not surprising that we have observed motoneuron degeneration in our experimental PD model as well. Our recent investigations also showed rotenone-induced toxicity and apoptotic and/or necrotic cell death in hybrid motor neuron cell cultures (Samantaray et al., 2006).
We further studied the inflammatory response in SC to assess the damage to SC in rotenone-induced neurotoxicity. Inflammation accompanying the parkinsonian neurodegeneration is well documented (Beal, 2003; McGeer and McGeer, 2004), in brain of PD patients (Hirsch et al., 2003; Ouchi et al., 2005) and in different experimental models including that of rotenone-induced experimental parkinsonism (Kurkowska-Jastrzebska et al., 1999; Cicchetti et al., 2002; Sherer et al., 2003). Hence, our observation of enhanced astrogliosis and microgliosis in both cervical and lumbar SC in experimental animals as compared to control is much anticipated. Our results indicated a gradient in astrogliosis that was more pronounced in cervical than lumbar SC, but no such gradient was seen in case of microgliosis. Evaluation of PD motor symptoms is distinct for forelimb versus hind limb impairment, which can be causally linked to dopaminergic degeneration in supraspinal midbrain nuclei (Meredith and Kang, 2006). As cervical and lumbar SC innervates fore limb and hind limb, respectively, a comparative account of degeneration of these two areas of the SC may provide valuable information. Furthermore, if the extent of inflammation is proportional to neuronal degeneration, we may expect a gradient in inflammatory response in cervical to lumbar SC. Our findings suggested a more robust astrogliosis in cervical than lumbar SC, but the trend was reversed in terms of microgliosis. It is important to note that although glial reaction is generally considered to be a consequence of the process of degeneration in PD, the toxic substances released by glial cells could significantly contribute to the progression and propagation of neuronal degeneration even when the initial cause of neuronal degeneration has disappeared (Hirsch et al., 2003; Ouchi et al., 2005).
Sustained mitochondrial dysfunction may give rise to aberrant Ca2+ homeostasis, which may built up a Ca2+ overload within the cells (Krieger and Duchen, 2002; Jacobson and Duchen, 2004) ultimately upregulating Ca2+ activated protease calpain (Ray and Banik, 2003). Early induction of calpain prior to activation of caspaspe-3 has been shown in vitro in primary cortical neurons exposed to rotenone (Chen et al., 2006). However, there are no reports on calpain involvement in an animal model of rotenone-induced experimental parkinsonism. Thus, we report for the first time calpain involvement in SC motoneuron death in rotenone-injected rats. In the present model, no active calpain reactivity was observed in SN at the time points studied. However, significant amount of active calpain was observed in both the cervical as well as lumbar SC sections of experimental rats. Calpain-involvement has convincingly been shown in MPTP-induced experimental parkinsonism (Ray et al., 2000b; Chera et al., 2002; Crocker et al., 2003; Chera et al., 2004). Active calpain in SN of the experimental animals may be present at an earlier time point or it may follow later depending upon the sequence of events during progression of rotenone-induced degeneration.
Calpain upregulation is implicated in both apoptotic and necrotic cell death (Harwood et al., 2005). In PD, damage to dopaminergic neurons may occur through oxidative stress and/or mitochondrial impairment and culminate in activation of an apoptotic cascade; some neurons experiencing energy failure may ultimately become necrotic. These events are reasonably well reflected in some of the PD animal models, notably those involving MPTP and rotenone. We present here a concomitant increase in active calpain and caspase-3 in SC of rotenone-administered experimental animals. Our present findings showing degeneration of neurons in both SN and SC confirm and corroborate our hypothesis. We previously reported SC neuron damage in another model of experimental parkinsonism induced by MPTP (Chera et al., 2002). Such degeneration of SC neuron has also been found in post-mortem tissue obtained from patients with PD (our unpublished data).
CONCLUSION
In conclusion, the present study strongly supports our hypothesis of SC involvement in experimental parkinsonism. It is important to mention that in clinical cases there is an overlap of PD with spinal motoneuron degeneration, athough rare, such cases have been reported (Williams et al., 1995; Klos et al., 2005). Moreover, midbrain dopaminergic neuronal loss has been confirmed in familial (Wolf et al., 1991) and sporadic ALS patients (Kato et al., 1993). Selective loss of midbrain dopaminergic neurons in transgenic mice model of ALS has been found to progress parallel to spinal motoneuron degeneration (Kostic et al., 1997). Parkinsonian toxin MPTP has been reported to exacerbate neurotoxicity in transgenic ALS mice via dopaminergic neurons sparing the serotonergic neurons (Andreassen et al., 2001). Thus, in light of these findings, the SC degeneration may be intricately linked with the midbrain dopaminergic degeneration, especially if common mechanisms like oxidative stress, mitochondrial dysfunction, aberrant Ca2+ homeostasis, interplay between calpain and caspase-3 (as suggested by the present study) drive the neurodegenerative process. In addition to nigrostriatal degeneration in PD, elaborate studies should focus on extranigral degeneration and information generated from such studies may help develop new therapies for the treatment of this complex neurodegenerative disease.
Acknowledgments
This investigation was supported in part by the R01 grants from the NINDS (NS-41088, NS-45967, and NS-57811), NCI (CA-91460) and NIH (C06 RRO15455). Varduhi Knaryan was a Fulbright Visiting Scholar in the Department of Neurosciences (Medical University of South Carolina, Charleston, SC, USA) under the Fulbright Visiting Scholar Program 2005–2006 awarded by the United States Department of State Bureau of Educational and Cultural Affairs (ECA) and Council for International Exchange of Scholars (CIES), Washington, DC, USA.
Abbreviations
- ChAT
choline acetyltransferase
- CNS
central nervous system
- GFAP
glial fibrillary acidic protein
- IR
immunoreactivity
- MBP
myelin basic protein
- NCP
nucleus caudate putamen
- NeuN
neuronal nuclei
- PD
Parkinson’s disease
- SC
spinal cord
- SNpc
substantia nigra pars compacta
- SBDP
spectrin breakdown product
- TH
tyrosine hydroxylase
- TUNEL
terminal deoxynucleotidyl transferase, recombinant (TdT)-mediated dUTP nick-end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmadi FA, Linseman DA, Grammatopoulos TN, Jones SM, Bouchard RJ, Freed CR, Heidenreich KA, Zawada WM. The pesticide rotenone induces caspase-3-mediated apoptosis in ventral mesencephalic dopaminergic neurons. J Neurochem. 2003;87:914–921. doi: 10.1046/j.1471-4159.2003.02068.x. [DOI] [PubMed] [Google Scholar]
- Alam M, Schmidt WJ. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res. 2002;136:317–324. doi: 10.1016/s0166-4328(02)00180-8. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Klivenyi P, Klein AM, Dedeoglu A, Albers DS, Kowall NW, Beal MF. Transgenic ALS mice show increased vulnerability to the mitochondrial toxins MPTP and 3-nitropropionic acid. Exp Neurol. 2001;168:356–363. doi: 10.1006/exnr.2001.7627. [DOI] [PubMed] [Google Scholar]
- Banik NL, Hogan EL, Jenkins MG, McDonald JK, McAlhaney WW, Sostek MB. Purification of a calcium-activated neutral proteinase from bovine brain. Neurochem Res. 1983;8:1389–1405. doi: 10.1007/BF00964996. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Biehlmaier O, Alam M, Schmidt WJ. A rat model of Parkinsonism shows depletion of dopamine in the retina. Neurochem Int. 2006 doi: 10.1016/j.neuint.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Bloch A, Probst A, Bissig H, Adams H, Tolnay M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol Appl Neurobiol. 2006;32:284–295. doi: 10.1111/j.1365-2990.2006.00727.x. [DOI] [PubMed] [Google Scholar]
- Braak E, Sandmann-Keil D, Rub U, Gai WP, de Vos RA, Steur EN, Arai K, Braak H. alpha-synuclein immunopositive Parkinson’s disease-related inclusion bodies in lower brain stem nuclei. Acta Neuropathol (Berl) 2001;101:195–201. doi: 10.1007/s004010000247. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages) J Neurol. 2002;249(Suppl 3):III/1–5. doi: 10.1007/s00415-002-1301-4. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Carvey PM, Punati A, Newman MB. Progressive dopamine neuron loss in Parkinson’s disease: the multiple hit hypothesis. Cell Transplant. 2006;15:239–250. doi: 10.3727/000000006783981990. [DOI] [PubMed] [Google Scholar]
- Chen MJ, Yap YW, Choy MS, Koh CH, Seet SJ, Duan W, Whiteman M, Cheung NS. Early induction of calpains in rotenone-mediated neuronal apoptosis. Neurosci Lett. 2006;397:69–73. doi: 10.1016/j.neulet.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Chera B, Schaecher KE, Rocchini A, Imam SZ, Ray SK, Ali SF, Banik NL. Calpain upregulation and neuron death in spinal cord of MPTP-induced parkinsonism in mice. Ann N Y Acad Sci. 2002;965:274–280. doi: 10.1111/j.1749-6632.2002.tb04169.x. [DOI] [PubMed] [Google Scholar]
- Chera B, Schaecher KE, Rocchini A, Imam SZ, Sribnick EA, Ray SK, Ali SF, Banik NL. Immunofluorescent labeling of increased calpain expression and neuronal death in the spinal cord of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. Brain Res. 2004;1006:150–156. doi: 10.1016/j.brainres.2004.01.065. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15:991–998. doi: 10.1046/j.1460-9568.2002.01938.x. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Smith PD, Jackson-Lewis V, Lamba WR, Hayley SP, Grimm E, Callaghan SM, Slack RS, Melloni E, Przedborski S, Robertson GS, Anisman H, Merali Z, Park DS. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J Neurosci. 2003;23:4081–4091. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dedov VN, Mandadi S, Armati PJ, Verkhratsky A. Capsaicin-induced depolarisation of mitochondria in dorsal root ganglion neurons is enhanced by vanilloid receptors. Neuroscience. 2001;103:219–226. doi: 10.1016/s0306-4522(00)00540-6. [DOI] [PubMed] [Google Scholar]
- Degli Esposti M. Inhibitors of NADH-ubiquinone reductase: an overview. Biochim Biophys Acta. 1998;1364:222–235. doi: 10.1016/s0005-2728(98)00029-2. [DOI] [PubMed] [Google Scholar]
- Diaz-Corrales FJ, Asanuma M, Miyazaki I, Miyoshi K, Ogawa N. Rotenone induces aggregation of gamma-tubulin protein and subsequent disorganization of the centrosome: relevance to formation of inclusion bodies and neurodegeneration. Neuroscience. 2005;133:117–135. doi: 10.1016/j.neuroscience.2005.01.044. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:111–119. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002;22:782–790. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, MacKenzie G, Peng TI, Stephans SE. Mitochondrial dysfunction in Parkinson’s disease. Biochem Soc Symp. 1999;66:85–97. doi: 10.1042/bss0660085. [DOI] [PubMed] [Google Scholar]
- Harwood SM, Yaqoob MM, Allen DA. Caspase and calpain function in cell death: bridging the gap between apoptosis and necrosis. Ann Clin Biochem. 2005;42:415–431. doi: 10.1258/000456305774538238. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:214–228. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Huang J, Liu H, Gu W, Yan Z, Xu Z, Yang Y, Zhu X, Li Y. A delivery strategy for rotenone microspheres in an animal model of Parkinson’s disease. Biomaterials. 2006;27:937–946. doi: 10.1016/j.biomaterials.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Jacobson J, Duchen MR. Interplay between mitochondria and cellular calcium signalling. Mol Cell Biochem. 2004:256–257. 209–218. doi: 10.1023/b:mcbi.0000009869.29827.df. [DOI] [PubMed] [Google Scholar]
- Kanki R, Nakamizo T, Yamashita H, Kihara T, Sawada H, Uemura K, Kawamata J, Shibasaki H, Akaike A, Shimohama S. Effects of mitochondrial dysfunction on glutamate receptor-mediated neurotoxicity in cultured rat spinal motor neurons. Brain Res. 2004;1015:73–81. doi: 10.1016/j.brainres.2004.04.044. [DOI] [PubMed] [Google Scholar]
- Karmakar S, Weinberg MS, Banik NL, Patel SJ, Ray SK. Activation of multiple molecular mechanisms for apoptosis in human malignant glioblastoma T98G and U87MG cells treated with sulforaphane. Neuroscience. 2006;141:1265–1280. doi: 10.1016/j.neuroscience.2006.04.075. [DOI] [PubMed] [Google Scholar]
- Kato S, Oda M, Tanabe H. Diminution of dopaminergic neurons in the substantia nigra of sporadic amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 1993;19:300–304. doi: 10.1111/j.1365-2990.1993.tb00444.x. [DOI] [PubMed] [Google Scholar]
- Klos KJ, Ahlskog JE, Josephs KA, Apaydin H, Parisi JE, Boeve BF, DeLucia MW, Dickson DW. Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology. 2006;66:1100–1102. doi: 10.1212/01.wnl.0000204179.88955.fa. [DOI] [PubMed] [Google Scholar]
- Klos KJ, Josephs KA, Parisi JE, Dickson DW. Alpha-synuclein immunohistochemistry in two cases of co-occurring idiopathic Parkinson’s disease and motor neuron disease. Mov Disord. 2005;20:1515–1520. doi: 10.1002/mds.20604. [DOI] [PubMed] [Google Scholar]
- Kostic V, Gurney ME, Deng HX, Siddique T, Epstein CJ, Przedborski S. Midbrain dopaminergic neuronal degeneration in a transgenic mouse model of familial amyotrophic lateral sclerosis. Ann Neurol. 1997;41:497–504. doi: 10.1002/ana.410410413. [DOI] [PubMed] [Google Scholar]
- Krieger C, Duchen MR. Mitochondria, Ca2+ and neurodegenerative disease. Eur J Pharmacol. 2002;447:177–188. doi: 10.1016/s0014-2999(02)01842-3. [DOI] [PubMed] [Google Scholar]
- Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61. doi: 10.1006/exnr.1998.6993. [DOI] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Li Z, Banik NL. The localization of mcalpain in myelin: immunocytochemical evidence in different areas of rat brain and nerves. Brain Res. 1995;697:112–121. doi: 10.1016/0006-8993(95)00949-q. [DOI] [PubMed] [Google Scholar]
- Litsky ML, Hohl CM, Lucas JH, Jurkowitz MS. Inosine and guanosine preserve neuronal and glial cell viability in mouse spinal cord cultures during chemical hypoxia. Brain Res. 1999;821:426–432. doi: 10.1016/s0006-8993(99)01086-0. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord. 2004;10(Suppl 1):S3–7. doi: 10.1016/j.parkreldis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Kang UJ. Behavioral models of Parkinson’s disease in rodents: A new look at an old problem. Mov Disord. 2006;21:1595–1606. doi: 10.1002/mds.21010. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun. 1989;163:1450–1455. doi: 10.1016/0006-291x(89)91141-8. [DOI] [PubMed] [Google Scholar]
- Mouatt-Prigent A, Karlsson JO, Agid Y, Hirsch EC. Increased M-calpain expression in the mesencephalon of patients with Parkinson’s disease but not in other neurodegenerative disorders involving the mesencephalon: a role in nerve cell death? Neuroscience. 1996;73:979–987. doi: 10.1016/0306-4522(96)00100-5. [DOI] [PubMed] [Google Scholar]
- Mytilineou C, McNaught KS, Shashidharan P, Yabut J, Baptiste RJ, Parnandi A, Olanow CW. Inhibition of proteasome activity sensitizes dopamine neurons to protein alterations and oxidative stress. J Neural Transm. 2004;111:1237–1251. doi: 10.1007/s00702-004-0167-2. [DOI] [PubMed] [Google Scholar]
- Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- Phinney AL, Andringa G, Bol JG, Wolters E, van Muiswinkel FL, van Dam AM, Drukarch B. Enhanced sensitivity of dopaminergic neurons to rotenone-induced toxicity with aging. Parkinsonism Relat Disord. 2006;12:228–238. doi: 10.1016/j.parkreldis.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA, Shrivastava S. A meta-analysis of Parkinson’s disease and exposure to pesticides. Neurotoxicology. 2000;21:435–440. [PubMed] [Google Scholar]
- Ray SK, Banik NL. Calpain and its involvement in the pathophysiology of CNS injuries and diseases: therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–189. doi: 10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- Ray SK, Schaecher KE, Shields DC, Hogan EL, Banik NL. Combined TUNEL and double immunofluorescent labeling for detection of apoptotic mononuclear phagocytes in autoimmune demyelinating disease. Brain Res Brain Res Protoc. 2000a;5:305–311. doi: 10.1016/s1385-299x(00)00027-1. [DOI] [PubMed] [Google Scholar]
- Ray SK, Wilford GG, Ali SF, Banik NL. Calpain upregulation in spinal cords of mice with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson’s disease. Ann N Y Acad Sci. 2000b;914:275–283. doi: 10.1111/j.1749-6632.2000.tb05202.x. [DOI] [PubMed] [Google Scholar]
- Ren Y, Liu W, Jiang H, Jiang Q, Feng J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J Biol Chem. 2005;280:34105–34112. doi: 10.1074/jbc.M503483200. [DOI] [PubMed] [Google Scholar]
- Rizzardini M, Lupi M, Mangolini A, Babetto E, Ubezio P, Cantoni L. Neurodegeneration induced by complex I inhibition in a cellular model of familial amyotrophic lateral sclerosis. Brain Res Bull. 2006;69:465–474. doi: 10.1016/j.brainresbull.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Rizzardini M, Mangolini A, Lupi M, Ubezio P, Bendotti C, Cantoni L. Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J Neurol Sci. 2005;232:95–103. doi: 10.1016/j.jns.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Samantaray S, Ray SK, Ali SF, Banik NL. Calpain Activation in Apoptosis of Motoneurons in Cell Culture Models of Experimental Parkinsonism. Ann N Y Acad Sci. 2006;1074:349–356. doi: 10.1196/annals.1369.034. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT. An in vitro model of Parkinson’s disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J Neurosci. 2002;22:7006–7015. doi: 10.1523/JNEUROSCI.22-16-07006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa SC, Maciel EN, Vercesi AE, Castilho RF. Ca2+-induced oxidative stress in brain mitochondria treated with the respiratory chain inhibitor rotenone. FEBS Lett. 2003;543:179–183. doi: 10.1016/s0014-5793(03)00421-6. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Ray SK, Banik NL. Estrogen prevents glutamate-induced apoptosis in C6 glioma cells by a receptor-mediated mechanism. Neuroscience. 2006;137:197–209. doi: 10.1016/j.neuroscience.2005.08.074. [DOI] [PubMed] [Google Scholar]
- Tada-Oikawa S, Hiraku Y, Kawanishi M, Kawanishi S. Mechanism for generation of hydrogen peroxide and change of mitochondrial membrane potential during rotenone-induced apoptosis. Life Sci. 2003;73:3277–3288. doi: 10.1016/j.lfs.2003.06.013. [DOI] [PubMed] [Google Scholar]
- Talpade DJ, Greene JG, Higgins DS, Jr, Greenamyre JT. In vivo labeling of mitochondrial complex I (NADH:ubiquinone oxidoreductase) in rat brain using [(3)H]dihydrorotenone. J Neurochem. 2000;75:2611–2621. doi: 10.1046/j.1471-4159.2000.0752611.x. [DOI] [PubMed] [Google Scholar]
- Uversky VN. Neurotoxicant-induced animal models of Parkinson’s disease: understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissue Res. 2004;318:225–241. doi: 10.1007/s00441-004-0937-z. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Li J, Bower K, Fink AL. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: implications for Parkinson’s disease. Neurotoxicology. 2002;23:527–536. doi: 10.1016/s0161-813x(02)00067-0. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Takahashi H. The intermediolateral nucleus and Clarke’s column in Parkinson’s disease. Acta Neuropathol (Berl) 1997;94:287–289. doi: 10.1007/s004010050705. [DOI] [PubMed] [Google Scholar]
- Wichmann T, DeLong MR. Pathophysiology of parkinsonian motor abnormalities. Adv Neurol. 1993;60:53–61. [PubMed] [Google Scholar]
- Williams TL, Shaw PJ, Lowe J, Bates D, Ince PG. Parkinsonism in motor neuron disease: case report and literature review. Acta Neuropathol (Berl) 1995;89:275–283. doi: 10.1007/BF00309344. [DOI] [PubMed] [Google Scholar]
- Wolf HK, Crain BJ, Siddique T. Degeneration of the substantia nigra in familial amyotrophic lateral sclerosis. Clin Neuropathol. 1991;10:291–296. [PubMed] [Google Scholar]
- Yanagisawa N. Natural history of Parkinson’s disease: From dopamine to multiple system involvement. Parkinsonism Relat Disord. 2006;12(Suppl 2):S40–46. [Google Scholar]
- Zhang X, Jones D, Gonzalez-Lima F. Neurodegeneration produced by rotenone in the mouse retina: a potential model to investigate environmental pesticide contributions to neurodegenerative diseases. J Toxicol Environ Health A. 2006;69:1681–1697. doi: 10.1080/15287390600630203. [DOI] [PubMed] [Google Scholar]