

Abstract
Steroidogenic acute regulatory protein (StAR) plays a critical role in steroid hormone biosynthesis, presumably by facilitating the delivery of cholesterol to P450scc in the inner mitochondrial membranes. StAR is synthesized as a 37-kDa preprotein that is processed to a 30-kDa mature form by cleavage of an N-terminal mitochondrial import sequence. To identify structural features required for StAR biological activity, we mutated the human StAR cDNA, including the deletion of N- and C-terminal sequences, and examined the ability of the mutants to promote steroidogenesis and enter the mitochondria of transfected COS-1 cells. Deletion of up to 62 residues from the N terminus (N-62) did not significantly affect steroidogenesis-enhancing activity. The N-terminal deletion mutants were associated with mitochondria-enriched fractions, but import and processing were progressively impaired with increasing length of the deletion. Immunogold electron microscopy and in vitro import assays showed that the active N-62 mutant was not imported into the mitochondria. Removal of the 28 C-terminal amino acids (C-28) inactivated StAR. Deletion of the C-terminal 10 amino acids (C-10) reduced steroidogenic activity by 53%, while truncation of the last 4 amino acids had no effect. The C-28 mutant StAR was not efficiently imported into mitochondria or processed, whereas some of the C-10 mutant was processed, indicating that import had occurred. We conclude that in the COS-1 cell system used, StAR does not need to enter into mitochondria to stimulate steroidogenesis and that residues in the C terminus are essential for steroidogenesis-enhancing activity. These findings imply that StAR acts via C-terminal domains on the outside of the mitochondria.
Steroidogenic acute regulatory protein (StAR) is essential for efficient gonadal and adrenal cortical steroidogenesis (1, 2). Human StAR is synthesized as a 285 amino acid preprotein (apparent molecular weight of 37 kDa) in the cytoplasm and then imported into mitochondria, where it is processed to yield a 30-kDa mature protein (2, 3, 4, 5, 6). StAR is thought to promote the movement of cholesterol from the outer to the inner mitochondrial membrane, where P450scc (the enzyme that catalyzes the first and rate-limiting step in steroidogenesis) resides (2, 7). The evidence that StAR is critical for this process has been derived, in part, from the demonstration that mutations in the StAR gene cause congenital lipoid adrenal hyperplasia in which gonadal and adrenal steroid syntheses are severely impaired at the cholesterol side-chain cleavage step, resulting in the massive accumulation of cholesterol in the adrenals (1, 8).
Although StAR plays a key role in steroidogenesis, its mechanism of action remains obscure. The N-terminal 26 amino acids in the preprotein are characteristic of proteins destined to be imported into mitochondria (5, 9, 10). In cell-free import assays, radiolabeled pre-StAR is incorporated into isolated mitochondria and processed to the mature protein (6, 9). Immunoelectron microscopy localized StAR to the intermembranous face of cristae and the intermembranous space (6). These observations led to the prediction that pre-StAR is the active molecule and that its entry into mitochondria is obligatorily linked to steroidogenesis (2).
To elucidate the structural features of human StAR that confer steroidogenesis-enhancing activity, we produced a series of StAR mutants and correlated their ability to stimulate steroidogenesis with their ability to enter into mitochondria and undergo processing.
MATERIALS AND METHODS
Construction of Mutant StAR Plasmids.
Mutations in the human StAR cDNA were produced by the polymerase chain reaction and specific oligonucleotide primers (10). The N-terminal deletions used internal methionines at residues 20 and 29 of wild-type StAR for translation initiation or entailed the mutations I49M and L63M. Mouse StAR N-terminal truncations of N-20 and N-47 were also produced. The A218V mutations were produced by site-directed mutagenesis. The mutant human StAR cDNAs were cloned into pSV-SPORT-1 and sequenced to verify the mutations. Murine StAR constructs were cloned into pCMV-5.
Evaluation of Steroidogenic Activity.
The steroidogenic activity of wild-type and mutant StAR proteins was assayed in COS-1 cells cotransfected with a vector for the cholesterol side-chain cleavage system (11) as described by Sugawara et al. (10) in the absence or presence of 5 μg/ml 22(R)-hydroxycholesterol. Each experiment included triplicate cultures for each treatment group, repeated on at least three separate occasions.
Western Blotting.
COS-1 mitochondria-enriched subcellular fractions were harvested for immunoblotting analysis of StAR (1) using an anti-peptide antibody against residues 89 to 99 of human StAR. The percentage of immunoreactive StAR in the preprotein and mature forms was estimated by densitometry. Mitochondria-enriched fractions were prepared by collecting cells into 0.25 M sucrose/10 mM Tris·HCl/10 mM EDTA, pH 7.4, and sonication for 5 sec. Disrupted cells were centrifuged at 600 × g for 15 min and the resulting supernatant was centrifuged at 13,000 × g for 20 min to isolate the mitochondria-enriched subcellular fraction. In some experiments, equal aliquots of suspended mitochondria were incubated without or with proteinase K (15 μg/ml) in the absence or presence of 0.05% Triton X-100 for 30 min on ice. Phenylmethylsulfonic acid was added to a final concentration of 1 mM to terminate the digestion.
Immunogold Electron Microscopy.
COS-1 cells transfected with empty vector or expression plasmids for wild-type StAR or the N-62 mutant were fixed for routine electron microscopy or with 4% paraformaldehyde, 0.2% glutaraldehyde and phosphate-buffered saline (12). Immunostaining was performed on thin sections on nickel grids (12). Sections were incubated with anti-StAR antibody (1:75) overnight at 4°C and the antibody was detected with 10 nm gold particles complexed to anti-rabbit Ig. Sections were stained with 2% uranyl acetate. Antigen recovery from sections prepared for routine electron microscopy entailed heating grids at 95°C for 10 min in 10 mM sodium citrate (pH 6.0), cooling them to room temperature, washing the grids, followed by incubation with normal guinea pig serum for 1 h and then with the anti-StAR antibody as descried above.
For quantitative analysis, 10 random fields were photographed at ×70,200 and gold particles associated with mitochondrial profiles (no. of particles/profile) were quantified. Gold particles lying within 15 nm of the midpoint of the outer mitochondrial membranes were considered to be on the mitochondrial surface; other particles were considered to be within the mitochondria.
In Vitro Import Assays.
Wild-type StAR and mutant plasmids were linearized with BamHI, and 35S-labeled proteins were synthesized using the Ribomax SP6 in vitro transcription/translation reagents (Promega). Protein import assays into mouse liver mitochondria were carried out as described by Gradi et al. (9). In some cases the mitochondria were treated with proteinase K without or with 0.25% Triton X-100 as described above after completion of the import incubation. Samples were subjected to SDS/PAGE and fluorography.
RESULTS AND DISCUSSION
The Mitochondrial Import Sequence of StAR Is Not Required for Steroidogenic Activity.
We prepared deletion mutations that initiate translation 20 (N-19), 29 (N-28), 49 (N-48), or 63 (N-62) amino acids downstream from the wild-type translation start site. The StAR cDNAs were introduced along with cholesterol side-chain cleavage enzyme system into COS-1 cells, and pregnenolone synthesis from endogenous cholesterol or 22(R)-hydroxycholesterol was determined. Wild-type StAR stimulated pregnenolone secretion from endogenous cholesterol more than 5-fold compared with the empty plasmid control (Fig. 1). The addition of 22(R)-hydroxycholesterol, which readily enters mitochondria and bypasses the requirement for StAR (10, 13), increased pregnenolone synthesis 2- to 3-fold above values produced from endogenous sterol. Under the latter incubation conditions, pregnenolone formation was similar in StAR-transfected cells and cells transfected with empty plasmid. Thus, cholesterol side-chain cleavage enzyme activity was equivalent in the two treatments.
Figure 1.
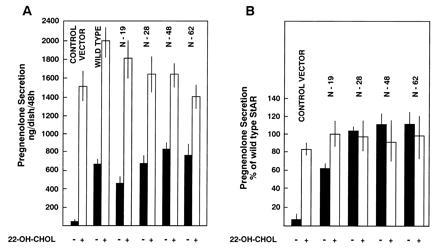
Deletions of the N terminus of StAR do not significantly impair its steroidogenic activity. The indicated constructs were expressed in COS-1 cells with the cholesterol side-chain cleavage system. Transfected cells were incubated without (solid bars) and with 22(R)-hydroxycholesterol (open bars) as described in the text. Pregnenolone secretion, determined by radioimmunoassay, was expressed as nanograms per dish per 48 h for a representative experiment (X ± SD, n = 3 triplicate culture dishes) (A) or as a percentage of that secreted by cells transfected with the wild-type StAR (B) where values are means ± SEM from three separate experiments with triplicate cultures in each treatment. (B) The mean secretion of pregnenolone by COS-1 cells transfected with wild-type StAR using endogenous cholesterol was 670 ng per dish per 48 h. The mean pregnenolone secretion by these cells in the presence of 22(R)-hydroxycholesterol was 1811 ng per dish per 48 h.
The N-19 mutant stimulated pregnenolone synthesis from endogenous cholesterol at a level of 62% of wild-type StAR, whereas the N-28, N-48, and N-62 mutants had 105%, 111%, and 116%, respectively, of the activity of the wild-type protein (Fig. 1). Steroidogenesis was equivalent to the control and wild-type StAR values for all the deletion mutants when 22(R)-hydroxycholesterol was provided as a substrate. However, if the alanine residue at codon 218 of wild-type StAR was mutated to a valine in the N-62 truncation [a mutation found in patients with lipoid CAH and which inactivates full-length StAR (14)], steroidogenic activity in COS-1 cells was lost when pregnenolone was synthesized from endogenous cholesterol (wild-type StAR, 100%; N-62, 138 ± 12; N-62/A218V, 28 ± 1.4%; empty vector, 18 ± 0.9%; n = 3 separate experiments with three replicate dishes per treatment group). Similarly, the N-19 and N-47 mouse StAR truncations displayed steroidogenic activity ranging from 70% to 80% and 57% to 71%, respectively, of mouse wild-type StAR, while simultaneous analysis of human StAR N-terminal truncations yielded 71%, 81%, 92%, and 152% of human wild-type StAR activity, respectively, for the N-19, N-28, N-48, and N-62 constructs.
Western blot analysis demonstrated that the N-19 and N-28 mutants were associated with mitochondria-enriched subcellular fractions and were processed to yield mature protein (Fig. 2A). Seventy percent of the wild-type StAR protein was in the mature form, whereas 48% and 30% of the N-19 and N-28 mutant proteins, respectively, were detected in the mature form. These observations suggest that there is redundancy in the mitochondrial import domain that permits deletion of N-terminal residues without complete impairment of entry and processing of the protein in mitochondria.
Figure 2.
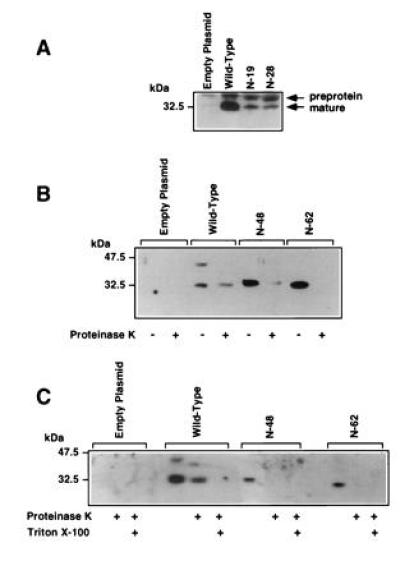
(A) Western blot analysis of N-19 and N-28 mutants expressed in COS-1 cells. Mitochondria-enriched fractions were prepared from COS-1 cells transfected with the indicated constructs expressing wild-type or mutant StAR proteins. (B and C) N-48 and N-62 StAR mutants are not imported into mitochondria or processed. Mitochondria-enriched subcellular fractions were prepared from COS-1 cells transfected with plasmids for wild-type StAR or the N-48 and N-62 mutants. One aliquot was incubated in buffer, the other in proteinase K; in some cases (C), an aliquot was incubated with 0.05% Triton X-100, before SDS/PAGE and Western blotting for StAR protein.
The N-48 and N-62 proteins were not processed, and the N-62 mutant migrated at approximately the same molecular weight as the wild-type mature protein (Fig. 2 B and C). To determine whether the N-48 and N-62 mutants were imported into mitochondria, we isolated mitochondria-enriched fractions from transfected cells and treated one aliquot with proteinase K to digest extra-mitochondrial proteins. The wild-type mature StAR protein was mostly resistant to the proteinase, but the wild-type preprotein was digested. The digestion of some mature StAR reflects the presence of some leaky or damaged mitochondria. However, virtually all of the N-48 and N-62 mutant proteins in the mitochondria-enriched fractions were digested by proteinase K, indicating that they were not imported into the mitochondrial matrix. The mature wild-type protein was only susceptible to proteinase K when Triton X-100 was included to permeabilize the mitochondria. The inability of the N-62 mutant to enter mitochondria was expected, as the N-terminal sequence of N-62 is enriched with acidic rather than basic amino acid residues and, hence, shows no similarity to classical mitochondrial import sequences (15).
COS-1 cells transfected with empty vector did not display significant Immunogold labeling (Fig. 3, Table 1). In contrast, wild-type StAR was detected both within and on the surface of mitochondria. The association of StAR antigen with the intermembranous space and cristae is similar to that reported by King et al. (6) for murine StAR in adrenal cortex. The majority of the gold particles associated with mitochondria from cells transfected with the N-62 mutant expression plasmid were on the outer mitochondrial membrane.
Figure 3.
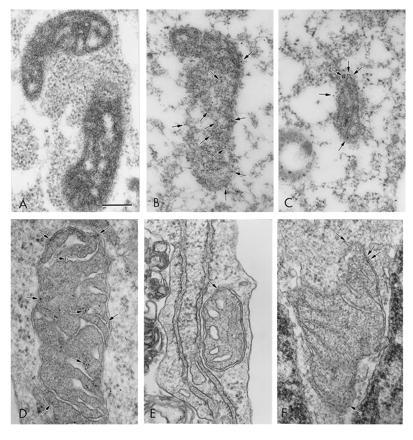
Immunogold electron microscopy detects wild-type StAR in the mitochondrial matrix, while the N-62 mutant is primarily localized to the mitochondrial outer membranes. (A) Mitochondrion from COS-1 cell transfected with empty plasmid; (B) mitochondrion from COS-1 cell transfected with wild-type StAR; and (C) mitochondrion from COS-1 cell transfected with N-62 mutant. (D) Mitochondrion from COS-1 cell transfected with wild-type StAR; (E and F) mitochondria from COS-1 cells transfected with N-62 mutant. (A–C) Fixation for immunoelectron microscopy; (D–F) fixation for routine electron microscopy with antigen recovery. Arrows indicate 10-nm gold particles. (Bar = 0.25 μm.)
Table 1.
Immunogold localization of wild-type StAR and N-62 mutant in mitochondria
| Plasmid | No. of gold particles/mitochondrial
profile
|
|
|---|---|---|
| Inside | Surface | |
| Empty vector | 0.7 ± 0.5 | 0.2 ± 0.1 |
| Wild-type StAR | 11.2 ± 1.9 | 4.4 ± 0.8 |
| N-62 | 2.8 ± 0.8 | 11.3 ± 2.4 |
COS-1 cells were transfected with the indicated plasmids and the cells were analyzed by immunogold electron microscopy. The number of gold particles associated with the mitochondrial surface or the mitochondrial matrix were quantitated in 10 random photomicrographs for each treatment. The number of gold particles inside mitochondria in the cells transfected with wild-type StAR is significantly greater (P < 0.001) than in cells transfected with the N-62 mutant. In contrast, the number of gold particles on the mitochondrial surface is greater (P < 0.01) in cells transfected with N-62 than wild-type StAR.
To confirm that the N-62 mutant is import-incompetent, we examined the ability of wild-type StAR and the N-62 mutant to be imported into and processed by isolated mouse liver mitochondria (Fig. 4). The in vitro translation system yielded two StAR proteins: the wild-type preprotein and a shorter protein produced from translation initiation at the methionine at residue 20 (9). The labeled wild-type preprotein was processed to the mature form of StAR in the mitochondrial matrix, as it was resistant to proteinase K digestion in the absence of detergent, but was degraded when Triton X-100 was included in the digestion reaction. The in vitro translation reaction generated the N-62 mutant as well as a shorter product initiated at methionine residue 83. The labeled N-62 mutant associated with the mitochondria, but was degraded by proteinase K in the absence of detergent, suggesting that the N-62 protein was not incorporated into the mitochondria.
Figure 4.
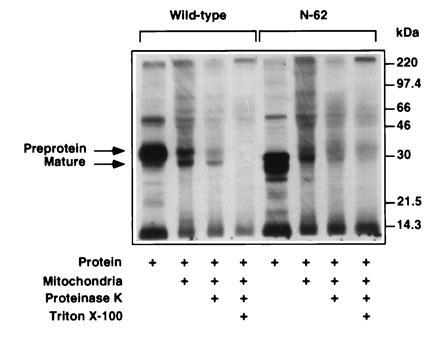
The wild-type StAR preprotein is imported and processed by mouse liver mitochondria, but the N-62 mutant is not. 35S-labeled wild-type StAR and N-62 mutant were incubated with mouse mitochondria as described. Imported and processed wild-type StAR is present in mitochondria following proteinase K digestion in the absence of Triton X-100.
Collectively, these data indicate that removal of the mitochondrial import sequence does not affect StAR’s function. Although the N-19 mutant displayed reduced steroidogenic activity compared with the wild-type StAR, it was processed to the mature form to a greater extent than the N-28 mutant, which had greater steroidogenesis-enhancing activity. Thus, processing of StAR and its functional activity can be dissociated. The basis for the reduced activity of the N-19 construct is unclear, but we speculate that aberrant folding may have compromised its ability to stimulate steroidogenesis. The fact that StAR can stimulate pregnenolone secretion without entering into mitochondria strongly suggests that StAR acts outside or on the surface of this organelle.
The site of cleavage of the mitochondrial import sequence of StAR has not been determined. Clark et al. (5) noted that the first 26 amino acids of the mouse preprotein are characterized by basic residues and form an amphipathic helix. They suggested that murine StAR undergoes two-step processing on the basis of the presence of a conserved sequence, R-X-φ-X-X-S, where X is any residue and φ is a hydrophobic residue. Thus, amino acid residue 48 might be the first residue of the mature murine StAR. However, the R-X-φ-X-X-S sequence is not present in human StAR (10). The rules of Gavel and von Heijne (16) suggest that the cleavage site of human StAR is at amino acid 64, which demarcates a region of the N terminus that is enriched in basic amino acids from a region that is enriched in acidic residues. The nearly identical migration of mature human StAR and the N-62 mutant on SDS/PAGE supports this model. A lack of conservation of the putative cleavage sites in murine and human StAR argues against a critical role for the N terminus in StAR action. Moreover, the fact that murine StAR retains steroidogenic activity following removal of significant portions of its predicted mitochondrial import sequence supports the idea that the N-terminal domain is not required for steroidogenic activity.
The StAR C Terminus Is Required for Mitochondrial Import and Steroidogenic Activity.
We previously reported that a nonsense mutation in the human StAR gene that results in the removal of the C-terminal 28 amino acids causes lipoid congenital adrenal hyperplasia (1). This observation raised the possibility that the association of StAR with mitochondria and its import are dependent upon the C-terminal domain. To test this notion, we prepared a series truncations in which 1, 4, 10, or 28 amino acid residues were removed from the StAR C terminus. We also mutated the C-terminal residue, a cysteine, to a serine. The mutant StAR proteins lacking the C-terminal 1 or 4 amino acid residues had steroidogenic activities greater than or equivalent to wild-type StAR, as did the C285S replacement mutant (Fig. 5). However, removal of the C-terminal 10 amino acids reduced steroidogenesis-enhancing activity to 53% of wild-type StAR, and removal of the last 28 amino acids completely abolished activity.
Figure 5.
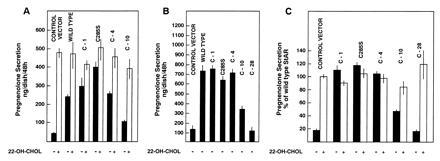
Truncations of the C terminus of StAR abrogate steroidogenesis-enhancing activity. COS-1 cells were transfected with plasmids expressing wild-type StAR and the indicated mutants with the cholesterol side-chain cleavage system. The transfected cells were incubated without (solid bars) or with 22(R)-hydroxycholesterol (open bars), and pregnenolone secretion was determined by radioimmunoassay. Values presented are nanograms per dish per 48 h (means ± SD, n = 3 dishes per treatment) from representative experiments (A and B) or relative to the pregnenolone secretion by cells transfected with the wild-type StAR as means ± SEM (n = 3 separate experiments with three dishes per treatment in each experiment) (C). Pregnenolone secretion by cells transfected with wild-type StAR averaged 439 ng per dish per 48 h from endogenous cholesterol and 850 ng per dish per 48 h in the presence of 22(R)-hydroxycholesterol.
The C-1, C-4, and C285S StAR mutants were all associated with mitochondria-enriched subcellular fractions and were processed to the mature form (Fig. 6A). The C-10 and C-28 mutants were less abundant in the mitochondria-enriched fraction. Some of the C-10 mutant was processed, but none of the C-28 mutant appeared to undergo processing (Fig. 6B). The in vitro import assays showed that the C-28 mutant was not efficiently incorporated into mitochondria or subjected to proteolytic processing (Fig. 7). This finding supports the idea that the C termini of certain proteins that enter into mitochondria are essential for the import process (17). Our observations demonstrate that C-terminal amino acids before residue 282 are essential for StAR action and efficient mitochondrial import, and that the C-terminal cysteine is either not linked by disulfide bonds to an internal cysteine, or if a disulfide linkage exists it is not critical for the maintenance of an active conformation of the molecule.
Figure 6.
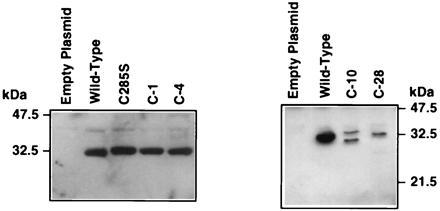
Western blot analysis of C-terminal truncation mutants. COS-1 cells were transfected with plasmids expressing wild-type StAR or the indicated mutants. Mitochondria-enriched subcellular fractions were subjected to Western blotting for StAR protein.
Figure 7.
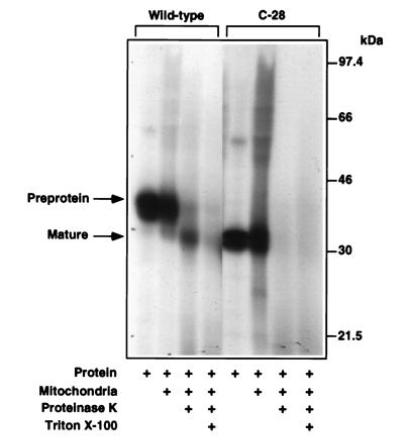
The C-28 mutant is not efficiently imported into mitochondria or processed. 35S-labeled wild-type StAR and C-28 mutant were analyzed in the in vitro import assay. Wild-type StAR is imported and processed yielding proteinase K resistant mature protein. The C-28 mutant is not imported.
Implications for the Mechanism of StAR Action.
Our conclusions must be considered in the context of the model system used to assess StAR activity, COS-1 cells transiently expressing the cholesterol side-chain cleavage enzyme system. Our transfected COS-1 cells secrete in the range of 4–9 μg of pregnenolone per 106 cells per 24 h in the presence of StAR, based on the measurement of pregnenolone secretion from 5 to 7.5 × 105 transfected cells and estimated transfection efficiencies of 5–10%. This secretion rate is greater than that of NCI-H295 human adrenal tumor cells (which secrete less than 3 μg per 106 cells per 24 h) (18), which are used as a model system for the study of adrenal steroidogenesis (19, 20). Although this documents the utility of the COS-1 cell system for studying steroidogenesis, these cells may not contain, quantitatively or qualitatively, all of the components of the steroidogenic machinery present in normal steroid producing cells. Hypothetically, such factors might influence StAR action. These caveats must be kept in mind in interpreting our data. Nevertheless, the possibility that the COS cell system yields artefactual results with respect to StAR action is remote, as they give very accurate correlations between StAR mutation genotypes and lipoid CAH phenotypes. Furthermore, other proteins that are targeted to mitochondria, including the peripheral benzodiazepine receptor, do not enhance steroid production when transfected into COS cells with the cholesterol side-chain cleavage system (unpublished observations).
Our observations dissociate the import and processing of StAR from its steroidogenic activity, indicating that StAR stimulates cholesterol side-chain cleavage activity outside the mitochondria, presumably by acting on the outer mitochondrial membrane. The C terminus of StAR seems to be critical and may contain sequences that are recognized by a receptor that binds StAR and possibly mediates its action. If the binding of StAR to this putative mitochondrial receptor via the C terminus is required for steroidogenic responses and efficient importation of the protein using the N-terminal import sequence, the observed correlation between steroidogenesis-enhancing activity and mitochondrial import of StAR would be expected for C-terminal mutations, but not N-terminal truncations. This model predicts a signal transduction cascade leading to augmented substrate delivery to P450scc. Such a model has been previously proposed for the interaction of endozepine with the mitochondrial peripheral benzodiazepine receptor (21). However, the putative receptor for StAR remains to be characterized. The possibility that StAR interacts with the peripheral benzodiazepine receptor should be examined. The suggestion that StAR may bind to a receptor that is distinct from the mitochondrial import machinery is also consistent with our current understanding of this process (22).
The conclusion that the C-terminal half of the StAR protein is essential for biological activity is consistent with the molecular analysis of the StAR gene in patients with congenital lipoid adrenal hyperplasia (1, 8, 14). All of the known amino acid replacements that cause this disease are located in exons 5–7; none are found in preceding exons. Moreover, the sequences of exons 5–7 are more similar in mouse and man than are those in exons 1–4. These observations further support the hypothesis that C-terminal sequences of StAR are essential for steroidogenesis-enhancing activity.
If StAR does not need to be imported into mitochondria and processed to stimulate steroidogenesis, what is the functional significance of the import/processing events? If StAR continuously promotes steroidogenesis while associated with the mitochondrial outer membrane, import into the mitochondrial matrix may be a mechanism to terminate its steroidogenic action. Thus, mitochondrial import may be the “off” switch, not an obligatory event in the “on” mechanism. Alternatively, StAR may have some other as yet unidentified function within the mitochondria following its initial action on steroidogenesis.
Acknowledgments
We thank Dr. Charles Strott (National Institutes of Health) for the gift of anti-pregnenolone antiserum, and Robert Smith (Electron Microscopy Core of the University of Pennsylvania Diabetes and Endocrinology Research Center, supported by U.S. Public Health Service Grant DK19525) for Immunogold electron microscopy. This work was supported by U.S. Public Health Service Grants HD06274 (J.F.S.), HD17481 (D.M.S.), and DK42154 (W.L.M.) and by a grant from the Lalor Foundation (F.A.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviation: StAR, steroidogenic acute regulatory protein.
References
- 1.Lin D, Sugawara T, Strauss J F, III, Clark B J, Stocco D M, Saenger P, Rogol A, Miller W L. Science. 1995;267:1828–1831. doi: 10.1126/science.7892608. [DOI] [PubMed] [Google Scholar]
- 2.Stocco D M, Clark B J. Endocr Rev. 1996;17:221–244. doi: 10.1210/edrv-17-3-221. [DOI] [PubMed] [Google Scholar]
- 3.Pon L A, Hartigan J A, Orme-Johnson N R. J Biol Chem. 1986;261:13309–13316. [PubMed] [Google Scholar]
- 4.Epstein L F, Orme-Johnson N. J Biol Chem. 1991;266:19739–19745. [PubMed] [Google Scholar]
- 5.Clark B J, Wells J, King S R, Stocco D M. J Biol Chem. 1994;269:28314–28322. [PubMed] [Google Scholar]
- 6.King S R, Ronen-Fuhrmann T, Timberg R, Clark B J, Orly J, Stocco D M. Endocrinology. 1995;136:5165–5176. doi: 10.1210/endo.136.11.7588255. [DOI] [PubMed] [Google Scholar]
- 7.Jefcoate C R, McNamara B C, Artemenko I, Yamazaki T. J Steroid Biochem Mol Biol. 1992;43:751–767. doi: 10.1016/0960-0760(92)90305-3. [DOI] [PubMed] [Google Scholar]
- 8.Tee M K, Lin D, Sugawara T, Holt J A, Guigen Y, Buckingham B, Strauss J F, III, Miller W L. Hum Mol Genet. 1995;4:2299–2305. doi: 10.1093/hmg/4.12.2299. [DOI] [PubMed] [Google Scholar]
- 9.Gradi A, Tang-Wai R, McBride H M, Chu L L, Shore G C, Pelletier J. Biochim Biophys Acta. 1995;1258:228–233. doi: 10.1016/0005-2760(95)00140-8. [DOI] [PubMed] [Google Scholar]
- 10.Sugawara T, Holt J A, Driscoll D, Strauss J F, III, Lin D, Miller W L, Patterson D, Clancy K P, Hart I M, Clark B J, Stocco D M. Proc Natl Acad Sci USA. 1995;92:4778–4782. doi: 10.1073/pnas.92.11.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harikrishna J A, Black S M, Szkaz G D, Miller W L. DNA Cell Biol. 1993;12:371–337. doi: 10.1089/dna.1993.12.371. [DOI] [PubMed] [Google Scholar]
- 12.Smith R M, Jarett L. In: Handbook of Endocrine Research Techniques. de Pablo F, Scanes C G, Weintraub B D, editors. San Diego: Academic; 1993. pp. 227–264. [Google Scholar]
- 13.Toaff M E, Schleyer H, Strauss J F., III Endocrinology. 1982;111:1785–1794. doi: 10.1210/endo-111-6-1785. [DOI] [PubMed] [Google Scholar]
- 14.Bose, H. S., Sugawara, T., Strauss, J. F., III, & Miller, W. L. for the International Lipoid CAH Consortium (1996) N. Engl. J. Med., in press.
- 15.Hendrick J P, Hodges P E, Rosenberg L E. Proc Natl Acad Sci USA. 1989;86:4056–4060. doi: 10.1073/pnas.86.11.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavel Y, von Heijne G. Protein Eng. 1990;4:33–37. doi: 10.1093/protein/4.1.33. [DOI] [PubMed] [Google Scholar]
- 17.Fenton W A. Am J Hum Genet. 1995;57:235–238. [PMC free article] [PubMed] [Google Scholar]
- 18.Gazdar A F, Oie H K, Shackleton C H, Chen T R, Triche T J, Myers C E, Chrousos G P, Brennan M F, Stein C A, La Rocca R V. Cancer Res. 1990;50:5488–5496. [PubMed] [Google Scholar]
- 19.Staels B, Hum D W, Miller W L. Mol Endocrinol. 1993;7:423–433. doi: 10.1210/mend.7.3.8387159. [DOI] [PubMed] [Google Scholar]
- 20.Rainey W E, Bird I M, Mason J I. Mol Cell Endocrinol. 1994;100:45–50. doi: 10.1016/0303-7207(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 21.Papadopoulos V. Endocr Rev. 1993;14:222–240. doi: 10.1210/edrv-14-2-222. [DOI] [PubMed] [Google Scholar]
- 22.Schatz G, Dobberstein B. Science. 1996;271:1519–1526. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]