

Abstract
The major human apurinic/apyrimidinic (AP) endonuclease (class II) is known to cleave DNA 5′ adjacent to an AP site, which is probably the most common DNA damage produced hydrolytically or by glycosylase-mediated removal of modified bases. p-Benzoquinone (pBQ), one of the major benzene metabolites, reacts with DNA to form bulky exocyclic adducts. Herein we report that the human AP endonuclease directly catalyzes incision in a defined oligonucleotide containing 3,N4-benzetheno-2′-deoxycytidine (pBQ-dC) without prior generation of an AP site. The enzyme incises the oligonucleotide 5′ to the adduct and generates 3′-hydroxyl and 5′-phosphoryl termini but leaves the pBQ-dC on the 5′ terminus of the cleavage fragment. The AP function of the enzyme is not involved in this action, as no preexisting AP site is present nor is a DNA glycosylase activity involved. Nicking of the pBQ-dC adduct also leads to the same “dangling base” cleavage when two Escherichia coli enzymes, exonuclease III and endonuclease IV, are used. Our finding of this unusual mode of action used by both human and bacterial AP endonucleases raises important questions regarding the requirements for substrate recognition and catalytic active site(s) for this essential cellular repair enzyme. We believe this to be the first instance of the presence of a bulky carcinogen adduct leading to this unusual mode of action.
Keywords: DNA repair; human carcinogen; 3,N4-benzetheno-2′-deoxycytidine; dangling base
The integrity of the human genome is constantly under attack by countless exogenous and endogenous DNA-damaging agents. This is clearly a major threat to the cell. To cope with this challenge, some cellular enzymes are able to recognize a wide range of substrates and/or have bi- or multiple functions toward different type of substrates.
Benzene is an ubiquitous human carcinogen and recognized as a major cause of acute myeloid leukemia and solid tumors as well, apparently resulting from widespread exposure to cigarette smoke and automobile exhaust (1). Benzene needs to be metabolized to exert its effects and one pathway (2) leads to p-benzoquinone (pBQ), the most potent mutagen of 12 structurally related simple benzoquinones (3). p-BQ is also recognized as an animal carcinogen (2).
Recently, we reported (4) that HeLa cells contained a repair activity for 3,N4-benzetheno-2′-deoxycytidine (pBQ-dC) and 1,N6-benzetheno-2′-deoxyadenosine (pBQ-dA) and we have now purified to apparent homogeneity a 38.1-kDa enzyme from human cells that specifically incises a defined oligonucleotide containing a pBQ-dC adduct derived from the reaction of pBQ with dC under mild conditions. We now find that the repair activity also cleaves apurinic/apyrimidinic (AP)-containing oligonucleotides, which raised the question regarding its relationship to the previously described major human AP endonuclease. Such types of multifunctional enzymes are present in both prokaryotic and eukaryotic cells (5, 6). In Escherichia coli, exonuclease III represents 80–90% of the total cellular AP activity, while endonuclease IV represents 5–10%. Both genes have been cloned and sequenced. Recently, the crystal structure of exonuclease III has been reported (7).
The human AP endonuclease [variously called Ape (8), HAP1 (9), or APEX (10)] is quite similar to E. coli exonuclease III in terms of protein sequence and function. The main function of the human enzyme is 5′ AP endonuclease activity, although a DNA 3′-diesterase and RNase H activities are also reported (5, 6). The human protein has also evolved a novel function (Ref-1) capable of regulating the activity of the AP-1 transcription factor (Jun–Fos heterodimer), which in turn controls expression of a number of genes (11, 12). The relationship between the repair activity and this redox activity is not clear.
Herein we report, on the basis of sequence homology and substrate specificities, that pBQ-dC nicking activity is, to our knowledge, a previously undescribed function of the human AP endonuclease in both HeLa and human leukemia HL-60 cells. The enzyme directly incises the oligonucleotide 5′ to the pBQ-dC adduct without prior generation of an AP site, resulting in the pBQ-dC left as a “dangling base” on the 5′ terminus. This unusual mechanism is also utilized by E. coli AP endonucleases, exonuclease III and endonuclease IV, in the incision of an intact oligonucleotide containing a single pBQ-dC adduct.
MATERIALS AND METHODS
Summary of Purification of pBQ-dC Endonuclease from HeLa and HL-60 Cells.
The detailed procedure of protein purification will be described in a separate publication. Briefly, the cell-free extracts after ammonium sulfate precipitation were desalted and then passed through a Whatman phosphocellulose P11 column. Purification by multiple column chromatography and FPLC fractionation was monitored by cleavage of a 5′ 32P-end-labeled pBQ-dC-containing 25-mer, annealed to a complementary strand with deoxyguanosine opposite pBQ-dC.
Oligonucleotide Substrates and Markers.
The synthesis of 3,N4-pBQ-deoxycytidine, its phosphoramidite and site-directed 25-mer-defined oligonucleotide was as described by Chenna and Singer (13). The sequence used in this study was 5′-CCGCTAG-pBQC-GGGTACCGAGCTCGAAT-3′. A uracil-containing oligomer used as a control was synthesized by placing the dU at the same position (position 8 from the 5′ end) in the same 25-mer sequence. Each of the oligomers was annealed to the same complementary 25-mer oligonucleotide.
The 5-, 7-, 17-, and 18-mer size markers with identical sequences to the putative cleavage products from both the dU and pBQ-dC oligomers were synthesized and purified by OPC cartridges (Applied Biosystems). The corresponding 5′-phosphorylated 17- and 18-mer markers were obtained using T4 polynucleotide kinase (United States Biochemical) and unlabeled ATP. These markers were finally purified through denaturing PAGE (14).
The unphosphorylated 18-mer marker containing pBQ-dC on the 5′ terminus was synthesized using the phosphoramidite technique as described (13). The presence of pBQ-dC in the oligomer was confirmed by HPLC and UV spectra of the peak corresponding to pBQ-dC (13).
The 5′- or 3′-End Labeling.
The 25-mer oligonucleotides with modified bases were 5′-end-labeled with [γ-32P]ATP (specific activity 6000 Ci/mmol; 1 Ci = 37 GBq; Amersham) and subsequently annealed to the complementary oligonucleotide as described by Rydberg et al. (15). The 3′-end labeling of oligonucleotides or 17-mer and 18-mer markers was carried out using the DNA 3′-end-labeling kit (Boehringer Mannheim) and 2′,3′-dideoxyadenosine 5′-[α-32P]triphosphate (ddATP) (specific activity 3000 Ci/mmol, Amersham) by following manufacturer’s instructions. The 3′-end-labeled oligonucleotides were then purified using denaturing PAGE in 18% gels. The full-length bands were cut out and eluted overnight at room temperature with 0.3 M sodium acetate (pH 7.25). The eluent was passed through a Sep-Pak column (Waters) and the collected purified oligonucleotides were then annealed to the complementary strand as described above.
Protein Microsequencing.
The pBQ-dC enzyme that was earlier well separated from other proteins on a 12% precast mini-SDS tris-glycine PAGE (Bio-Rad), was stained with a dye solution containing Coomassie blue (0.1%). Sequencing of two internal tryptic peptides (13 and 20 amino acids, respectively) of the 38.1-kDa target band (Fig. 1) was carried out by the Protein Structure Laboratory, University of California, Davis, using automated Edman degradation and subsequent identification of the phenylthiohydantoin amino acid derivatives by HPLC.
Figure 1.
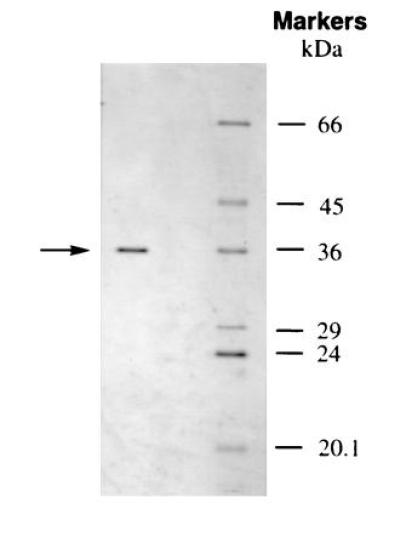
SDS/PAGE of the purified pBQ-dC repair enzyme (left lane) and protein size markers (right lane). Sixty-seven nanograms of the peak active fraction from the final purification step (Superdex 75) was loaded on the 12% gel, which was subsequently stained using a silver staining kit (Pharmacia). The calculated molecular mass of the pBQ-dC enzyme indicated by the arrow is 38.1 kDa. The molecular weight standards were BSA (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (29 kDa), trypsinogen (24 kDa), and trypsin inhibitor (20.1 kDa).
DNA Nicking Assay.
The nicking assay in this work was performed essentially as described by Rydberg et al. (15, 16). The nicking reaction was carried out in a total volume of 10 μl in 25 mM Hepes·KOH, pH 7.8/0.5 mM EDTA/0.5 mM dithiothreitol (DTT)/0.5 μg of poly(dI-dC)/0.5 mM spermidine/1 mM MgCl2/250 μg of BSA/10% glycerol for 1 hr at 37°C. Usually up to 2.5 μl (33 ng) of homogeneous pBQ-dC enzyme was used. As the result of a generous gift of Ape, recombinant Ape proteins (17), E. coli endonuclease IV from Bruce Demple (Harvard University), a comparison could be made of substrate specificity. In addition, exonuclease III (GIBCO/BRL) was also used in this assay. This is illustrated in Fig. 2.
Figure 2.
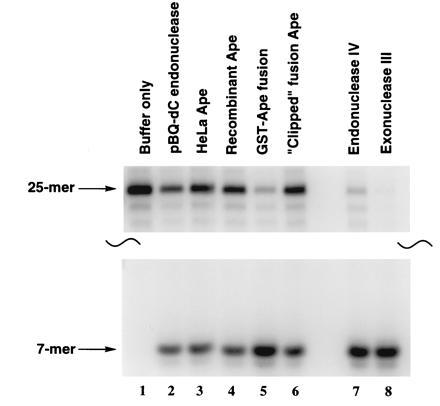
Ability of various human and E. coli 5′ AP endonucleases to cleave the 5′-32P-end labeled pBQ-dC-containing 25-mer oligonucleotide annealed to a complementary strand. Sixteen femtomoles was incubated with various units of the enzymes (lanes 2–8) at 37°C for 1 h. The cleavage products were then resolved on a 12% denaturing PAGE gel. The purified HeLa pBQ-dC endonuclease is in lane 2. In all cases the 5′ cleavage product is a 7-mer (lanes 2–8).
End-Group Determination After Enzymatic Cleavage.
To determine the nature of the 5′-end group of the nick, 5′-end-labeled pBQ-dC-oligomer was reacted with pBQ-dC enzyme as described above and subsequently treated with 10 units of terminal deoxynucleotidyltransferase (United States Biochemical) and 2 μM unlabeled ddATP in a buffer containing 0.5 mM CoCl2 for 30 min at 37°C. Reactions were stopped and electrophoresed as described above with appropriate markers.
Similarly, to examine the 3′-end group of the nick, the pBQ-dC enzyme-treated 3′-end-labeled pBQ-dC oligomer was incubated with 0.25 unit of calf intestinal phosphatase (Pharmacia) for 20 min at 37°C and then run on 18% denaturing PAGE with various 3′-end-labeled size markers.
RESULTS
Substrate Specificity of Human Cell Preparations.
The original premise was that we were dealing with a specific DNA glycosylase since the nicking pattern (5′ to the adduct) was the same when pBQ-dC was the adduct as when a series of human glycosylase substrates were tested using crude HeLa extracts. As purification proceeded, separation was achieved between glycosylases cleaving dU, dI, ɛA, ɛC, thymine glycol, G/T mismatch, and an activity cleaving pBQ-dC. Thus, it appeared that pBQ-dC incision was due to a distinct enzymatic activity. Fig. 3 shows partial or complete separation of several of these activities on a Mono S column. At this stage of purification, it was apparent from complete separation of activity that ɛA and ɛC were repaired by separate glycosylases (18).
Figure 3.
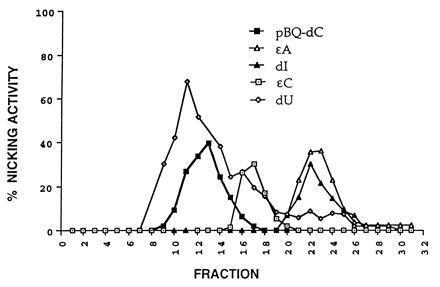
Distribution of five nicking activities after a Mono S HR 5/5 FPLC column (Pharmacia) in the purification of the pBQ-dC enzyme from HeLa cells. Additional specific enzymatic activities such as thymine glycol nicking activity were completely separated during earlier purification steps (data not shown). The nicking assay was used to identify these enzymatic activities.
pBQ-dA nicking activity was also identified in cell-free HeLa extracts (4) but became marginal during attempted purification. This activity was found to be heat-unstable compared with pBQ-dC activity and is presently considered to be a different enzyme and is under investigation.
Identification of the Purified pBQ-dC Nicking Enzyme With the Human AP Endonuclease.
After purification of pBQ-dC nicking activity to apparent homogeneity (Fig. 1), microsequencing was feasible and two internal tryptic peptides of 13 and 20 amino acids were found to be identical with peptides of the major human AP endonuclease (Fig. 4 and refs. 8, 9, 10). This was evidence that the pBQ-dC enzyme was the same as the human AP endonuclease since no other protein in the protein database except eukaryotic AP endonucleases showed absolute homology to the two peptides sequenced.
Figure 4.

Comparison of amino acid sequences in two tryptic peptides from the purified pBQ-dC endonuclease with those of described major mammalian AP endonucleases. Ape, human AP endonuclease (also called HAP1 and APEX) (8, 9, 10); BAP, bovine AP endonuclease (19); APEX, mouse AP endonuclease (20).
With both the purified human AP endonuclease and the recombinant human AP proteins, we were also able to compare the nicking activity toward pBQ-dC of these characterized human AP endonuclease and the recombinant AP proteins, as well as two E. coli AP endonucleases, exonuclease III and endonuclease IV. The data obtained indicated that all the enzymes used were able to efficiently cleave the pBQ-dC oligomer 5′ to the adduct (Fig. 2).
Mechanism of Cleavage of pBQ-dC by AP Endonucleases.
The accepted mechanism of AP endonucleases is to recognize an AP site, regardless of its origin, and then cleave the phosphodiester bond 5′ to the AP site. To examine the possible involvement of a DNA glycosylase activity that could be either associated or coeluted with the purified enzyme and released the pBQ residue, resulting in an AP site, 3′-end labeling was carried out. In Fig. 5, 3′-end-labeled oligomers were used to examine the nature of the 3′ cleavage product from the nick. The 3′ pBQ-dC cleavage product (Fig. 5 Left, lane 8) migrated slower in the gel than both unphosphorylated and 5′-phosphorylated 18-mer markers (lanes 5 and 6). In contrast, when a 25-mer with dU placed at the same position as pBQ-dC was used as substrate for uracil DNA glycosylase (UDG, GIBCO/BRL), the electrophoretic mobility of the cleavage fragment (lane 2) is identical to that of the phosphorylated 17-mer (lane 4). The latter is consistent with the established mechanism of action of simple glycosylases. The addition of a 5′ AP endonuclease to the UDG reaction did not change the UDG-mediated cleavage pattern on the gel (data not shown).
Figure 5.
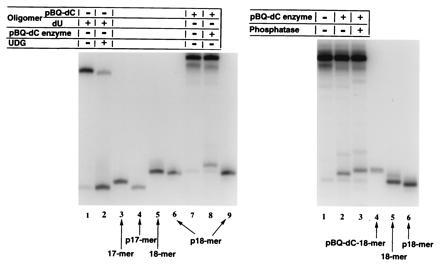
(Left) Denaturing PAGE (20% gels) of 3′-end-labeled fragments obtained from uracil-DNA glycosylase (UDG) activity on a 25-mer oligonucleotide containing dU at the same position as used for pBQ-dC. Lane 2 shows that after UDG treatment the fragment migrates with p17-mer (lane 4) and not with the 17-mer (lane 3). The same procedures for identification of the pBQ-dC endonuclease-treated oligomer (lane 8) did not produce the 17-mer, p17-mer, the 18-mer (lane 5), or p18-mer (lanes 6 and 9). (Right) Size and charge markers to identify the 3′ cleavage product of the pBQ-dC oligomer are in lanes 4–6. Only a synthetic pBQ-dC-18-mer (lane 4) coincided in migration mobility with the pBQ-dC endonuclease-produced 3′ fragment after dephosphorylation (lane 3). This gel shows that it is possible to distinguish between phosphorylated (lane 2) and dephosphorylated (lane 3) cleavage products.
Thus, the unexpected mobility pattern of the pBQ-dC enzyme-mediated cleavage product seen in Fig. 5 strongly suggested that the pBQ-dC still remained on the 5′ end of the cleavage product and was due to a unique mechanism that left a 5′-terminal “dangling base.” This was confirmed by synthesizing a pBQ-dC-containing 18-mer as a marker as shown in Fig. 5 Right. The 32P-3′-end-labeled cleavage fragment from the 25-mer (lane 3) comigrated only with the pBQ-dC-18-mer marker (lane 4) and not with the unmodified 18-mer either with or without 5′-terminal phosphate (lanes 5 and 6).
The 3′-end labeling also showed that the nicking of the pBQ-dC adduct by E. coli endonuclease IV and exonuclease III led to the same “dangling base” cleavage (data not shown).
Determination of End Groups.
The determination of end groups at the nick was carried out using 5′- or 3′-end-labeled oligomers and modifying enzymes. The treatment of the 5′-end-labeled cleavage product with terminal deoxynucleotidyltransferase and ddATP resulted in a DNA band that migrated to a position corresponding to an 8-mer, one nucleotide longer than the cleavage fragment, indicating the presence of a hydroxyl group at the 3′ terminus of the cleavage product (Fig. 6).
Figure 6.
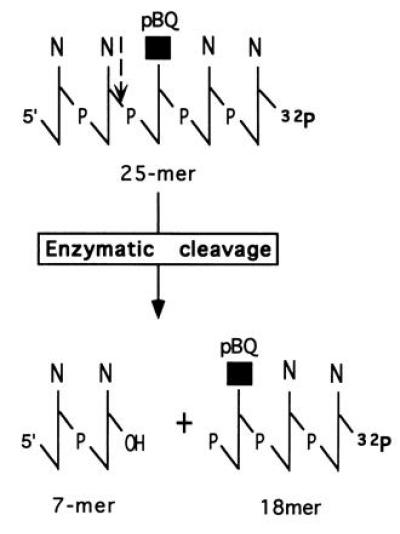
Schematic representation of the proposed mechanism of cleavage by the pBQ-dC endonuclease using the 25-mer oligonucleotide with pBQ-dC at position 8 from the 5′ end. The 5′ cut yields a 7-mer fragment with a 3′-hydroxyl group plus an 18-mer with a 5′-terminal phosphate group with the pBQ-dC residue attached as shown in the drawing. The vertical dashed arrow (top) indicates the site of incision.
The presence of a 5′-terminal phosphoryl group in the 3′-end-labeled cleavage product was confirmed by treatment of the cleavage product with calf intestinal phosphatase, which removes a 5′-terminal phosphoryl group, producing a hydroxyl-containing terminus. The addition of calf intestinal phosphatase resulted in a shift of the cleavage band to a slight slower position, indicating the presence of the phosphoryl group, as expected (Fig. 6).
DISCUSSION
The major AP endonuclease is a multifunctional enzyme with its primary biological role as a class II AP endonuclease (5, 6). Herein we describe a previously undescribed activity of the enzyme acting on a bulky exocyclic DNA adduct produced by the reaction of pBQ with dC. This activity appears to be specific for pBQ-dC, as a series of DNA lesions were tested and found not to be substrates for the enzyme.
In this work we demonstrated that the purified pBQ-dC endonuclease and the major human AP endonuclease appears to be identical. (i) Microsequencing data show complete homology between the two internal sequences tested in this work and the corresponding peptide sequences of the human AP endonuclease. (ii) Homogeneous pBQ-dC endonuclease possesses an 5′ AP activity. (iii) The purified Ape is also able to incise pBQ-dC adduct. (iv) Recombinant human Ape proteins incise the pBQ-dC adduct. Note that these proteins were purified through two different approaches (17). (v) Both enzyme preparations have same molecular weight (within the range of standard error).
The major finding is that the pBQ-dC nicking activity/Ape is unusual since no AP site is measurable prior to the treatment of oligomer with the enzyme. This is supported by the study on the stability of pBQ-dC as a nucleotide and after incorporation into oligomers (13). No chemical cleavage of the pBQ-dC containing oligomer was observed after denaturing PAGE involving heat and alkali, which normally breaks the AP site by β-elimination. The likelihood that a DNA glycosylase activity is involved that releases the pBQ residue, resulting in an AP site, can be ruled out by the findings from 3′-end labeling as described in the Results. All the data presented herein indicate that the enzyme acts as a DNA endonuclease and incises the oligonucleotide 5′ to the adduct, generating 3′-hydroxyl and 5′-phosphoryl termini. However, pBQ-dC is still attached to the 5′ terminus of the cleavage fragment, for which we use the term, “dangling base.” A schematic representation of the mechanism of action is summarized in Fig. 6.
Finally, this unusual mode of initiation of repair of a bulky carcinogen-derived adduct by both human and E. coli AP endonucleases raises new questions regarding factors in enzyme–substrate recognition. The structural requirements for this mechanism of enzyme action are presently under investigation using a variety of related exocyclic carcinogen adducts.
Acknowledgments
The HL-60 cells were a gift from the Cell Culture Center, Endotronics, Inc. Minneapolis, MN, a National Institutes of Health-sponsored facility. This work was supported by Grants CA 47723 and ES 07363 from the National Institutes of Health and was administered by Lawrence Berkeley National Laboratory under Department of Energy Contract DE-AC03-76SF00098.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: AP, apurinic/apyrimidinic; pBQ, p-benzoquinone; pBQ-dC, 3,N4-benzetheno-2′-deoxycytidine; UDG, uracil DNA glycosylase; ddATP, 2′,3′-dideoxyadenosine 5′-triphosphate; Ape, human AP endonuclease.
References
- 1.International Agency for Research on Cancer. IARC Monographs on the Evaluation of the Carcinogenic Risks to Humans. 1987; 1987. (International Agency for Research on CancerLyonFrance)Suppl. 7120122. [PMC free article] [PubMed] [Google Scholar]
- 2.Huff J E, Haseman J K, DeMarini D M, Eustis S, Maronpot R R, Peters A C, Persing R L, Chrisp C E, Jacobs A C. Environ Health Perspect. 1989;82:125–163. doi: 10.1289/ehp.8982125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hricko A. Environ Health Perspect. 1994;102:276–81. doi: 10.1289/ehp.94102276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chenna A, Hang B, Rydberg B, Kim E, Pongracz K, Bodell W J, Singer B. Proc Natl Acad Sci USA. 1995;92:5890–5894. doi: 10.1073/pnas.92.13.5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demple B, Harrison L. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 6.Barzilay G, Hickson I D. BioEssays. 1995;17:713–719. doi: 10.1002/bies.950170808. [DOI] [PubMed] [Google Scholar]
- 7.Mol C D, Kuo C-F, Thayer M M, Cunningham R P, Tainer J A. Nature (London) 1995;374:381–386. doi: 10.1038/374381a0. [DOI] [PubMed] [Google Scholar]
- 8.Demple B, Herman T, Chen D S. Proc Natl Acad Sci USA. 1991;88:11450–11454. doi: 10.1073/pnas.88.24.11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robson C N, Hickson I D. Nucleic Acids Res. 1991;19:5519–5523. doi: 10.1093/nar/19.20.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seki S, Hatsushika M, Watanabe S, Akiyama K, Nagao K, Tsutsui K. Biochim Biophys Acta. 1992;1131:287–299. doi: 10.1016/0167-4781(92)90027-w. [DOI] [PubMed] [Google Scholar]
- 11.Xanthoudakis S, Miao G, Wang F, Pan Y C, Curran T. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker L J, Robson C N, Black E, Gillespie D, Hickson I D. Mol Cell Biol. 1993;13:5370–5376. doi: 10.1128/mcb.13.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chenna A, Singer B. Chem Res Toxicol. 1995;8:865–874. doi: 10.1021/tx00048a007. [DOI] [PubMed] [Google Scholar]
- 14.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Mannual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 15.Rydberg B, Dosanjh M K, Singer B. Proc Natl Acad Sci USA. 1991;88:6839–6842. doi: 10.1073/pnas.88.15.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rydberg B, Qiu Z-H, Dosanjh M K, Singer B. Cancer Res. 1992;52:1377–1379. [PubMed] [Google Scholar]
- 17.Wilson D M, III, Takeshita M, Grollman A P, Demple B. J Biol Chem. 1995;270:16002–16007. doi: 10.1074/jbc.270.27.16002. [DOI] [PubMed] [Google Scholar]
- 18.Hang B, Chenna A, Rao S, Singer B. Carcinogenesis. 1996;17:155–157. doi: 10.1093/carcin/17.1.155. [DOI] [PubMed] [Google Scholar]
- 19.Robson C N, Milne A M, Pappin D J C, Hickson I D. Nucleic Acids Res. 1991;19:1087–1092. doi: 10.1093/nar/19.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seki S, Akiyama K, Watanabe S, Hatsushika M, Ikeda S, Tsutsui K. J Biol Chem. 1991;266:20797–20802. [PubMed] [Google Scholar]