

Abstract
Peroxisomes in human liver contain two distinct acyl-CoA oxidases with different substrate specificities: (i) palmitoyl-CoA oxidase, oxidizing very long straight-chain fatty acids and eicosanoids, and (ii) a branched-chain acyl-CoA oxidase (hBRCACox), involved in the degradation of long branched fatty acids and bile acid intermediates. The accumulation of branched fatty acids and bile acid intermediates leads to severe mental retardation and death of the diseased children. In this study, we report the molecular characterization of the hBRCACox, a prerequisite for studying mutations in patients with a single enzyme deficiency. The composite cDNA sequence of hBRCACox, derived from overlapping clones isolated via immunoscreening and hybridization of human liver cDNA expression libraries, consisted of 2225 bases and contained an open reading frame of 2046 bases, encoding a protein of 681 amino acids with a calculated molecular mass of 76,739 Da. The C-terminal tripeptide of the protein is SKL, a known peroxisome targeting signal. Sequence comparison with the other acyl-CoA oxidases and evolutionary analysis revealed that, despite its broader substrate specificity, the hBRCACox is the human homolog of rat trihydroxycoprostanoyl-CoA oxidase (rTHCCox) and that separate gene duplication events led to the occurrence in mammals of acyl-CoA oxidases with different substrate specificities. Northern blot analysis demonstrated that—in contrast to the rTHCCox gene—the hBRCACox gene is transcribed also in extrahepatic tissues such as heart, kidney, skeletal muscle, and pancreas. The highest levels of the 2.6-kb mRNA were found in heart, followed by liver. The enzyme is encoded by a single-copy gene, which was assigned to chromosome 3p14.3 by fluorescent in situ hybridization. It was absent from livers of Zellweger patients as shown by immunoblot analysis and immunocytochemistry.
In mammals, more than half of the enzymes present in peroxisomes is associated intimately with lipid metabolism (1, 2, 3). Thus, peroxisomes are involved in the β-oxidation of very long straight-chain fatty acids and branched-chain fatty acids, dicarboxylic fatty acids, and eicosanoids. They are also responsible for the β-oxidation of the side chain of the bile acid intermediates di- and trihydroxycoprostanic acids, resulting in the formation of the primary bile acids (chenodeoxycholic and cholic acid, respectively). Most likely, the different substrates are degraded by distinct β-oxidation pathways (1). In the human, the first and rate-limiting step of these pathways is carried out by (at least) two acyl-CoA oxidases (4, 5): (i) palmitoyl-CoA oxidase (hPALMCox),** oxidizing the CoA esters of straight-chain fatty acids, dicarboxylic acids, and eicosanoids and (ii) a branched-chain acyl-CoA oxidase (hBRCACox), oxidizing the CoA esters of 2-methyl-branched fatty acids (e.g., pristanic acid) and of the bile acid intermediates di- and trihydroxycoprostanic acids, which also possess a 2-methyl substitution in their side chain. In the rat, branched fatty acids and bile acid intermediates appear to be oxidized by two separate enzymes, pristanoyl-CoA oxidase (rPRISCox) and trihydroxycoprostanoyl-CoA oxidase (rTHCCox), respectively (11, 12, 13). The former enzyme is expressed in liver and extrahepatic tissues, whereas the latter one is found only in liver and to a very low extent in kidney (11, 12, 13, 14, 15). The hBRCACox, which appears to combine the function of the two rat enzymes, is expressed in liver and kidney, the only tissues examined thus far (5).
Patients, with single enzyme defects, apparently affecting one of the two β-oxidation pathways (1), exhibit elevated serum levels of either very long straight-chain fatty acids or long branched fatty acids and unusual bile acid intermediates (e.g., di- and trihydroxycoprostanic acids) (16, 17). The primary structure of hPALMCox and its corresponding gene has been described recently (8, 9, 18). However, no information is available concerning the molecular nature of the hBRCACox. Molecular characterization of the enzyme and chromosomal mapping of its corresponding gene are prerequisites—as has been shown for mitochondrial β-oxidation defects (19) and the hPALMCox defects in pseudoneonatal adrenoleukodystrophy (8)—to elucidate and prove the nature of hBRCACox deficiencies. Presumably this deficiency is associated with severe mental retardation and early death during childhood (17). In this paper we describe the molecular cloning and further characterization of the hBRCACox, an enzyme consisting of most probably two identical subunits of approximately 70 kDa (4, 5).
EXPERIMENTAL PROCEDURES
Tissue Samples.
Human liver, kidney, and heart tissue was kindly provided by F. Penninckx and R. Aerts, L. Baert and H. Van Poppel, and W. Flameng (all at the Universitair Ziekenhuis, Gasthuisberg, Leuven, Belgium), respectively. Liver samples from patients with peroxisomal disorders were provided by R. J. A. Wanders (Academisch Ziekenhuis, Amsterdam). Approval was granted by the Institutional Ethics Committee.
Isolation of Acyl-CoA Oxidases and Generation of Polyclonal Antibodies.
Isolation of the hBRCACox (5) and the other acyl-CoA oxidases (15, 20) and preparation of antibodies against hBRCACox (21), rat palmitoyl-CoA oxidase (rPALMCox) subunits B and C (14), rPRISCox (14), rTHCCox (15, 20), and peptides containing a C-terminal SKL sequence (22) have been described. The antibody against the A subunit of rPALMCox was kindly provided by A. Völkl (University of Heidelberg, Heidelberg).
Amino Acid Sequencing of hBRCACox Peptides.
The proteins present in the peak fractions from the hydroxylapatite column (see above) were precipitated with trichloroacetic acid and subjected to SDS/PAGE (23). Bands of interest were excised, and material was eluted and concentrated in an agarose gel (24). Trypsin digestion was carried out in the molten gel at 40°C and the agarose was subsequently removed by precipitation (24). Peptides present in the supernatant were separated on a C18 reverse-phase column (Vydac, 250 × 4.6 mm, 300 Å; 5 μm) by using a linear gradient of acetonitrile (0–70%) containing 0.1% trifluoroacetic acid, generated over a 70-min period (1 ml/min). Aliquots of the peptides were taken for analysis by matrix-assisted-laser-desorption-ionization time-of-flight mass spectrometry (Bruker Bioflex instrument, Bruker, Billerica, MA). Some of the peptides with a unique mass were selected for automated gas-phase or pulsed liquid-phase sequencing (Applied Biosystems 470A and 477A sequencers).
Immunoblot Analysis.
Human liver, kidney, and heart tissue was homogenized in 4 vol of 0.25 M sucrose/1 mM EDTA, pH 7.2/0.1% ethanol. Aliquots of the homogenates corresponding to 25 μg of protein (or 200 ng of purified enzyme) were applied to ready-to-use gradient gels (ExcelGel SDS, 8–18%, Pharmacia). After electrophoresis and semi-dry blotting (25), acyl-CoA oxidase-specific bands were detected with different oxidase antisera and visualized via chemoluminescence (ECL, Amersham).
Isolation of the cDNAs for the Human Acyl-CoA Oxidases.
The cDNA for hBRCACox was isolated by immunoscreening of 1 × 106 plaque-forming units of a λ-UniZap human liver cDNA library [oligo(dT)-primed, Stratagene]. Fusion proteins were expressed overnight by overlaying the plates with isopropyl β-d-thiogalactoside-soaked nitrocellulose membranes. The plaques of interest were detected with the polyclonal rabbit antibody against the hBRCACox, followed by incubation with an alkaline phosphatase-labeled sheep anti-rabbit IgG and color detection with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate. The cDNAs of six strongly cross-reactive clones were sequenced from their 5′ ends with vector-specific primers (26). Computer alignment of the cDNA-deduced amino acid sequences with that of hPALMCox (8, 10) revealed that the longest clone (1.9-kb insert) in the 5′ direction started at residue 71 of hPALMCox. To obtain the complete open reading frame, 5 × 105 plaque-forming units of a 5′-stretch λ-gt11 human liver cDNA library [random- and oligo(dT)-primed, CLONTECH] were screened by hybridization with a radioactively labeled 5′ PCR fragment of the longest clone (bases 266–428). Twenty intensely cross-hybridizing clones were purified, the phage DNA was isolated (Lambdaprep, Pharmacia), and further characterized by PCR with oxidase-specific primers and a λ-gt11 primer in the opposite direction. Three clones with the largest extensions in the 5′ direction were used for further studies. Sequencing was done according to Sanger et al. (26) by using cycle sequencing kits (Perkin–Elmer or Pharmacia). The complete cDNA was sequenced at least once in both directions.
A partial cDNA encoding hPALMCox, containing 2098 bases of the hPALMCox cDNA sequence (from base 82 in the open reading frame to base 2180) was isolated as described (15). Plasmid DNA from this clone was used for all hybridization experiments.
Expression of the hBRCACox cDNA.
Since no compatible restriction sites were present in the different cDNA clones for ligation into the expression vector (pTrxFus; Invitrogen), the full-length cDNA was generated by fusion PCR. In the first reaction, separate 5′ (bases −4 to 1064) and 3′ (bases 492 to 2077) fragments were amplified from two different clones by using hBRCACox-specific primers. After a short clean-up to remove the primers, both fragments were annealed with each other and the single-stranded 5′ overhangs were filled up with Taq polymerase (Perkin–Elmer). The full-length construct was amplified in a second PCR with primers from both ends. The resulting 2081-bp fragment was intermediately subcloned by “TA” cloning into pGEMT (Promega). A recombinant clone with insert orientation in sense direction was cut with BamHI (modified restriction site in 5′ primer) and SalI (in pGEMT) and the fragment was ligated into pTrxFus. After controlling the reading frame by sequencing, hBRCACox was overexpressed in Escherichia coli GI698 or GI724 by addition of 100 μM tryptophan to the growth medium. To visualize the expressed oxidase, bacterial lysates (20 μg of protein) were separated on homogeneous SDS/10% gels and transferred to nitrocellulose by semi-dry blotting (25). The thioredoxin–hBRCACox fusion protein was detected with anti-hBRCACox or anti-thioredoxin (Invitrogen) antibodies.
Northern Blot Hybridization.
Human multiple tissue Northern blots (CLONTECH) were hybridized according to the manufacturer’s instructions except that 1.5% SDS instead of 2% was present in the hybridization solution and that more stringent washing conditions were used [up to 0.1× standard saline citrate (SSC)/0.5% SDS, 68°C] to prevent cross-hybridization of the different oxidases. As probe, the complete hBRCACox cDNA (combined from different clones), labeled with 32P (ReadyToGo dCTP labeling kit; Pharmacia), was used. After stripping, the membrane was reprobed with 32P-labeled hPALMCox cDNA, followed by a human β-actin probe (CLONTECH). Blots were exposed to Kodak X-Omat films or to an imaging plate that was scanned with a PhosphorImager (Molecular Dynamics).
Fluorescent in Situ Hybridization of Metaphase Spreads.
Biotinylated probes of the complete hBRCACox and hPALMCox cDNAs were generated by incorporation of biotin-16-dUTP during nick translation (GIBCO/BRL kit). The preparation of metaphase spreads of white blood cells and subsequent treatment with proteases prior to hybridization was done according to ref. 27. Denaturation of the metaphases was carried out in 70% formamide/2× SSC for 3 min, followed by dehydration of the preparations in a series of ice-cold graded (70–100%) ethanol. After denaturation (5 min at 75°C) of the specific probe [10 μl containing 30 ng of biotin-labeled oxidase cDNA in 50% formamide/2× SSC/50 mM Na3PO4/10% dextran sulfate/salmon sperm DNA (5 μg/μl)/CotI DNA (4 μg/μl)], hybridization was carried out overnight at 37°C. After several washing steps and blocking of the nonspecific protein binding sites, the hybridized cDNA was visualized with avidin-coupled fluorescein isothiocyanate (FITC) and signal amplification was obtained by using biotinylated anti-avidin followed by avidin-FITC (27). The preparations were stained with 4′,6-diamidino-2-phenylindole and examined in a Leica fluorescence microscope equipped with a cooled charge-coupled device camera. The collected signals were analyzed with smartcapture software (Vysis, Stuttgart, Germany).
Postembedding Immunocytochemistry with the Protein A-Gold Technique.
Tissue samples were cut into small blocks and immersed overnight at 4°C with a fixative containing 4% paraformaldehyde, 0.05% glutaraldehyde, and 2% sucrose in 0.1 M cacodylate buffer (pH 7.4). After preparation of 100-μm-thick sections with a microslicer (Dosaka, Kyoto, Japan) and a short rinse in 0.1 M cacodylate buffer, the sections were embedded in LR white according to standard protocols (28). Ultrathin sections (50–70 nm) were cut on Formvar-coated nickel grids. After blocking of the nonspecific binding sites with 4% BSA in TBS, hBRCACox and hPALMCox were detected with the appropriate primary antibodies and protein A-gold (15 nm) labeling (29). After contrasting with uranyl acetate and lead citrate, the sections were inspected in a Philips 301 electron microscope.
RESULTS AND DISCUSSION
The hBRCACox cDNA.
After consecutive screening of a λ-UniZap human liver cDNA expression library and a λ-gt11 human liver cDNA library, the sequence of a cDNA encoding hBRCACox was obtained. The composite cDNA sequence contained 2225 bases: 92 bases of the 5′ leader sequence, an open reading frame of 2046 bases, and 87 bases of the 3′ trailer sequence ending at an adenine-rich region (no “AATAAA” polyadenylylation signal was detected in the 3′ trailer) (Fig. 1). The open reading frame encodes a protein of 681 amino acids, with a nominal molecular mass of 76,739 Da. The protein’s C-terminal tripeptide is SKL (see also Fig. 2), which serves as a peroxisome targeting signal (PTSI) (30).
Figure 1.

Nucleotide and deduced amino acid sequence of hBRCACox. The nucleotide sequence shown is derived from overlapping clones of two different libraries and numbered in 5′ → 3′ direction. Numbers on the left of the sequences indicate the appropriate nucleotide or amino acid position. Nucleotides in the 5′ leader sequence are indicated by negative numbers. The position of the stop codon is indicated by ∗∗∗. The amino acid sequences of tryptic peptides are underlined. Their experimentally determined masses agreed with their calculated masses. In addition, some other peptides could be positioned according to their mass [measured and calculated mass (M+H)+ given within parentheses]: amino acids 102–108 (871.2; 871.9), 109–120 (1265.2; 1266.4), 395–409 (1697.9; 1696), and 544–552 (1111.2; 1112.3).
Figure 2.
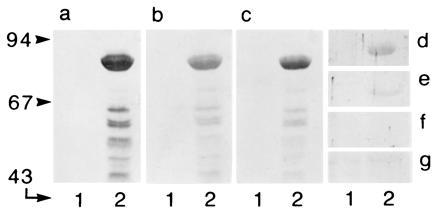
Relationship between the different acyl-CoA oxidases visualized in Western blots. Aliquots of bacterial lysate (20 μg of protein), containing thioredoxin–hBRCACox fusion protein (theoretical molecular mass, 91 kDa), were analyzed for cross-reactivity with different antisera. (a) Anti-thioredoxin. (b) Anti-hBRCACox. (c) Anti-rTHCCox. (d) Anti-SKL. (e) Anti-rPALMCox (B subunit). (f) Anti-rPALMCox (C subunit). (g) Anti-rPRISCox. Lanes 1 and 2 represent a noninduced culture and an induced culture, respectively. Migration of molecular weight markers (expressed in kDa) is indicated at the left.
The identity of the cloned cDNA was confirmed by aligning the peptide sequences with the deduced amino acid sequence (Fig. 1). All peptide sequences (six in total), which were distributed along the whole protein, matched exactly. In addition, four peptides, the mass of which was obtained by matrix-assisted, laser desorption-ionization time-of-flight mass spectrometry, could also be positioned (Fig. 1). Two methionines (the second ATG at bases 52–54) were present in the same reading frame near the translation start site, with the second methionine at the same position as the hPALMCox start site. The first methionine, however, was defined as the start codon, since its flanking nucleotides fitted the Kozak rule (31) (adenine at position −3 and guanine at position +4) and a tryptic peptide of the hBRCACox could be mapped to amino acids 8–14.
The BRCACox Is the Human Counterpart of rTHCCox.
Computer alignment of all cloned mammalian acyl-CoA oxidases revealed that hBRCACox shares 74% overall amino acid identity (76% homology) with rTHCCox (15). Two stretches of low amino acid identity were present at amino acids 58–126 (45% identity) and 466–526 (51% identity) of the two proteins. The remaining part of the two proteins was 80% identical. The amino acid identity of the hBRCACox with the h- and rPALMCox was 45% and 43%, respectively, whereas the identity with rPRISCox (32) was only 21%, a value that is comparable to the identities of the hBRCACox and the yeast acyl-CoA oxidases (20–25%) (data not shown). This relationship between the distinct acyl-CoA oxidases is also reflected by the cross-reactivity of the different oxidase antibodies with the overexpressed hBRCACox fusion protein. Whereas hBRCACox was recognized equally well by the anti-hBRCACox antibody and the anti-rTHCCox antibody, only a weak cross-reactivity was detectable with an antiserum against the B subunit of rPALMCox. No reaction was visible with antibodies against rPRISCox or against the C subunit of rPALMCox (Fig. 2).
Interestingly, the first region of low hBRCACox/rTHCCox identity (see above) partly overlaps (20 amino acids) the differentially spliced exon 3I and 3II sequences of PALMCox (7, 8) (amino acids 90–144 corresponding to hBRCACox amino acids 107–159). The second region of low identity is also not conserved between hBRCACox and hPALMCox or rTHCCox and rPALMCox and overlaps the cleavage site of the rPALMCox (6) (Gln–Val-468 and Ala-469–Val). It should be noted that the hBRCACox and rTHCCox are not cleaved into subunits. Overall, the comparison of the amino acid sequences of the different oxidases clearly shows that the hBRCACox, which combines the function of rTHCCox and rPRISCox, is much more closely related to rTHCCox than to rPRISCox and is in fact the human homolog of rTHCCox. This is also reflected by the N-ethylmaleimide sensitivity (4, 11) and the substrate spectrum‡‡ of the hBRCACox and rTHCCox, and the evolutionary analysis, shown in Fig. 3, where the enzymes cluster in the same branch of the phylogenetic tree.
Figure 3.
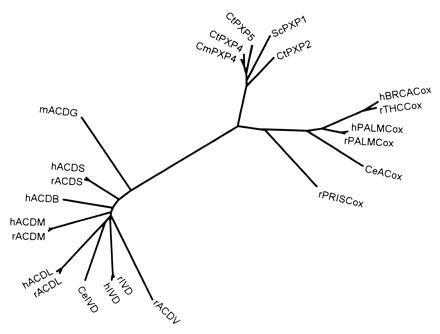
Evolutionary analysis of acyl-CoA oxidases and acyl-CoA dehydrogenases. Multiple sequence alignment of the protein sequences was performed using the pileup program (34) from the GCG package as well as with clustal w (35). The phylogenetic tree was constructed according to the neighbor-joining method (36), as implemented in the neighbor program while the distance matrices needed were calculated with protdist, both from the phylip package (37). The reliability of the tree was assessed by bootstrap analysis. Bootstrap values were in general higher than 80%. Whereas all known acyl-CoA oxidases were aligned, a selection of acyl-CoA dehydrogenases, possessing distinct substrate specificities, was made (data bank accession numbers are also given) as follows: hBRCACox/rTHCCox, X95189X95189; h/rPALMCox I, X71440X71440 and P07872P07872; rPRISCox, X95188X95188; CeACox (Caenorhabditis elegans acyl-CoA oxidase), P34355P34355; ScPXP1 (S. cerevisiae POX1 protein), P13711P13711; CtPXP2/4/5 (Candida tropicalis POX2/4/5 proteins), P06598P06598, P11356P11356, and P08790P08790; CmPXP4 (Candida maltosa POX4 protein), P05335P05335; mACDG (mouse glutaryl-CoA dehydrogenase), U18992U18992; h/rACDS (human and rat short-chain acyl-CoA dehydrogenases), P16219P16219 and P15651P15651; hACDB (human short branched-chain acyl-CoA dehydrogenase), P45954P45954; h/rACDM (human and rat medium-chain acyl-CoA dehydrogenases), P11310P11310 and P08503P08503; h/rACDL (human and rat long-chain acyl-CoA dehydrogenases), P28330P28330 and P15650P15650; rACDV (rat very-long-chain acyl-CoA dehydrogenase), P45953P45953; Ce/h/rIVD (C. elegans, human, and rat isovaleryl-CoA dehydrogenases), P34275P34275, P26440P26440, and P12007P12007. The h/rPALMCoxs II are not shown since the exon duplication, giving rise to the type I and II mRNAs, is a recent event and the branches are hardly visible. Similar topologies were seen in trees obtained by other programs, the tree-construction option in clustal w (35) or the quartet puzzling method (38) (data not shown).
The Acyl-CoA Oxidases and Acyl-CoA Dehydrogenases Originate from a Common Ancestor Gene.
Based on the sequences of rPALMCox and Candida tropicalis acyl-CoA oxidases, Tanaka and Ido (39) suggested that these peroxisomal enzymes and their mitochondrial counterparts, the acyl-CoA dehydrogenases, originated from a common ancestral gene. Multiple alignment of all available acyl-CoA oxidase sequences, including the recently cloned rPRISCox (32) and rTHCCox (15) and hBRCACox, and evolutionary analysis, depicted in Fig. 3, further strengthens this idea and provides additional information on this superfamily. rPRISCox (32) does not share a larger amino acid identity with the other mammalian oxidases than with the yeast oxidases, suggesting that the divergence of the rPRISCox gene was an early event in evolution. This notion is substantiated by the fact that the Caenorhabditis elegans acyl-CoA oxidase is closer to the other mammalian oxidases than rPRISCox is. Interestingly, like the yeast acyl-CoA oxidases, rPRISCox is a homooctamer (14), whereas the other mammalian acyl-CoA oxidases appear to be homodimers. The much larger amino acid identity between the PALMCoxs and rTHCCox/hBRCACox points to a second duplication of a common PALMCox and rTHCCox/hBRCACox ancestor gene later in evolution. The exon duplication leading to PALMCox I and II (not shown in Fig. 3) clearly occurred after the divergence of the PALMCox and rTHCCox/hBRCACox genes.
The hBRCACox Gene Is Transcribed in Extrahepatic Tissues.
Whereas rTHCCox is a hepatic enzyme, hBRCACox, although being the counterpart of rTHCCox, is present not only in liver but also in kidney (5). To investigate whether the hBRCACox gene is transcribed in extrahepatic tissues other than kidney, we hybridized a multiple-tissue Northern blot with the radioactively labeled hBRCACox cDNA probe and for the sake of comparison with the hPALMCox cDNA probe. The Northern blot analysis revealed that the size of the hBRCACox mRNA is approximately 2.6 kb (Fig. 4a), similar to that of the rTHCCox mRNA (15). The size of the hPALMCox mRNA (3.7 kb) was also identical to that of its rat counterpart (Fig. 4b). However, three additional bands, estimated at 8, 6, and 5 kb were present in the human tissues. Similar bands have been described also by Varanasi et al. (18) and were explained as differently spliced precursors of the hPALMCox mRNA. In contrast to their results, however, the specific 3.7-kb band for hPALMCox could be visualized also in extrahepatic tissues. The relative abundance of the precursor forms seems to vary between the different tissues, which might indicate a distinct mRNA processing.
Figure 4.
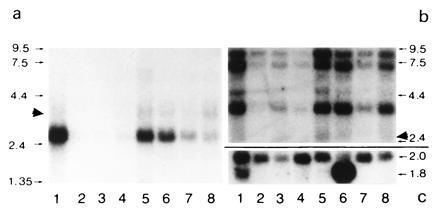
Northern blot analysis of BRCACox in human tissues. A human multiple tissue blot (CLONTECH) containing ≈1 μg of purified mRNA of different tissues was analyzed for the presence of hBRCACox mRNA (a). Lanes: 1, heart; 2, brain; 3, placenta; 4, lung; 5, liver; 6, skeletal muscle; 7, kidney; 8, pancreas. After longer exposures, signals corresponding to BRCACox mRNA became also visible in lanes 2–4 (data not shown). For comparison, in b, the signals obtained for hPALMCox mRNA are shown, while the amount of β-actin mRNA is shown in c. Despite the stringent washing conditions, a very slight cross-reactivity between the mRNAs for hBRCACox and hPALMCox was noticed (marked by solid arrowheads). This is probably due to a stretch of bases with high homology in the middle portion of the mRNA sequences. The migration of standards (expressed in kb) are indicated on both sides (a and b). The values at the right of c correspond to the length (in kb) of the mRNAs for the distinct β-actin isoforms.
More importantly, the Northern blot analysis revealed that the hBRCACox gene is transcribed not only in liver and kidney, as had been suggested by enzyme activity measurements, but also in all other tissues studied (Fig. 4a). Because of the proposed function of the hBRCACox in the oxidation of 2-methyl-branched fatty acids, this observation was not unexpected. Surprising, however, were the high hBRCACox mRNA levels in heart, suggesting that the enzyme may have an important function in cardiac muscle. In agreement with this contention, BRCACox activity appeared to be as high as PALMCox activity in human heart (E.B., G.P.M., and P.P.V.V., unpublished data).
The hBRCACox Gene Is Present in a Single Copy on Chromosome 3p14.3.
Southern blot analysis of human genomic DNA revealed single bands in several lanes of the blot after hybridization with distinct hBRCACox probes generated by PCR from different regions of the cDNA (data not shown). The presence of a single gene encoding BRCACox in the human genome was verified by fluorescence in situ hybridization. The hBRCACox gene was assigned to chromosome 3p14.3 (Fig. 5a). No cross-reactivity with the hPALMCox gene (Fig. 5b), which in agreement with the data of Varanasi et al. (18) was assigned to chromosome 17q25.1, was detected.
Figure 5.
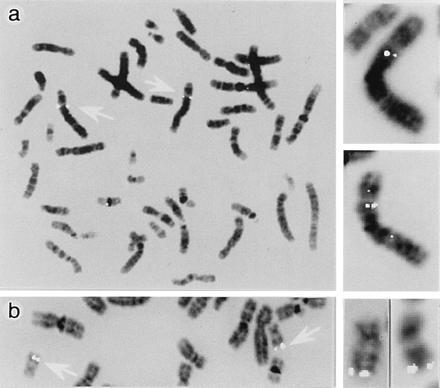
Chromosomal assignment of the hBRCACox gene. The chromosomal mapping of the hBRCACox gene was investigated by means of fluorescent in situ hybridization of metaphase spreads of white blood cells with biotinylated probes. Labeling was confined to chromosome 3p14.3 (a) and no cross-reactivity of the cDNA probe with the hPALMCox gene was found. In agreement with earlier reports (18), the latter gene was assigned to chromosome 17q25.1 (b). The signals, associated with the alleles of the oxidase genes on chromosomes 3 and 17 of the metaphase spreads are indicated by open arrows. The inserts at the right side show several examples of chromosomes 3 (a) and 17 (b) at a higher magnification, demonstrating specific hybridization of the probes with both alleles.
The hBRCACox Is Not Detectable in the Liver of Zellweger Patients.
Generalized peroxisomal disorders such as the Zellweger syndrome are the consequence of a deficiency of the import of peroxisomal matrix proteins (40, 41). These proteins are synthesized at their normal rates in the cytosol but not imported into peroxisomes. Some of the proteins including PALMCox and other β-oxidation proteins are rapidly degraded in the cytosol, so that they become hardly detectable on immunoblot analysis. To verify whether the same would hold for the BRCACox, we performed an immunoblot analysis of control livers and livers from Zellweger patients with antibodies raised against the hBRCACox and with antibodies raised against the subunits of rPALMCox. As can be seen from Fig. 6, the hBRCACox was immunologically not detectable in livers from Zellweger patients, as was the case for hPALMCox. These findings are in agreement with the previously reported enzyme deficiencies in Zellweger liver (4, 5). The biochemical results were corroborated by postembedding immunoelectron microscopy. In Zellweger patients, no specific signals for hBRCACox and hPALMCox were found, whereas even in autolytic samples of human control livers both acyl-CoA oxidases were present (Fig. 7).
Figure 6.
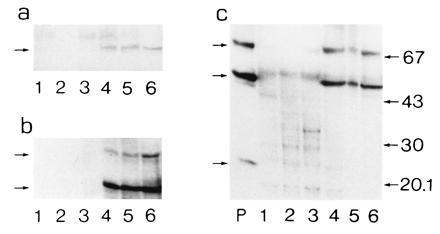
Western blot analysis of hBRCACox in liver of control subjects and Zellweger patients. Aliquots of liver homogenates (25 μg of protein) from different Zellweger patients (lanes 1–3) and healthy individuals (lanes 4–6) were subjected to SDS/PAGE followed by blotting (in triplicate). The blots were incubated with anti-hBRCACox (a), anti-rPALMCox (B subunit) (b), or anti-rPALMCox (A subunit) (c). Lane P in c represents 200 ng of purified rPALMCox. No specific signals for either hBRCACox or PALMCox protein were detectable in Zellweger patients. The small arrows at the left of a refer to the position of the 70-kDa hBRCACox subunit band, of b to the positions of the 70- and 51-kDa subunits of hPALMCox, and of c to the position of the 70-, 51-, and 23-kDa subunits of hPALMCox. Migration of the molecular mass standards (expressed in kDa) is indicated only at the right of c.
Figure 7.
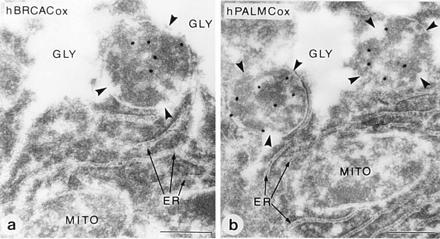
Immunocytochemical localization of BRCACox in human liver peroxisomes. Pieces of human liver were processed for immunoelectron microscopy and stained for hBRCACox (a) and hPALMCox (b). Peroxisomes (marked with arrowheads) are specifically labeled with gold particles depicting the different acyl-CoA oxidase antigens. Other cell organelles such as mitochondria (MITO), endoplasmic reticulum (ER), lysosomes, or nuclei were not labeled. Peroxisomes in this sample are often associated with glycogen deposits (GLY). The sample was derived from a ruptured and, therefore, nontransplanted liver lobe of a liver prepared for transplantation. Due to delay between excision of the liver and fixation, the sections show signs of autolysis. In contrast to the positive results (despite the autolysis) obtained with the liver of the healthy individual, no specific labeling was obtained in sections of a well-preserved liver biopsy of a Zellweger patient, either with antibodies against hBRCACox or with the different antibodies against rPALMCox (data not shown).
Conclusions.
The data reported in this paper show that the BRCACox is actually the human counterpart of rTHCCox. In contrast to the rat enzyme, which is expressed only in liver (and to a very small extent in kidney) (11, 15), the hBRCACox is expressed also in extrahepatic tissues. This multiple-tissue expression is not surprising since the human enzyme is responsible not only for the degradation of the side chain of cholesterol but also for the degradation of 2-methyl-branched fatty acids and since PRISCox, the enzyme responsible for the degradation of branched-chain fatty acids in the rat, is apparently not expressed in the human (14, 32). As is the case for other peroxisomal β-oxidation enzymes, the hBRCACox is severely deficient in livers from Zellweger patients.
Evolutionary analysis, after comparison of the different deduced amino acid sequences of the mammalian and yeast acyl-CoA oxidases, indicates that the PRISCox gene diverged early in evolution from a common ancestral gene followed later by a divergence of the PALMCox and rTHCCox/hBRCACox genes.
Acknowledgments
The skillful technical assistance of Mrs. C. Brees and Mrs. P. Van Rompuy (Leuven) and Mrs. A. Appel (Heidelberg) is gratefully acknowledged. This work was supported by grants from the Geconcerteerde Onderzoeksacties van de Vlaamse Gemeenschap and the Fonds voor Geneeskundig Wetenschappelijk Onderzoek. The peptide sequencing (Gent) was supported by grants from the Nationaal Fonds voor Wetenschappelijk Onderzoek and from the Commission of the European Union. One of us (E.B.) was partly supported by the Deutsche Forschungsgemeinschaft.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: hBRCACox, human branched-chain acyl-CoA oxidase; hPALMCox, human palmitoyl-CoA oxidase; rPALMCox, rat palmitoyl-CoA oxidase; rPRISCox, rat pristanoyl-CoA oxidase; rTHCCox, rat trihydroxycoprostanoyl-CoA oxidase.
Data deposition: The sequence reported in this paper has been deposited in the GenBank data base (accession no. X95190X95190).
In man, as in rat (6, 7), two mRNAs for palmitoyl-CoA oxidase have been described which are derived from a common gene via differential splicing (8). The corresponding oxidases have not been purified to homogeneity, but the one derived from the type I mRNA has been produced in expression systems (9, 10).
References
- 1.Mannaerts, G. P. & Van Veldhoven, P. P. (1995) Ann. N.Y. Acad. Sci. 804, in press. [DOI] [PubMed]
- 2.van den Bosch H, Schutgens R B H, Wanders R J A, Tager J. Annu Rev Biochem. 1992;61:157–197. doi: 10.1146/annurev.bi.61.070192.001105. [DOI] [PubMed] [Google Scholar]
- 3.Reddy J K, Mannaerts G P. Annu Rev Nutr. 1994;14:343–370. doi: 10.1146/annurev.nu.14.070194.002015. [DOI] [PubMed] [Google Scholar]
- 4.Casteels M, Schepers L, Van Veldhoven P P, Eyssen H J, Mannaerts G P. J Lipid Res. 1990;31:1865–1872. [PubMed] [Google Scholar]
- 5.Vanhove G F, Van Veldhoven P P, Fransen M, Denis S, Eyssen H J, Wanders R J A, Mannaerts G P. J Biol Chem. 1993;268:10335–10344. [PubMed] [Google Scholar]
- 6.Miyazawa S, Hayashi H, Hijikata M, Ishii N, Furuta S, Kagamiyama H, Osumi T, Hashimoto T. J Biol Chem. 1987;262:8131–8137. [PubMed] [Google Scholar]
- 7.Osumi T, Ishii N, Miyazawa S, Hashimoto T. J Biol Chem. 1987;262:8138–8143. [PubMed] [Google Scholar]
- 8.Fournier B, Saudubray J-M, Benichou B, Lyonnet S, Munnich A, Clevers H, Poll-The B T. J Clin Invest. 1994;94:526–531. doi: 10.1172/JCI117365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aoyama T, Tsushima K, Souri M, Kamijo T, Suzuki Y, Shimozawa N, Orii T, Hashimoto T. Biochem Biophys Res Commun. 1994;198:1113–1118. doi: 10.1006/bbrc.1994.1158. [DOI] [PubMed] [Google Scholar]
- 10.Chu R, Varanasi U, Chu S, Lin Y, Usuda N, Rao S, Reddy J K. J Biol Chem. 1995;270:4908–4915. doi: 10.1074/jbc.270.9.4908. [DOI] [PubMed] [Google Scholar]
- 11.Schepers L, Van Veldhoven P P, Casteels M, Eyssen H J, Mannaerts G P. J Biol Chem. 1990;265:5242–5246. [PubMed] [Google Scholar]
- 12.Van Veldhoven P P, Vanhove G, Vanhoutte F, Dacremont G, Parmentier G, Eyssen H J, Mannaerts G P. J Biol Chem. 1991;266:24676–24683. [PubMed] [Google Scholar]
- 13.Van Veldhoven P P, Vanhove G, Asselberghs S, Eyssen H J, Mannaerts G P. J Biol Chem. 1992;267:20065–20074. [PubMed] [Google Scholar]
- 14.Van Veldhoven P P, Van Rompuy P, Fransen M, De Bethune B, Mannaerts G P. Eur J Biochem. 1994;222:795–801. doi: 10.1111/j.1432-1033.1994.tb18926.x. [DOI] [PubMed] [Google Scholar]
- 15.Baumgart E, Vanhooren J C T, Fransen M, Van Leuven F, Fahimi H D, Van Veldhoven P P, Mannaerts G P. Biochem J. 1996;320:115–121. doi: 10.1042/bj3200115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poll-The B T, Roels F, Ogier H, Scotto J, Vamecq J, Schutgens R B H, Wanders R J A, Van Roermund C W T, Van Wijland M J A, Schram A W, Tager J M, Saudubray J M. Am J Hum Genet. 1988;42:422–434. [PMC free article] [PubMed] [Google Scholar]
- 17.Przyrembel H, Wanders R J A, Van Poermund C W T, Schutgens R B H, Mannaerts G P, Casteels M. J Inher Metab Dis. 1990;13:367–370. doi: 10.1007/BF01799397. [DOI] [PubMed] [Google Scholar]
- 18.Varanasi U, Chu R, Chu S, Espinosa R, LeBeau M M, Reddy J K. Proc Natl Acad Sci USA. 1994;91:3107–3111. doi: 10.1073/pnas.91.8.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coates P M, Tanaka K, editors. Recent Developments in Fatty Acid Oxidation. New York: Wiley–Liss; 1992. [Google Scholar]
- 20.Van Veldhoven P P, Van Rompuy P, Vanhooren J C T, Mannaerts G P. Biochem J. 1994;303:195–200. doi: 10.1042/bj3040195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanhove, G. (1994) Ph. D. thesis (Leuven Univ., Leuven, Belgium).
- 22.Fransen M, Brees C A E, de Bethune B, Van Veldhoven P P, Mannaerts G P. Biochim Biophys Acta. 1994;1201:157–164. doi: 10.1016/0304-4165(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 23.Matsudaira P, Burgess D R. Anal Biochem. 1978;87:386–396. doi: 10.1016/0003-2697(78)90688-7. [DOI] [PubMed] [Google Scholar]
- 24.Rider M H, Puype M, Van Damme J, Gevaert K, De Boeck S, D’Alayer S, Rasmussen H-H, Celis J, Vandekerckhove J. Eur J Biochem. 1995;230:258–265. doi: 10.1111/j.1432-1033.1995.0258i.x. [DOI] [PubMed] [Google Scholar]
- 25.Khyse-Andersen J. J Biochem Biophys Methods. 1984;10:203–209. doi: 10.1016/0165-022x(84)90040-x. [DOI] [PubMed] [Google Scholar]
- 26.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaffanet M, Baens M, Aerssens J, Schoenmakers E, Cassiman J-J, Marynen P. Cytogenet Cell Genet. 1995;69:27–32. doi: 10.1159/000133930. [DOI] [PubMed] [Google Scholar]
- 28.Newman G R, Jasani B, Williams E D. Histochem J. 1983;15:543–555. doi: 10.1007/BF01954145. [DOI] [PubMed] [Google Scholar]
- 29.Slot J W, Geuze H J. Eur J Cell Biol. 1985;38:87–93. [PubMed] [Google Scholar]
- 30.Gould S J, Keller G A, Hosken N, Wilkinson J, Subramani S. J Cell Biol. 1989;108:1657–1664. doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozak M. Nucleic Acids Res. 1981;9:5233–5252. doi: 10.1093/nar/9.20.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanhooren J C T, Fransen M, De Bethune B, Baumgart E, Baes M, Torrekens S, Van Leuven F, Mannaerts G P, Van Veldhoven P P. Eur J Biochem. 1996;239:302–309. doi: 10.1111/j.1432-1033.1996.0302u.x. [DOI] [PubMed] [Google Scholar]
- 33.Van Veldhoven P P, Croes K, Asselberghs S, Herdewijn P, Mannaerts G P. FEBS Lett. 1996;388:80–84. doi: 10.1016/0014-5793(96)00508-x. [DOI] [PubMed] [Google Scholar]
- 34.Devereux J, Haeberli P, Smithies O. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson J D, Higgins D G, Gibbson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saitou N, Nei M. Mol Biol Evol. 1988;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 37.Felsenstein, J. (1993) phylip: Phylogeny Inference Package (Department of Genetics, Univ. Washington, Seattle), Version 3.5c.
- 38.Strimmer, K. & Von Haeseler, A. (1996) Mol. Biol. Evol., in press.
- 39.Tanaka K, Ido Y. In: New Developments in Fatty Acid Oxidation. Coates M, Tanaka K, editors. New York: Wiley–Liss; 1992. pp. 95–110. [Google Scholar]
- 40.Moser H W. In: Advances in Human Genetics. Harris H, Hirschhorn K, editors. New York: Plenum; 1993. pp. 1–106. [Google Scholar]
- 41.Wanders R J A, Barth P G, Schutgens R B H, Tager J M. In: Peroxisomes: Biology and Importance in Toxicology and Medicine. Gibson G, Lake B, editors. London: Taylor and Francis; 1993. pp. 63–98. [Google Scholar]