

Abstract
The DNA damage checkpoint response delays cell cycle progression upon DNA damage and prevents genomic instability. Genetic analysis has identified sensor, mediator, signal transducer, and effector components of this global signal transduction pathway. Here we describe an in vitro system with purified human checkpoint proteins that recapitulates key elements of the DNA damage checkpoint. We show that the damage sensor ATR in the presence of topoisomerase II binding protein 1 (TopBP1) mediator/adaptor protein phosphorylates the Chk1 signal-transducing kinase in a reaction that is strongly dependent on the presence of DNA containing bulky base lesions. The dependence on damaged DNA requires DNA binding by TopBP1, and, indeed, TopBP1 shows preferential binding to damaged DNA. This in vitro system provides a useful platform for mechanistic studies of the human DNA damage checkpoint response.
Keywords: Chk1 kinase, damage recognition, signal transduction, topoisomerase II binding protein 1
Most of our knowledge about the DNA damage checkpoint response is based on genetic data from model organisms, including budding and fission yeasts and humans and the Xenopus egg extract system. These studies have identified damage sensors, mediators, signal transducers, and effectors as key components of this signal-transduction pathway (1–3). The phosphatidylinositol 3-kinase-related protein kinase (PIKK) family members have been shown to be key DNA damage sensors and signal transducers in the checkpoint response. Of these, ATM is mainly responsible for initiation of the checkpoint response elicited by double-strand breaks caused by ionizing radiation or radiomimetic agents. Some semidefined systems for the ATM-mediated checkpoint response have been described recently (4–8). Another PIKK family member, ATR, initiates the DNA damage checkpoint response caused by UV radiation and UV-mimetic agents that produce base damage such as N-acetoxy-2-acetylaminofluorene (N-Aco-AAF). Although this important signal-transduction pathway has been characterized in some detail by using Xenopus egg extracts (9–15) and in human cell-free systems (16, 17), only recently have partial in vitro systems been developed with a subset of either Xenopus (14, 15) or yeast (18) checkpoint proteins. Currently, there is no well defined system for ATR-mediated DNA damage checkpoint response in humans. Recently, it has been shown that the multifunctional XtopBP1 protein, which is known to be required for the ATR-mediated checkpoint (19), activates the ATR kinase on downstream targets, in particular the Chk1 signal-transducing kinase, in the absence of DNA or any other checkpoint protein except the ATR-interacting protein (ATRIP) (14). Here we describe a human in vitro system in which ATR phosphorylates Chk1 kinase in a reaction that depends on topoisomerase II binding protein 1 (TopBP1) and is strongly stimulated by DNA containing bulky DNA adducts. We believe this is a useful system for the ultimate development of an in vitro human checkpoint response dependent on all checkpoint proteins identified by genetic methods. Such a system would open new opportunities for mechanistic studies of the human DNA damage checkpoint response.
Results
TopBP1-Dependent Stimulation of ATR Kinase Activity by DNA.
We were interested in developing a human ATR-mediated checkpoint system in vitro. To this end, we purified human ATR–ATRIP, TopBP1, and Chk1 proteins. The ATR–ATRIP complex was purified from HeLa cell-free extracts (Fig. 1 A and B) to a high degree of purity, free of other PIKK kinases that could possibly complicate the interpretation of the results. TopBP1 and Chk1 were purified from Escherichia coli and baculovirus-infected insect cells, respectively [supporting information (SI) Fig. 7]. Fractions containing highly purified ATR–ATRIP were tested for the ability to phosphorylate Chk1-S345 in the presence and absence of TopBP1 (Fig. 1C). We found that in reaction mixtures that were of low ionic strength, ATR phosphorylated Chk1 in a manner virtually dependent on TopBP1, which is in agreement with previous reports (14, 20) (Fig. 2, lane 3). However, when the ionic strength was increased to more physiologically relevant levels, the stimulation of ATR kinase by TopBP1 was, by and large, eliminated (Fig. 2, lanes 7 and 11). We reasoned that under these conditions, the checkpoint-signaling reaction may be reestablished by including DNA in the reaction. Fig. 2 shows the phosphorylation of Chk1 by ATR in the absence and presence of DNA with and without TopBP1. Under higher ionic strength reaction conditions, TopBP1 has no measurable effect on the kinase activity of ATR (lane 7). Significantly, however, the addition of DNA to this reaction resulted in a 7-fold stimulation of ATR activity (lane 8), restoring the Chk1 phosphorylation to levels observed in the low ionic strength buffer (lane 3).
Fig. 1.

Purification of human ATR–ATRIP and activation of ATR kinase activity by TopBP1. (A) Scheme of ATR–ATRIP purification from HeLa cell-free extracts. (B) Purification of ATR–ATRIP. Fractions from the Mono S column were separated on a 5/10% discontinuous SDS/polyacrylamide gel and analyzed by Western blotting using the indicated antibodies (α-) (upper four blots) and by silver staining (lower gel). ATM and DNA-dependent PK catalytic subunit (DNAPKcs) were separated from fractions containing ATR–ATRIP during chromatography on CM Affi-Gel Blue and DEAE columns, respectively. The peak of ATR–ATRIP eluted at 220 mM KCl (fraction 8) from the Mono S column. NE, nuclear extract; L, load; E, eluate; FT, flow-through; 1–12, fraction numbers; asterisks, ATR and ATRIP. The line between the 100- and 150-kDa markers is the boundary between the 5% and 10% acrylamide regions of the gel. (C) Activation of purified ATR kinase activity by TopBP1. Kinase assays were performed with the Mono S column fractions to test for TopBP1-dependent ATR kinase activity. Mono S load (lanes 1 and 2), flow-through (lanes 3 and 4), and eluted fractions (lanes 5–12) were incubated under low ionic strength reaction conditions (45 mM total salt concentration) with His-Chk1-kd in the presence or the absence of GST-TopBP1-His. An equal volume of each fraction was preincubated for 15 min at 30°C with 5 nM GST-TopBP1-His. Chk1-kd (20 nM) was then added into the reaction and incubated for 20 min at 30°C. Reactions were analyzed by immunoblotting for phosphorylated Chk1 (P-Chk1, which is phosphorylated at S345) (Upper) and total Chk1 (Lower).
Fig. 2.

TopBP1-dependent stimulation of ATR kinase activity by DNA and effects of ionic strength on this stimulation. (Upper) Kinase assays were carried out with ATR–ATRIP, His-Chk1-kd, GST-TopBP1-His, and double-stranded DNA under different ionic strength reaction conditions. ATR–ATRIP (1 nM) was preincubated with or without 5 nM pUC19 plasmid in the presence and absence of 1 nM GST-TopBP1-His. Chk1-kd (20 nM) was then added to the reaction and incubated for 20 min at 30°C. (Lower) Levels of Chk1 phosphorylation were quantified, and the results from three independent experiments were averaged and graphed. Phosphorylation was normalized to the control reaction in lane 1 (without TopBP1 and DNA).
Effects of Various DNA Substrates on TopBP1-Dependent ATR Kinase Activity.
The dramatic TopBP1-dependent stimulation of ATR kinase activity that we observed in Fig. 2 was achieved with double-stranded plasmid DNA. We reasoned that a more physiologically relevant form of DNA might result in further stimulation of ATR in our in vitro reactions. As a model for both primer templates and excision repair gaps, deca-primed DNA (ten 30-mer primers, roughly equally spaced along the DNA circle) has been reported to be optimal for stimulating Saccharomyces cerevisiae Mec1ATR kinase (18). The addition of deca-primed DNA to our reactions results in up to a 5-fold stimulation of Chk1 phosphorylation by ATR in the presence of TopBP1 (Fig. 3, lanes 6–8). However, the stimulatory effect was not specific to deca-primed DNA, because we observed essentially the same levels of stimulation of the TopBP1-dependent ATR kinase activity by both single-stranded (lanes 3–5) and double-stranded (lanes 9–11) plasmid DNAs. Because a number of studies have indicated that replication protein A (RPA)-covered single-stranded DNA is a common, if not universal, signal for ATR-mediated checkpoint activation (21, 22), we tested the effect of RPA on the reaction by using both deca-primed and double-stranded DNA. We observed only a slight stimulation upon the addition of RPA with both types of DNA (SI Fig. 8). Although these results do not eliminate RPA-covered single-stranded DNA or template/primer structures as checkpoint signals, they do show that under our experimental conditions of physiological ionic strength and limiting concentrations of TopBP1, all forms of DNA tested act similarly in activating ATR in a TopBP1-dependent manner.
Fig. 3.

Stimulation of ATR kinase activity by various DNA substrates in the presence of TopBP1. (Upper) Kinase assays were performed with ATR–ATRIP, His-Chk1-kd, GST-TopBP1-His, and different forms of DNA substrates under high ionic strength reaction conditions (85 mM total salt concentration) as described in Materials and Methods. ATR–ATRIP (1 nM) was incubated with 1.25–5 nM single-stranded, deca-primed, or double-stranded pIBI25 plasmid in the presence and absence of 1 nM GST-TopBP1-His. (Lower) The average levels of Chk1 phosphorylation from three independent experiments were quantitated and graphed.
TopBP1-Dependent Stimulation of ATR Kinase Activity by DNA Containing Bulky Base Lesions.
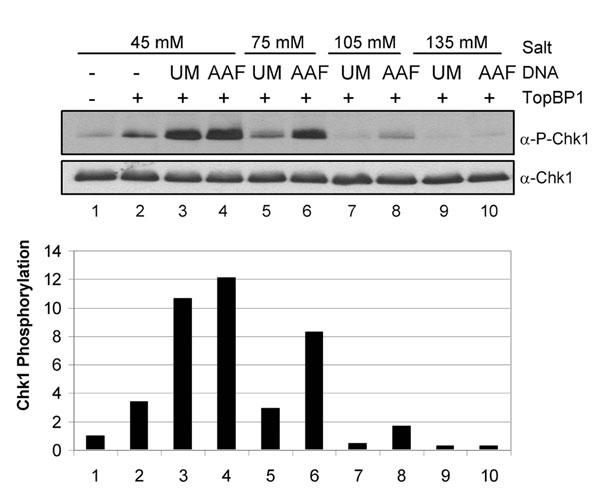
Next, we tested DNA damaged by N-Aco-AAF and UV light. Both of these agents produce bulky base adducts, AAF-guanine and pyrimidine dimers, respectively. Moreover, evidence has been obtained indicating that checkpoint proteins bind directly to these lesions, leading to checkpoint activation (23, 24). Hence, we reasoned that DNA damaged by these agents may specifically activate ATR in the presence of TopBP1. In Fig. 4A, we show the effect of various concentrations of N-Aco-AAF-damaged DNA on the kinase activity of ATR. As is apparent from this figure, damaged DNA stimulates the ATR kinase 20-fold more than the level of TopBP1 alone, and more importantly, damaged DNA stimulates the TopBP1-dependent ATR kinase activity approximately 3- to 4-fold higher than undamaged DNA. We conducted kinetic experiments to analyze the reaction in more detail to determine the initial reaction rates (Fig. 4B). From these experiments, we determined that the initial rate of the ATR kinase reaction was 5 times faster with N-Aco-AAF-damaged DNA than with undamaged DNA. Qualitatively similar but quantitatively less significant effects were observed with UV-damaged DNA as well (SI Fig. 9). Once again, we observed increased specificity of the reaction under higher ionic strength conditions (SI Fig. 10).
Fig. 4.

TopBP1-dependent stimulation of ATR kinase activity by N-Aco-AAF-damaged DNA. (A) (Upper) Kinase assays were performed with 85 mM final salt concentration as described in the legend of Fig. 3, except with 0.62–5 nM unmodified (UM) or N-Aco-AAF-damaged (AAF) pUC19 plasmid DNA. (Lower) The average levels of Chk1 phosphorylation from four independent experiments are quantitated in the graph. (B) (Upper) Time course of Chk1 phosphorylation. Kinase assays as in A were carried out with 0.62 nM DNA for the times indicated. (Lower) The average levels of Chk1 phosphorylation from two independent experiments are quantitated in the graph.
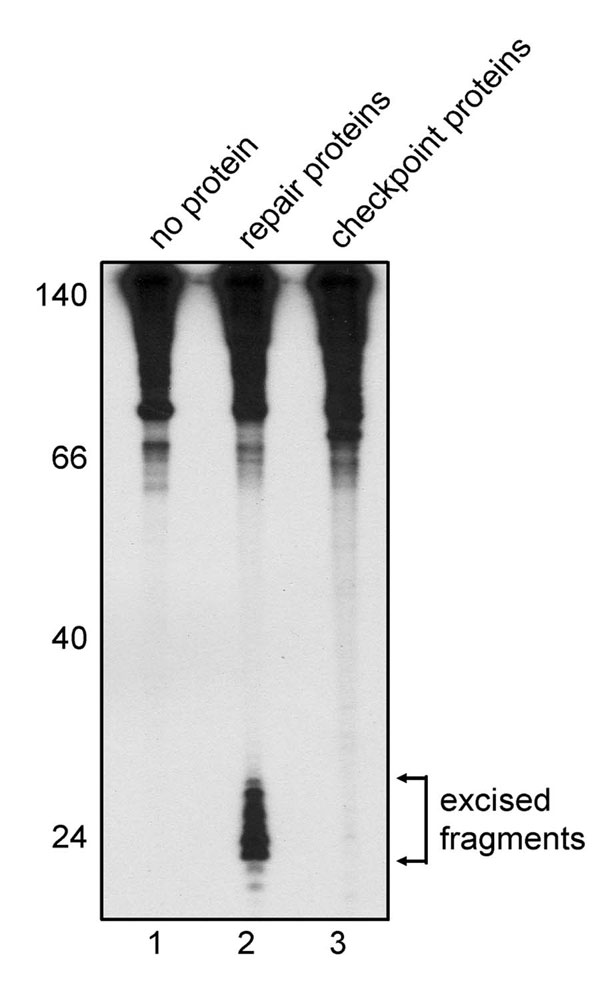
Because several reports indicate that the ≈30-nt gap produced by excision repair constitutes a strong signal for the ATR-mediated checkpoint response (25–27), we considered the possibility that impurities in our ATR, Chk1, or TopBP1 preparations were at a sufficient level to excise the base damage and create excision gaps that would constitute a signal for checkpoint response. To test for this eventuality, we conducted an “excision assay” (28, 29) with a mixture of the proteins used for checkpoint response. No excision was observed (SI Fig. 11), suggesting that the base damage itself is the signal in our in vitro checkpoint system.
Dependence of DNA-Binding Activity of TopBP1 for ATR Activation.
The results presented so far are consistent with the idea that TopBP1 recruits ATR to DNA damage sites and in so doing stimulate its kinase activity. To test this idea, we measured the DNA-binding activity of TopBP1 for undamaged vs. N-Aco-AAF-damaged DNA (Fig. 5A). Full-length TopBP1 bound DNA with relatively high affinity and with ≈5-fold preference for damaged DNA (lanes 5 and 6). Interestingly, the ≈5-fold TopBP1-dependent stimulation of ATR kinase activity by N-Aco-AAF-damaged DNA over undamaged DNA (Fig. 4) correlates with the ≈5-fold higher binding affinity of TopBP1 to damaged DNA.
Fig. 5.

Dependence of DNA-binding activity of TopBP1 for ATR activation. (A) Preferential binding of TopBP1 to N-Aco-AAF-damaged DNA. (Upper) The indicated amounts of GST-TopBP1-His or the GST-TopBP1-(978–1192) fragment on glutathione-Sepharose beads was incubated at 37°C for 10 min with 1 ng of unmodified or N-Aco-AAF-damaged DNA that had been 5′-end-labeled. Bound DNA was eluted and analyzed by agarose gel electrophoresis. Fifty percent of the DNA added into the reaction was loaded in the input lanes. (Lower) Data from three independent experiments are presented as means ± SD in the graph. (B) Contribution of DNA-binding activity of TopBP1 to stimulation of ATR kinase activity by double-stranded DNA. (Upper) Kinase assays were performed as in Fig. 2, except with the TopBP1-(978–1192) fragment compared with full-length TopBP1 under low ionic strength reaction conditions (35 mM total salt concentration). The TopBP1-(978–1192) fragment (15 nM) was used to obtain a level of Chk1 phosphorylation comparable to that observed with 1 nM full-length TopBP1 (compare lanes 3 and 7). (Lower) The average levels of Chk1 phosphorylation from two independent experiments are quantitated in the graph. (C) Requirement of DNA-binding activity of TopBP1 for stimulation of ATR kinase activity by N-Aco-AAF-damaged DNA. (Upper) Kinase assays were performed with 85 mM final salt concentration as in Fig. 4A, except with the TopBP1-(978–1192) fragment (15 nM) compared with full-length TopBP1 (1 nM). For the reactions containing full-length TopBP1, 5 nM unmodified or N-Aco-AAF-damaged DNA was added. (Lower) The average levels of Chk1 phosphorylation from two independent experiments are quantitated in the graph.
To determine whether DNA binding by TopBP1 was required for the DNA-dependent stimulation of ATR kinase activity, we used a C-terminal TopBP1 fragment encompassing amino acids 978-1192, which is known to activate ATR kinase (14) but lacks DNA-binding activity (30). In fact, the amino acids 978-1192 fragment did not bind DNA at moderate enzyme concentrations (Fig. 5A, lanes 7 and 8) and bound only weakly and nonspecifically at 10-fold higher concentrations (lanes 9 and 10). We conducted ATR kinase assays to compare the ability of the full-length and the 978-1192 fragment of TopBP1 to stimulate ATR in the presence or absence of DNA. In low ionic strength ATR kinase reactions, both the full-length and the 978-1192 fragment of TopBP1 had similar basal ATR-stimulatory activities (Fig. 5B, lanes 3 and 7); significantly, however, only in the presence of the full-length TopBP1 was ATR kinase activity further stimulated 3- to 4-fold by DNA (lanes 4 vs. 8). Next, we performed ATR kinase assays with the 978-1192 fragment of TopBP1 at higher ionic strength to compare the stimulatory effect of undamaged vs. N-Aco-AAF-damaged DNA (Fig. 5C). The results reveal that damaged DNA, in agreement with data in Fig. 4, stimulates full-length TopBP1-activated ATR kinase ≈5-fold more than undamaged DNA (lanes 3 and 4). In contrast, no or only marginal stimulation by the 978-1192 fragment of TopBP1 is observed with damaged or undamaged DNA (lanes 6–13), which supports the model that TopBP1 binding to damaged DNA is essential for optimal stimulation of the ATR kinase activity on Chk1.
Discussion
Data from this study suggest that bulky DNA lesions are recognized by TopBP1, which recruits ATR to the damage site and potentiates its kinase activity on the checkpoint signal transduction kinase, Chk1 (Fig. 6). This human in vitro checkpoint system recapitulates a number of key features of the checkpoint response. However, the system in its present form has certain limitations. First, there are extensive data that primer/template structures, in which the single-stranded region is covered by RPA, generated by replication blocks or by nucleotide excision repair are strong signals for checkpoint activation (22). However, those data and our in vitro findings in this paper and in vivo data published previously (24) are not necessarily mutually exclusive because it is quite likely that more than one type of DNA/protein structure may constitute a signal for checkpoint activation. Second, in vivo data indicate that in addition to TopBP1, several other proteins, such as Rad17-RFC, the 9-1-1 complex, Claspin, and Timeless are required for phosphorylation of Chk1 by ATR (3, 22). Indeed, a recent study indicates that the Rad9 subunit of the 9-1-1 complex plays a role in recruitment of TopBP1 to chromatin and thus facilitates ATR activation by this mechanism (31). Clearly, establishing an in vitro checkpoint response dependent on all known checkpoint components, including damaged DNA and checkpoint proteins, is undoubtedly a challenging problem. However, we believe that the in vitro system we describe here constitutes an important framework for incorporation of the other known checkpoint proteins, which may be required for amplification of the checkpoint signal observed under our reaction conditions or may be essential for checkpoint signaling under different and perhaps more stringent reaction conditions, resulting in reconstitution of a bona fide human DNA damage checkpoint-signaling pathway in an entirely defined in vitro system.
Fig. 6.

Model of TopBP1-dependent stimulation of ATR kinase activity by damaged DNA. Data from this study suggest that bulky DNA lesions are recognized by TopBP1, which recruits ATR to the damage site and potentiates its kinase activity on the checkpoint signal transduction kinase, Chk1.
Materials and Methods
Antibodies and Checkpoint Proteins.
The Chk1 P345 antibody was purchased from Cell Signaling Technology (Beverly, MA). ATRIP, ATM, and DNA-dependent PK antibodies were purchased from Novus Biologicals (Littleton, CO). Chk1 and ATR (N-19) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The DNA-dependent PK catalytic subunit (Ab-4) antibody was purchased from NeoMarkers (Fremont, CA). GST-TopBP1-His and the GST-TopBP1-(978–11920 fragment were induced in Escherichia coli BL21 RIPL (Stratagene, La Jolla, CA) at 16°C for 5 h. GST-TopBP1-His was purified with glutathione-Sepharose (GE Healthcare, Piscataway, NJ), and nickel nitrilotriacetate-agarose (Qiagen, Valencia, CA), and GST-TopBP1-(978–1192) fragment was purified with glutathione-Sepharose by standard procedures. His-Chk1 kinase-dead (Chk1-kd) was produced by baculovirus infection of Sf21 insect cells (Invitrogen) and purified with nickel nitrilotriacetate-agarose beads as described (16). The p11-tRPA plasmid was obtained from M. S. Wold (University of Iowa, Iowa City, IA), and RPA was purified as described (32).
Purification of ATR–ATRIP from HeLa Cell-Free Extracts.
Native ATR–ATRIP complex was isolated from 70 liters of HeLa cells. HeLa cell-free extracts [80 ml (8.3 mg/ml)] were prepared as described (33) and applied onto a DE52 column (Whatman, Clifton, NJ). The flow-through was then loaded onto a CM Affi-Gel Blue (Bio-Rad, Hercules, CA) column and eluted with 1 M NaCl, and fractions were collected and analyzed by Western blotting and silver-staining. The ATR-containing fractions were pooled and dialyzed against buffer D (20 mM Hepes, pH 7.9/20% glycerol/0.2 mM EDTA/0.5 mM DTT) containing 100 mM KCl. It should be noted that this column separates RPA from ATR–ATRIP quantitatively (34), and as a consequence our highly purified ATR–ATRIP was free of RPA. The dialyzed fractions were applied onto a 50-ml DEAE-Sepharose column (GE Healthcare), and the flow-through was collected and loaded onto a 20-ml Mono Q column (GE Healthcare) and then eluted with a 0.1–0.5 M KCl gradient. The ATR-containing fractions were pooled and dialyzed against buffer D containing 25 mM KCl. The dialyzed fractions were then loaded onto a 1-ml Mono S column (GE Healthcare) and eluted with a 25–400 mM KCl gradient. The peak of ATR–ATRIP complex eluted at 220 mM KCl. This fraction (0.4 ml, 44 μg) was free of DNA-dependent PK catalytic subunit, and ATM, as determined by Western blotting, contained ≈4 μg of ATR–ATRIP and was stored frozen in buffer D containing 220 mM KCl.
Preparation of DNA Substrates for Kinase Assays.
Single-stranded or double-stranded pIBI25 plasmid was purified by standard procedures. For preparation of deca-primed DNA, ten 30-mers were hybridized to single-stranded pIBI25 DNA at regular intervals. The annealing reaction (100 μl) contained 10 mM Hepes (pH 7.9), 100 mM NaCl, 9 pmol of each oligomer, and 5 pmol of single-stranded pIBI25 DNA. The reaction was incubated at 90°C for 3 min and cooled gradually. For N-Aco-AAF-damaged DNA, pUC19 plasmid was treated with N-Aco-AAF (National Cancer Institute Chemical Carcinogen Reference Standard Repository, Midwest Research Institute, Kansas City, MO) as described previously (35) to introduce ≈20–30 adducts per plasmid. Briefly, pUC19 (50 μg/ml) was treated with 0.3 mM N-Aco-AAF in 2 mM sodium citrate, pH 7.0, at room temperature for 3 h. The reaction was followed by ether extraction and ethanol precipitation to remove the nonreacted excess N-Aco-AAF. For UV irradiation, pUC19 plasmid was irradiated with 3,000 J/m2 UVC from a 254-nm germicidal lamp, as determined with a UVX radiometer. After N-Aco-AAF treatment or UV irradiation, the DNA was analyzed on a 0.8% agarose gel to determine that no additional nicking was introduced by the treatment.
Kinase Assays.
Kinase assay reaction mixtures contained 1 μM microcystin, 15 mM Hepes (pH 7.9), 35 mM KCl, 50 mM NaCl, 3 mM MgCl2, 1 mM ATP, 5 mM creatine phosphate, 8 μg/ml creatine kinase, 0.5 mM DTT, 1% polyethylene glycol, and 5% glycerol in a 10-μl final volume. Where indicated, the kinase assays were carried out with different salt concentrations. Purified ATR (1 nM) was preincubated for 15 min at 30°C with 1 nM GST-TopBP1-His and 0.62–5 nM DNA. After the preincubation, 20 nM Chk1-kd was added to the reaction, the mixture was incubated for 20 min at 30°C, unless otherwise indicated, the reaction was terminated by the addition of SDS/PAGE loading buffer, and the products were separated by SDS/PAGE. Chk1 phosphorylation was detected by immunoblotting using Chk1 phospho-S345 antibody. For kinase assays with RPA, 5 nM DNA was first incubated with 0.24–1.92 μM RPA for 10 min on ice. The reaction was then followed by the procedures described above. For quantification purposes, levels of Chk1 phosphorylation were quantified by using ImageQuant 5.2 software (GE Healthcare) after scanning. The level of Chk1 phosphorylation in the control reaction (without TopBP1 and DNA) was quantitated and set equal to 1. The levels of Chk1 phosphorylation in the other lanes were determined relative to this value.
DNA-Binding Assays.
The procedure was a modification of a previously described method (30). pUC19 plasmid was linearized with BamHI and 5′-end-labeled with [γ-32P]ATP by T4 polynucleotide kinase. The labeled DNA was then mock-treated or treated with N-Aco-AAF as described above. Purified proteins (3–30 pmol) on glutathione beads were incubated at 37°C for 10 min with the DNA (1 ng) in buffer B [10 mM Tris·Cl (pH7.7)/1 mM EDTA/0.5% Nonidet P-40) containing 200 mM NaCl. After the incubation, the beads were washed three times with buffer B, and bound DNA was eluted by incubation with 0.1 μg/μl proteinase K at 37°C for 15 min. The DNA was resolved in a 0.8% agarose gel, visualized by autoradiography, and quantitated by using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA) and ImageQuant 5.2 software.
DNA Excision Assays.
The internally radiolabeled DNA substrate was a 148-bp duplex prepared by ligating a N-Aco-AAF-damaged oligomer with five other partially overlapping oligomers as described previously (29). Human excision repair proteins were purified from various sources as described (29, 32). Substrate DNA (7.5 fmol) was incubated at 30°C for 2 h with purified excision repair proteins [27 nM xeroderma pigmentosum (XP)A/9 nM XPC–HR23B/100 nM RPA/300 ng partially purified TFIIH/4 nM XPF–ERCC1/5 nM XPG] or with purified checkpoint proteins (1 nM ATR–ATRIP/1 nM GST-TopBP1-His/20 nM His-Chk1-kd) in 12.5 μl of reaction buffer containing 32 mM Hepes (pH 7.9), 64 mM KCl, 6.4 mM MgCl2, 5% glycerol, 4 mM ATP, and 200 μg/ml BSA. After the incubation, the DNA was purified, resolved in a denaturing 10% polyacrylamide gel, and visualized by autoradiography.
Supplementary Material
Acknowledgments
We thank J. Reardon for assistance with the ATR–ATRIP purification, excision assays, and critical reading and M. Kemp, J. Hurwitz, and P. Modrich for useful comments on the manuscript. This work was supported by National Institutes of Health Grant GM32833 (to A.S.).
Abbreviations
- N-Aco-AAF
N-acetoxy-2-acetylaminofluorene
- ATRIP
ATR-interacting protein
- kd
kinase-dead
- PIKK
phosphatidylinositol 3-kinase-related protein kinase
- RPA
replication protein A
- TopBP1
topoisomerase II binding protein 1.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0706013104/DC1.
References
- 1.Abraham RT. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 2.Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Annu Rev Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- 3.Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 4.Lee JH, Paull TT. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 5.Lee JH, Paull TT. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 6.Costanzo V, Robertson K, Ying CY, Kim E, Avvedimento E, Gottesman M, Grieco D, Gautier J. Mol Cell. 2000;6:649–659. doi: 10.1016/s1097-2765(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 7.Dupre A, Boyer-Chatenet L, Gautier J. Nat Struct Mol Biol. 2006;13:451–457. doi: 10.1038/nsmb1090. [DOI] [PubMed] [Google Scholar]
- 8.Bakkenist CJ, Kastan MB. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 9.Kumagai A, Guo Z, Emami KH, Wang SX, Dunphy WG. J Cell Biol. 1998;142:1559–1569. doi: 10.1083/jcb.142.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michael WM, Ott R, Fanning E, Newport J. Science. 2000;289:2133–2137. doi: 10.1126/science.289.5487.2133. [DOI] [PubMed] [Google Scholar]
- 11.Guo Z, Kumagai A, Wang SX, Dunphy WG. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. Genes Dev. 2007;21:898–903. doi: 10.1101/gad.1522607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan S, Lindsay HD, Michael WM. J Cell Biol. 2006;173:181–186. doi: 10.1083/jcb.200601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumagai A, Lee J, Yoo HY, Dunphy WG. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto Y, Tsujimura T, Sugino A, Takisawa H. Genes Cells. 2006;11:993–1007. doi: 10.1111/j.1365-2443.2006.00998.x. [DOI] [PubMed] [Google Scholar]
- 16.Yoshioka K, Yoshioka Y, Hsieh P. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke CA, Clarke PR. Biochem J. 2005;388:705–712. doi: 10.1042/BJ20041966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majka J, Niedziela-Majka A, Burgers PM. Mol Cell. 2006;24:891–901. doi: 10.1016/j.molcel.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parrilla-Castellar ER, Karnitz LM. J Biol Chem. 2003;278:45507–45511. doi: 10.1074/jbc.C300418200. [DOI] [PubMed] [Google Scholar]
- 20.Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. Mol Cell Biol. 2007;27:3367–3377. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou L, Elledge SJ. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 22.Zou L. Genes Dev. 2007;21:879–885. doi: 10.1101/gad.1550307. [DOI] [PubMed] [Google Scholar]
- 23.Ünsal-Kaçmaz K, Makhov AM, Griffith JD, Sancar A. Proc Natl Acad Sci USA. 2002;99:6673–6678. doi: 10.1073/pnas.102167799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang G, Sancar A. Mol Cell Biol. 2006;26:39–49. doi: 10.1128/MCB.26.1.39-49.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 26.Marini F, Nardo T, Giannattasio M, Minuzzo M, Stefanini M, Plevani P, Falconi MM. Proc Natl Acad Sci USA. 2006;103:17325–17330. doi: 10.1073/pnas.0605446103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumoto M, Yaginuma K, Igarashi A, Imura M, Hasegawa M, Iwabuchi K, Date T, Mori T, Ishizaki K, Yamashita K, et al. J Cell Sci. 2007;120:1104–1112. doi: 10.1242/jcs.03391. [DOI] [PubMed] [Google Scholar]
- 28.Huang JC, Svoboda DL, Reardon JT, Sancar A. Proc Natl Acad Sci USA. 1992;89:3664–3668. doi: 10.1073/pnas.89.8.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reardon JT, Sancar A. Methods Enzymol. 2006;408:189–213. doi: 10.1016/S0076-6879(06)08012-8. [DOI] [PubMed] [Google Scholar]
- 30.Yamane K, Tsuruo T. Oncogene. 1999;18:5194–5203. doi: 10.1038/sj.onc.1202922. [DOI] [PubMed] [Google Scholar]
- 31.Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henricksen LA, Umbricht CB, Wold MS. J Biol Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- 33.Manley JL, Fire A, Samuels M, Sharp PA. Methods Enzymol. 1983;101:568–582. doi: 10.1016/0076-6879(83)01038-1. [DOI] [PubMed] [Google Scholar]
- 34.Mu D, Hsu DS, Sancar A. J Biol Chem. 1996;271:8285–8294. doi: 10.1074/jbc.271.14.8285. [DOI] [PubMed] [Google Scholar]
- 35.Hess MT, Gunz D, Naegeli H. Nucleic Acids Res. 1996;24:824–828. doi: 10.1093/nar/24.5.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}