

Abstract
We previously showed that in the perfused rat heart, the capacity of n-fatty acids to generate mitochondrial acetyl-CoA decreases as their chain length increases. In the present study, we investigated whether the oxidation of a long-chain fatty acid, oleate, is inhibited by short-chain fatty acids, acetate or propionate (which do and do not generate mitochondrial acetyl-CoA, respectively). We perfused rat hearts with buffer containing 4 mM glucose, 0.2 mM pyruvate, 1 mM lactate, and various concentrations of either (i) [U-13C]acetate, (ii) [U-13C]acetate plus [1-13C]oleate, or (iii) unlabeled propionate plus [1-13C]oleate. Using mass isotopomer analysis, we determined the contributions of the labeled substrates to the acetyl moiety of citrate (a probe of mitochondrial acetyl-CoA) and to malonyl-CoA. We found that acetate, even at low concentration, markedly inhibits the oxidation of [1-13C]oleate in the heart, without change in malonyl-CoA concentration. We also found that propionate, at a concentration higher than 1 mM, decreases (i) the contribution of [1-13C]oleate to mitochondrial acetyl-CoA, and (ii) malonyl-CoA concentration. The inhibition by acetate or propionate of acetyl-CoA production from oleate probably results from a competition for mitochondrial CoA between the CoA-utilizing enzymes.
Introduction
In non-ruminant mammals, the arterial acetate concentration is usually very low (0.05 to 0.15 mM). The main endogenous sources of acetate are (i) gut flora fermentation in the lower digestive tract, (ii) acetone metabolism [1], (iii) decarboxylation of pyruvate by reactive oxygen species [2], and (iv) deacetylation of acetyl-CoA, acetylcholine and acetylated proteins. Streptozotocin-diabetic rats [3] and humans with type II diabetes [4] have elevated blood acetate concentrations. The main exogenous sources of acetate are ethanol oxidation in the liver, and hemodialysis against solutions containing up to 40 mM acetate1. In patients dialyzed against such solutions, the capacity to oxidize acetate can be exceeded, and arterial acetate concentrations can increase up to 11 mM [5]. Adverse effects of acetate include hypoxemia and cardiovascular effects which have been ascribed to uncontrolled AMP degradation to adenosine [6]. In normal human volunteers, ingestion of ethanol results in a major decrease in whole-body lipid oxidation [7], and, in some cases to alcoholic hypoglycemia [8]. It is not clear whether the decrease in fatty acid oxidation results from ethanol per se, or from acetate derived from ethanol oxidation.
In the normal heart, 60–90% of mitochondrial acetyl-CoA derives from long-chain fatty acid oxidation [9,10]. Our previous study demonstrated that in the perfused rat heart, the capacity of n-fatty acids to generate mitochondrial acetyl-CoA decreases as their chain length increases (see Figs 8A and 8B of ref. [11]). Acetate was much more effective at generating mitochondrial acetyl-CoA than oleate. Since acetate can accumulate in body fluids after ethanol ingestion or during hemodialysis, one could hypothesize that acetate could compete with long-chain fatty acids for the generation of mitochondrial acetyl-CoA, the oxidation of which is the main source of energy in the heart [9,12,13].
Randle et al perfused rat hearts with 5.5 mM glucose, 5 mM [14C]acetate, and no long-chain fatty acids. From the production of 14CO2, they concluded that most of mitochondrial acetyl-CoA was derived from acetate [14]. In the same hearts, glucose oxidation was suppressed via inhibition of pyruvate dehydrogenase.
Contradictory conclusions were reported on the interaction between short-chain and long-chain fatty acid metabolism in perfused rat hearts. Bethencourt et al. perfused rat hearts with recirculating buffer containing 0.72 mM [1-14C]palmitate + 0, 0.5 or 5 mM unlabeled acetate. Since the 14CO2 production and the total 14C-lipid content of the hearts were the same in all three groups, they concluded that the oxidation of [1-14C]palmitate and its incorporation into tissue lipids were unaltered by acetate [15].
In contrast, O’Donnell et al. perfused rabbit hearts with recirculating buffer containing 5 mM glucose, 0.5 mM [2,4,6,8,10,12,14,16-13C]palmitate ± 0.1 mM unlabeled butyrate, a four carbon short chain fatty acid, the oxidation of which is not regulated by the carnitine palmitoyltransferase system. Using the NMR-assayed labeling of C4 of glutamate as a proxy for the labeling of mitochondrial acetyl-CoA, they found that palmitate oxidation was completely inhibited by 0.1 mM butyrate [16].
We hypothesized that, in the heart, acetate or propionate competes with long-chain fatty acids, such as oleate, the most abundant long-chain fatty acid in plasma, for the production of mitochondrial acetyl-CoA. We took advantage of mass isotopomer analysis2 which allows measuring the contribution of a 13C-substrate to mitochondrial acetyl-CoA from the labeling of the acetyl moiety of citrate. In addition, this technique allows measuring, in the same experiment, the contribution to mitochondrial acetyl-CoA of two substrates which generate singly-labeled (M1) and doubly-labeled (M2) acetyl-CoA, for example, [1-13C]oleate and [U-13C]acetate, respectively. Also, we wanted to test whether acetate interferes with the regulation of fatty acid oxidation by malonyl-CoA. In one of the control conditions, we used propionate which is not a precursor of mitochondrial acetyl-CoA but which, like acetate, affects the distribution of CoA species.
Previous work from our group has demonstrated that a sizeable fraction of the acetyl moiety of heart malonyl-CoA derives from the partial oxidation of fatty acids in peroxisomes [17]. The present project expands on this topic in the framework of the competition between long-chain fatty acids and acetate for the formation of mitochondrial acetyl-CoA. Because of the wealth of information derived from the mass isotopomer distribution of the acetyl moiety of citrate and of malonyl-CoA, and because of the small amount of heart tissue available, we gave priority to assays of labeling patterns and the tissue content of malonyl-CoA over assays of concentrations of other metabolites.
Experimental procedures
Materials
Chemicals and biochemicals were obtained from Sigma-Aldrich. [1-13C]Oleic acid, sodium [U-13C]acetate, sodium [1-13C]acetate, [U-13C]citric acid, [1,5-13C]citric acid, and [U-13C]malonic acid were purchased from Isotec (Miamisburg, OH). [U-13C]malonyl-CoA was prepared from [U-13C]malonic acid as described previously [11], and purified by high pressure liquid chromatography. ATP-citrate lyase was prepared from the livers of rats that had been starved for 2 days and then re-fed for 3 days with a high glucose diet. The enzyme, isolated from the Bio-Gel column [18] was precipitated with 50% ammonium sulfate, and aliquots of the suspension (2 U/0.1 ml) were kept frozen at -80°C. Before use, the enzyme was dissolved in 1 ml of 250 mM Tris-HCl, pH 8.7 containing 5 mM dithiothreitol.
Heart Perfusion Experiments
Male Sprague-Dawley rats (250–310g) were fed ad libitum for 10–12 days with standard laboratory chow. Hearts from overnight-fasted rats were perfused in the Langendorff mode (12 ml/min) with non-recirculating bicarbonate buffer containing 3% dialyzed bovine serum albumin (Intergen, fatty acid-free), 50 μM L-carnitine, 8 nM insulin, 4 mM glucose, 1 mM lactate and 0.2 mM pyruvate. After 15 min equilibration, the following protocols were conducted for additional 40 min before quick-freezing the hearts; in protocol 1, 2 and 3, 0–2 mM [U-13C]acetate (low range of plasma (acetate) in hemodialyzed patients), and either 0.4 mM [1-13C]oleate (n = 5, physiological range of long-chain fatty acid concentration), or 0.8 mM [1-13C]oleate (n = 3, high range of long-chain fatty acid concentration) were added to the perfusate. In protocol 4 and 5, 0.4 mM [1-13C]oleate, and either 0–2 mM [U-13C]acetate or 0 – 2 mM unlabeled propionate were infused into the perfusate respectively. Lastly, in protocol 6, 0.8 mM [1-13C]oleate and 0–2 mM [U-13C]acetate was infused into the perfusate.
Analytical Procedures
The 13C-labeling of malonyl-CoA was assayed as described previously [17], except that (i) we isolated the short-chain CoA esters on an Oasis cartridge (Waters), and (ii) malonate was assayed as the bis-TBDMS derivative (monitoring m/z 333–336). For the assay of the malonyl-CoA concentration, other samples of frozen tissue were spiked with an internal standard of [U-13C]malonyl-CoA before extraction. The 13C-labeling of the acetyl moiety of citrate (a probe of mitochondrial acetyl-CoA) was assayed by cleaving citrate with ATP-citrate lyase isolated from rat liver [18] and analyzing acetyl-CoA by ion trap liquid chromatography-mass spectrometry (LC-MS) [19]. The assay was set up using standards of [1,5-13C]citrate and [U-13C]citrate which generate M1 and M2 acetyl-CoA, respectively. In the calculations of molar percent enrichments (MPE) all raw data were corrected for natural abundance of 13C.
Data Presentations and Statistics
Mass isotopomers of metabolites containing 1 to n 13C atoms are identified as Mi with i = 1, 2,... n. The molar percent enrichment (MPE) of individual 13C-labeled mass isotopomers (Mi) was calculated as:
where AM and AMi represent the peak areas from ion chromatograms corrected for natural abundance, corresponding to unlabeled (M) and 13C-labeled (Mi) mass isotopomers, respectively.
We present data from about 40 heart perfusion experiments. For each of the six experimental conditions chosen, we ran 5–8 perfusions in the presence of unlabeled or selected 13C-labeled substrate(s) with the concentration parameters being allowed to vary. Data shown in figures are the 13C-enrichments and the malonyl-CoA concentration assayed in the heart. The control data of protocols 2 and 3 are presented as means ± S.E. The data of the other 4 protocols are means of duplicate GC-MS or LC-MS injections, respectively, which differed by < 2%.
Results
In hearts perfused with 0–2 mM [U-13C]acetate, in the absence of long-chain fatty acids, the M2 enrichment of the acetyl moiety of citrate increased to about 90% (Fig 1A). This enrichment represents the contribution of [U-13C]acetate to mitochondrial acetyl-CoA. In the same hearts, the M2 enrichment of malonyl-CoA increased to 30–40% (Fig 2A). The concentration of malonyl-CoA remained between 4.4 and 6.3 nmol g.w.w−1. at [U-13C]acetate concentrations 0–0.6 mM, and increased to about 7.5 nmol g.w.w−1. at 1 and 2 mM [U-13C]acetate (Fig 3, solid squares).
Fig 1.
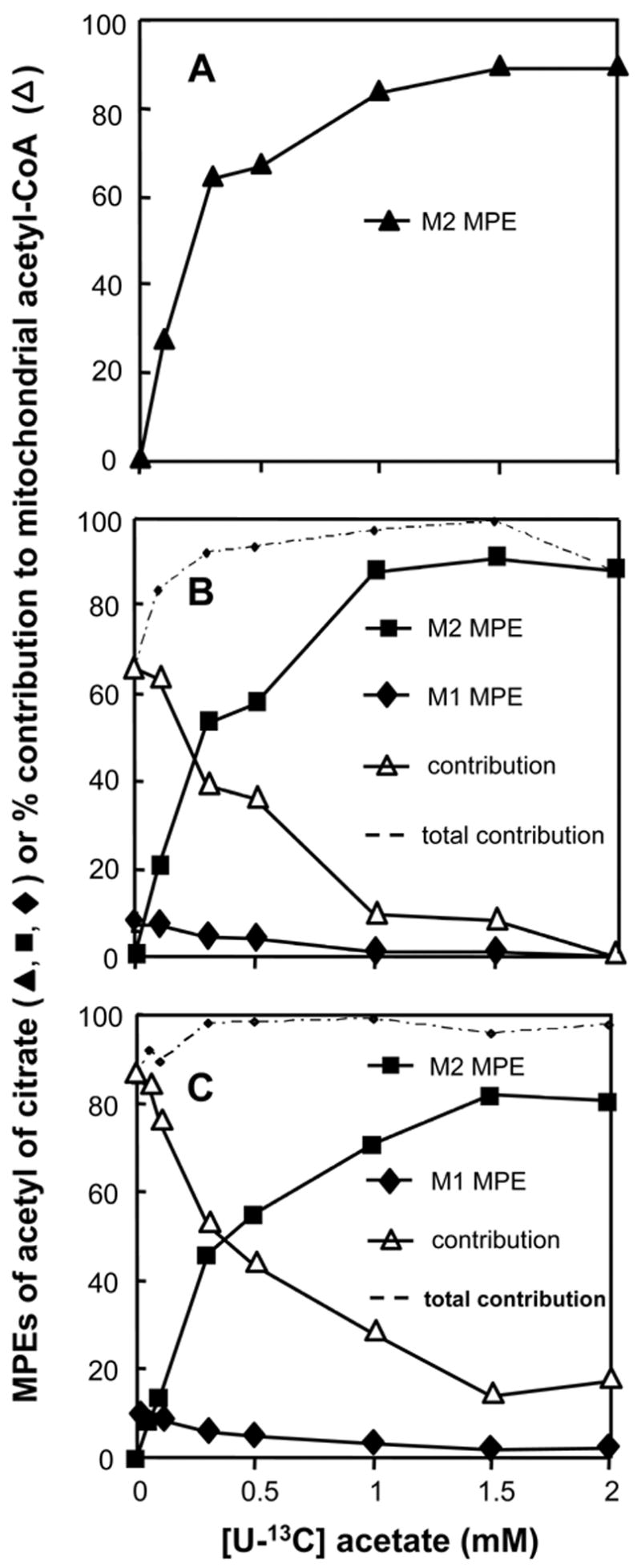
Profiles of the M1 and M2 molar percent enrichments (MPE) of the acetyl moiety of citrate in hearts perfused with increasing concentrations of [U-13C]acetate, and either no [1-13C]oleate (panel A), 0.4 mM [1-13C]oleate (panel B), or 0.8 mM [1-13C]oleate (panel C). The M2 MPE derived from [U-13C]acetate also represent the percent contribution of [U-13C]acetate to mitochondrial acetyl-CoA. The percent contribution of [1-13C]oleate to mitochondrial acetyl-CoA (open triangles in panels B and C) was calculated as 9 times the M1 enrichment of the acetyl moiety of citrate (solid diamonds). The total contribution of [U-13C]acetate and [1-13C]oleate to mitochondrial acetyl-CoA is shown as dotted lines in panels B and C.
Fig 2.
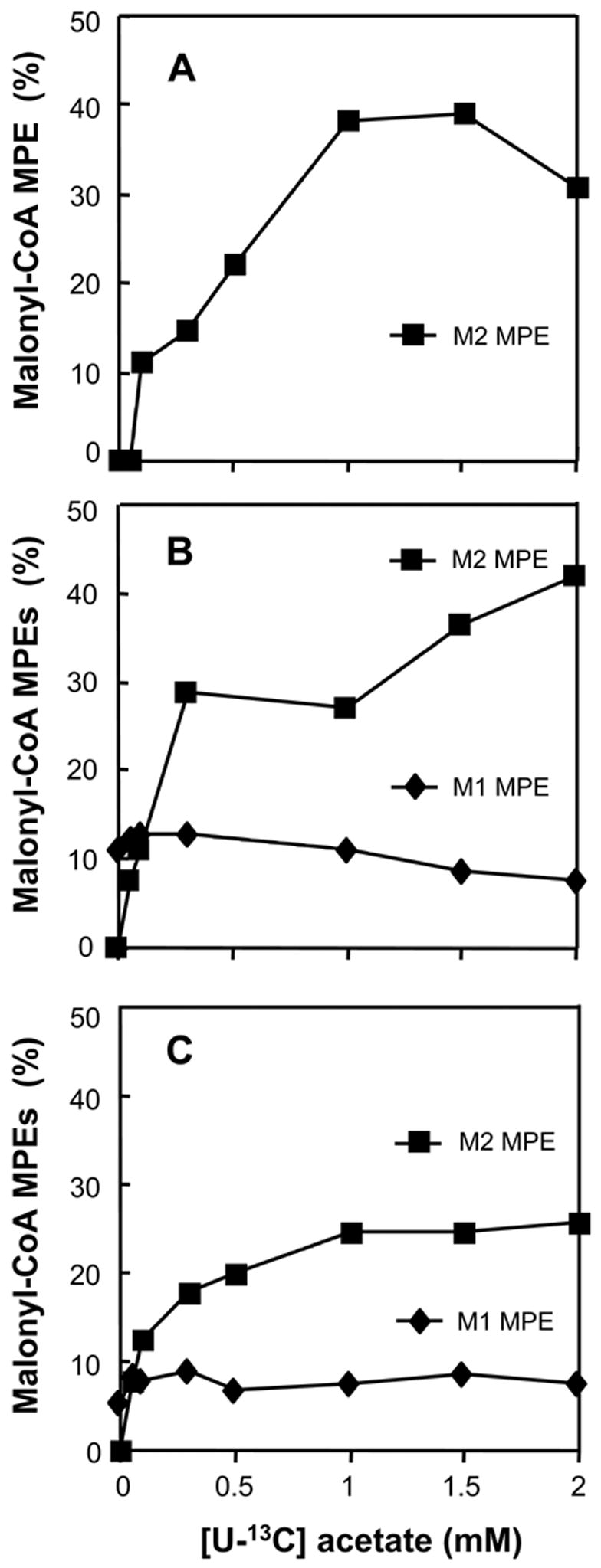
Profiles of the M1 and M2 MPE of malonyl-CoA in hearts perfused with increasing concentrations of [U-13C]acetate (M2), and either no [1-13C]oleate (panel A), 0.4 mM [1-13C]oleate (panel B, M1), or 0.8 mM [1-13C]oleate (panel C, M1).
Fig 3.
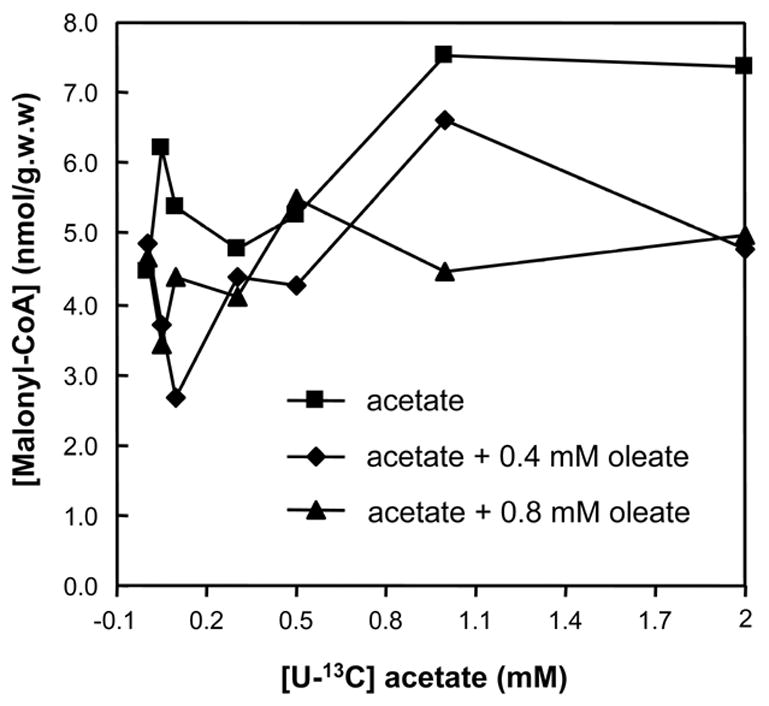
The malonyl-CoA concentration of hearts perfused with (i) [U-13C]acetate (0-2mM), (ii) [U-13C]acetate (0-2mM) + 0.4mM [1-13C]oleate, or (iii) [U-13C]acetate (0–2mM) + 0.4mM [1-13C]oleate.
In hearts perfused with a constant 0.4 mM [1-13C]oleate and 0-2 mM [U-13C]acetate, the M2 enrichment of the acetyl moiety of citrate (derived from [U-13C]acetate) increased to about 90% as in the absence of [1-13C]oleate (compare Figs 1B and 1A). At 0 mM [U-13C]acetate, the M1 enrichment of the acetyl moiety of citrate (derived from [1-13C]oleate) was 7.3 ± 0.64 % (S.E., n = 5, Fig 1B, solid diamonds). As the [U-13C]acetate concentration was increased to 2 mM, the M1 enrichment of the acetyl moiety of citrate decreased to zero. Since only one of the acetyl moieties of [1-13C]oleate was labeled, the contribution of [1-13C]oleate to mitochondrial acetyl-CoA was calculated by multiplying the M1 enrichment of the acetyl moiety of citrate by 9. Thus, the contribution of 0.4 mM [1-13C]oleate to mitochondrial acetyl-CoA decreased from 66 ± 13 % (S.E., n = 5) to zero as the [U-13C]acetate concentration was increased to 2 mM (Fig 1B, open triangles). Fig 1B also shows that the total contribution of acetate and oleate to mitochondrial acetyl-CoA (dotted line) ranged from 90 to 100% at acetate concentrations from 0.2 to 2 mM. Thus, the contribution of other acetyl-CoA sources (glucose, lactate, pyruvate from perfusate + endogenous sources) was very small.
In the same hearts, the M2 enrichment of malonyl-CoA from [U-13C]acetate increased to 42 % (Fig 2B, solid squares). In parallel, the M1 MPE of malonyl-CoA from [1-13C]oleate decreased from 11 ± 0.9 % (S.E., n = 5) to 7.7 % (Fig 2B, solid diamonds). The malonyl-CoA concentration did not follow any clear pattern, ranging between 2.5 to 6.7 nmol g.w.w−1. (Fig 3, solid diamonds).
In hearts perfused with a constant 0.8 mM [1-13C]oleate and 0–2 mM [U-13C]acetate (Figs 1C and 2C) the data were similar as those obtained with 0.4 mM [1-13C]oleate and 0–2 mM [U-13C]acetate (Figs 1B and 2B), with the following exceptions. First, the contribution of [U-13C]acetate to mitochondrial acetyl-CoA increased to 80% (Fig 1C, solid squares). Second, the contribution of [1-13C]oleate to mitochondrial acetyl-CoA decreased from 90 ± 0.94 % (S.E., n = 3) to 17% (Fig 1C, open triangles). Third, the total contribution of acetate and oleate to mitochondrial acetyl-CoA was greater than 95% at acetate concentrations greater than 0.2 mM. The M2 and M1 enrichments of malonyl-CoA were somewhat lower than in the presence of 0.4 mM [1-13C]oleate (compare Figs 2C and 2B). Again, the malonyl-CoA concentration did not follow a clear pattern (Fig 3, solid triangles).
In the above experiments, both [1-13C]oleate and [U-13C]acetate contributed to mitochondrial acetyl-CoA. To test whether a fatty acid that does not generate acetyl-CoA can influence the oxidation of oleate, we perfused a group of hearts with a constant 0.4 mM [1-13C]oleate and 0-2 mM unlabeled propionate. At 0 mM propionate, the M1 enrichment of the acetyl moiety of citrate (derived from [1-13C]oleate) was 7.3 ± 0.64% (S.E., n = 5, Fig 4A, solid diamonds). When the propionate concentration was increased to 2 mM, the M1 enrichment of the acetyl moiety of citrate decreased to 3.9%. Multiplying these percentages by 9 yields the contribution of 0.4 mM [1-13C]oleate to mitochondrial acetyl-CoA which decreased from 66 ± 13% (S.E., n = 5) to 35% as the propionate concentration was increased to 2 mM (Fig 4A, solid squares). In the same hearts, the M1 enrichment of malonyl-CoA remained between 8 and 11% (Fig 4B, solid triangles). The malonyl-CoA concentration decreased from 5 ± 0.5 nmol g.w.w−1 (S.E., n = 5) to 2.8 nmol g.w.w−1. (Fig 4B, solid diamonds).
Fig 4.
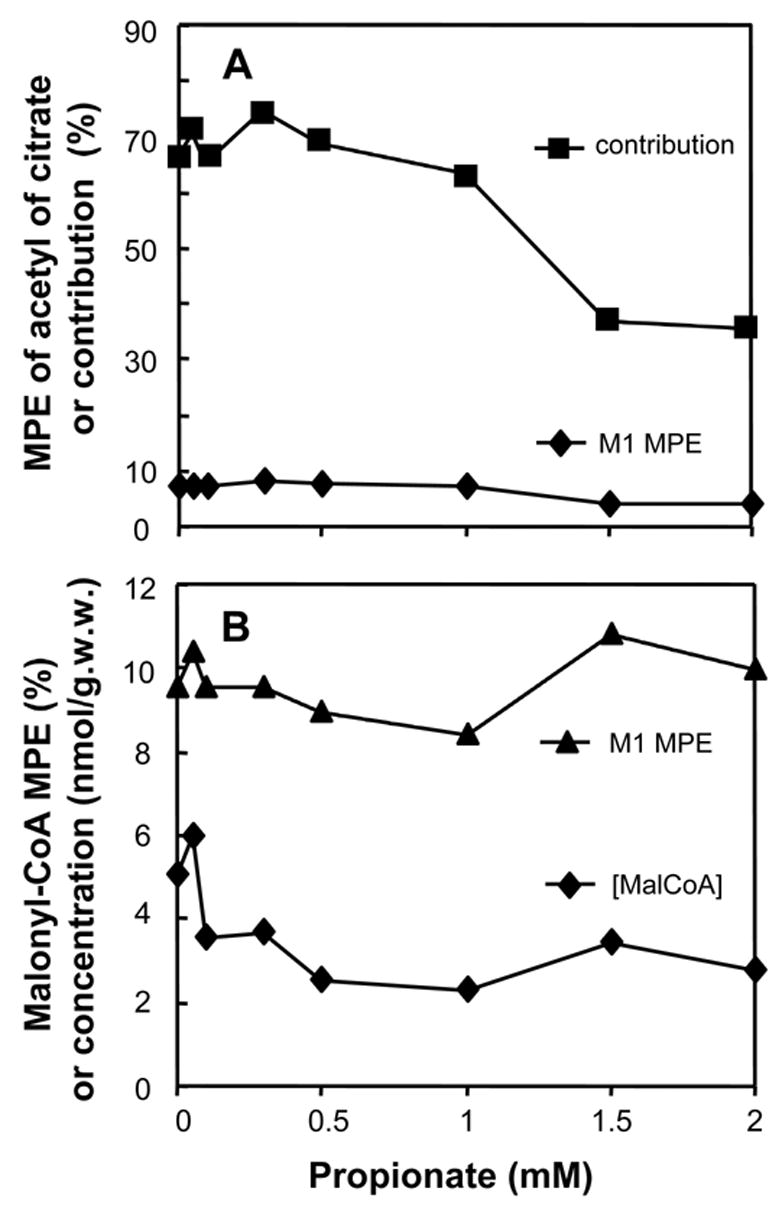
Data from hearts perfused with 0.4 mM [1-13C]oleate and increasing concentrations of unlabeled propionate. Panel A: M1 enrichment of the acetyl moiety of citrate (solid diamonds), and contribution of [1-13C]oleate to mitochondrial acetyl-CoA (solid squares)calculated as 9 times the M1 enrichment of the acetyl moiety of citrate (solid diamonds). Panel B: M1 MPE and the concentration of malonyl-CoA
Discussion
Perfusions with [U-13C]acetate
In hearts perfused with [U-13C]acetate, and no oleate, the contribution of [U-13C]acetate to mitochondrial acetyl-CoA reached 90% at 2 mM substrate concentration (Fig 1A). This, in spite of the presence of other acetyl-CoA generating substrates in the perfusate (glucose, lactate, pyruvate) (Fig 1A). Thus, acetate was the main source of energy in the heart. Although, in vivo, the heart of the rat (a non-ruminant mammal) is not in contact with substantial concentrations of acetate, the activity of mitochondrial acetyl-CoA synthetase 2 (heart form, AceCS2) is high (about 1 U/g) [20], with a fairly low Km for acetate of 0.06 mM [21]. Thus, the heart can rapidly switch to acetate as an energy source.
Perfusions with [U-13C]acetate and [1-13C]oleate
In the absence of acetate, [1-13C]oleate, 0.4 or 0.8 mM, contributed 65 or 85% of mitochondrial acetyl-CoA, respectively (left-hand side of Figs 1B and 1C). We chose oleate concentrations of 0.4 and 0.8 mM oleate to simulate the range of physiological conditions. With increasing concentrations of [U-13C]acetate, the contribution of [1-13C]oleate to mitochondrial acetyl-CoA decreased to low levels (Figs 1B and 1C, open triangles). Establishing that in spite of the presence of abundant [1-13C]oleate, [U-13C]acetate still contributed most of mitochondrial acetyl-CoA. Thus, low concentrations of acetate strongly inhibit the oxidation of oleate. Our findings differ from those of Bethencourt et al who reported that 14CO2 production from 0.72 mM [1-14C]palmitate by the perfused rat heart was not affected by 0.5 or 5 mM acetate [15]. Bethencourt et al acknowledged the surprising character of their finding, for which we cannot provide an explanation.
While the M2 enrichment of malonyl-CoA (derived from [U-13C]acetate) was about one-half that of the acetyl moiety of citrate (Fig 2B), its M1 enrichment (derived from [1-13C]oleate) was, in almost all cases, greater than that of the acetyl moiety of citrate. This higher M1 labeling of malonyl-CoA derives from partial peroxisomal beta-oxidation of [1-13C]oleate [11,17], which generates highly labeled [1-13C]acetyl-CoA. The latter was transferred to the cytosol, and used for malonyl-CoA synthesis. The transfer of peroxisomal acetyl-CoA to the cytosol may involve (i) carnitine acetyltransferase (although this enzyme was reported absent in the extra-mitochondrial space of the heart [22]), and/or (ii) direct transfer through the peroxisomal membrane [23,24]. Mitochondrial acetyl-CoA also contributes to malonyl-CoA synthesis, after transfer to the cytosol via citrate. Highly labeled acetyl-CoA derived from peroxisomes mixes with low-labeled acetyl-CoA transferred from mitochondria in the cytosol. This combined acetyl-CoA pool, more labeled than the acetyl moiety of citrate, provides the substrate pool for the synthesis of malonyl-CoA.
Perfusions with propionate and [1-13C]oleate
To test whether a fatty acid that does not generate acetyl-CoA can influence the oxidation of oleate, we perfused hearts with a constant 0.4 mM [1-13C]oleate and 0–2 mM unlabeled propionate. In the absence of propionate, [1-13C]oleate contributed about 66% of mitochondrial acetyl-CoA (Fig 4A). When propionate was added, the M1 enrichment of mitochondrial acetyl-CoA decreased from 7.5% to 4% (Fig 4A). This corresponds to a decrease of the contribution of [1-13C]oleate to mitochondrial acetyl-CoA from 66 to 35%. This inhibition results probably from the decreased availability of mitochondrial CoA which is partly trapped as propionyl-CoA. However, the inhibition of oleate oxidation is less pronounced with propionate than with acetate, although both are activated by heart AceCS2 [25]. A likely explanation of this difference is the lower affinity of AceCS2 for propionate than for acetate, i.e., Km of 4.1 mM vs. 0.06 mM [21]. Similar differences in affinity for propionate and acetate were also found in guinea-pig heart mitochondria [25]. An additional explanation may involve the inhibition of the 3-ketoacyl-CoA thiolase involved in oleate oxidation by acetyl-CoA [26] formed from acetate.
In spite of the decrease in the M1 enrichment of mitochondrial acetyl-CoA, the M1 enrichment of malonyl-CoA remained between 9 and 11%, i.e., higher than the enrichment of mitochondrial acetyl-CoA. This confirms that part of the M1 acetyl-CoA used for malonyl-CoA synthesis derived from the partial peroxisomal oxidation of [1-13C]oleate [17]. Also, the constancy of the M1 enrichment of malonyl-CoA, while the enrichment of mitochondrial acetyl-CoA decreased, shows that the contribution of peroxisomal oxidation of [1-13C]oleate to cytosolic acetyl-CoA increased while the contribution of [1-13C]oleate to mitochondrial acetyl-CoA decreased.
Mechanisms involved in the inhibition of oleate oxidation by acetate or propionate
Our previous study showed that the capacity of a single fatty acid to contribute to mitochondrial acetyl-CoA increases as its chain length decreases [11]. The present study showed that the competition between a short-chain and a long-chain fatty acid for the production of mitochondrial acetyl-CoA favors the short-chain compound. Also, a short-chain fatty acid which does not generate acetyl-CoA (propionate at concentration > 1 mM) inhibits the production of mitochondrial acetyl-CoA from a long-chain fatty acid (oleate).
First, consider the inhibition of oleate oxidation by propionate concentrations greater than 1 mM
In another study3, we found that in rat hearts perfused with 0 to 1 mM propionate, the total concentration of propionyl-CoA + methylmalonyl-CoA + succinyl-CoA increased from 38 to 70 nmol g.w.w.−1. Although the concentration of free CoA was not measured, this suggested substantial trapping of CoA in the three metabolites of propionate. This is supported by reports that (i) the total concentration of free CoA decreased four- and five-fold when 1 or 2 mM propionate was added to the perfusate of rat hearts [27,28]. Thus, in the present study, propionate probably decreased the amount of free CoA in the mitochondrial matrix available to the malonyl-CoA insensitive carnitine palmitoyltransferase II (CPT II) and 3-ketoacyl-CoA thiolase reactions involved in the mitochondrial phase of oleate oxidation as illustrated in Fig 5. The decreased availability of free CoA with increasing propionate concentrations would explain the lower contribution of oleate to mitochondrial acetyl-CoA at propionate concentrations greater than 1 mM (Fig 4A).
Fig 5.
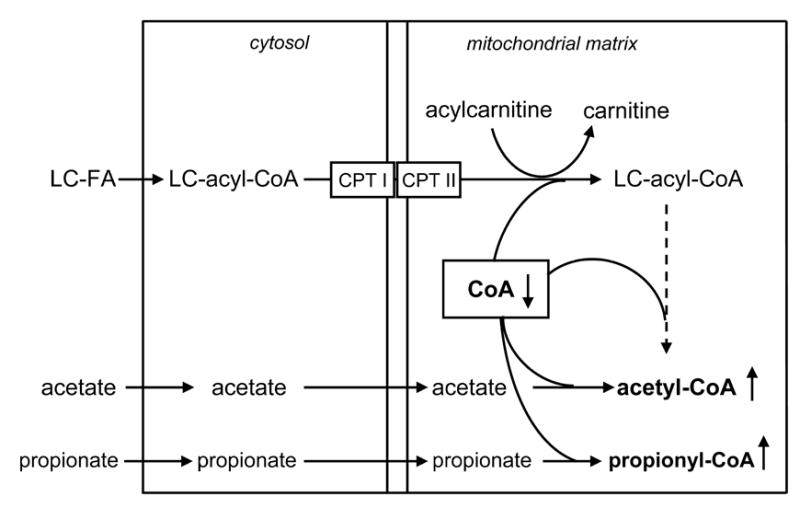
Hypothetical competition for mitochondrial CoA between acetate or propionate activation and the β-oxidation of long-chain acyl-CoAs derived from long-chain fatty acids.
The inhibition of oleate oxidation by propionate did not result from an increase in malonyl-CoA concentration which, actually, decreased as the concentration of propionate increased (Fig 4B, solid diamonds). Liedtke et al reported that infusion of 2 mM propionate into a branch of the coronary artery of pigs did not affect the heart concentrations of ATP, ADP, or AMP [29]. Therefore, it is unlikely that, in our study, the activity of acetyl-CoA carboxylase was affected via modulation of AMP-activated protein kinase. A more likely explanation is that the acetyl-CoA carboxylase flux was decreased by a lowering of the cytosolic concentration of acetyl-CoA. In support of this explanation, two studies conducted in perfused rat hearts [27] and in live pig hearts [29] reported that 1 or 2 mM propionate decreased the myocardial concentration of acetyl-CoA by half. Since 90% of heart acetyl-CoA is mitochondrial [30], it is likely that in our study, propionate decreased the concentration of cytosolic acetyl-CoA. The decrease in long-chain fatty acid oxidation and malonyl-CoA concentration by propionate shows that, even in the heart, the rate of long-chain fatty acid oxidation is not absolutely linked to malonyl-CoA concentration.
Although propionate decreased the labeling of mitochondrial acetyl-CoA from [1-13C]oleate (Fig 4A), it did not affect the M1 enrichment of malonyl-CoA (Fig 4B, solid triangles). The most likely explanation of this finding is that the contribution of peroxisomal oxidation of [1-13C]oleate to [1-13C]acetyl-CoA used for malonyl-CoA synthesis increased as the transfer of mitochondrial acetyl-CoA to the cytosol decreased. We previously showed [17], that the peroxisomal oxidation of fatty acids contributes a substantial fraction to the cytosolic acetyl-CoA used for malonyl-CoA synthesis.
Second, consider the inhibition of oleate oxidation by acetate
This inhibition may result from a competition for free mitochondrial CoA between AceCS, CPT II and 3-ketoacyl-CoA thiolases. The concentration of free mitochondrial CoA in the heart (40 to 60 μM) is lower than the Km for CoA of AceCS (570 μM [31]), CPT II (121 μM[32]), but higher than the Km of general 3-ketoacyl-CoA thiolase (18 μM [32]). Therefore, the competition for free CoA between AceCS and CPT II is set by the relative activities of the two enzymes. The activity of rat heart AceCS2 is about 100 nmol min−1 mg protein−1 [31]. The total CPT (CPT I + CPT II) activity in rat heart is about 8.9 nmol min−1 mg protein−1 [33]. So, the activity of AceCS is much higher than that of CPT II in rat heart. Thus, in the presence of acetate, AceCS outcompetes CPT II for free CoA, and hinders the conversion of oleoylcarnitine to oleoyl-CoA, and thus oleate oxidation.
The trapping of mitochondrial CoA into esters plays a role in a number of physiological and pathological processes, such as the oxidation of acetoacetate in isolated heart mitochondria [34] and perfused hearts [35], propionate metabolism in the heart [27,28], inborn errors of long-chain fatty acid oxidation [36], valproate intoxication [37,38], etc. Stanley et al. showed that a moderate elevation of β-hydroxybutyrate suppressed palmitate oxidation in pig hearts [39], without changes in concentrations of malonyl-CoA, acetyl-CoA, or free CoA. The authors hypothesized that the elevation in β-hydroxybutyrate may reduce the delivery of fatty acyl-CoA to the mitochondria. It is also possible that the concentration of acetoacetate formed via a low β–hydroxybutyrate concentration (0.8 mM) is not sufficient to drain the pool of mitochondrial CoA.
The inhibition of oleate oxidation by acetate probably does not involve the inhibition of CPT I by malonyl-CoA, since the addition of acetate did not increase the malonyl-CoA concentration in the hearts (Fig 3). Indeed, the inhibition of oleate oxidation by butyrate is not due to the decreased entry of long-chain acyl-CoA into the mitochondria [40].
The data on the labeling of malonyl-CoA yield information on the old question as to whether an AceCS is present in heart cytosol. Barth et al and Ballard et al reported the presence of cytosolic AceCS [20,41,42], while Scholte et al claimed that acetyl-CoA synthetase is only localized in the mitochondria [43]. Recently, Fujino et al reported that most of heart acetyl-CoA synthetase activity is mitochondrial, with a very small fraction of the activity in the cytosol [21]. If cytosolic activation of acetate occurred in the heart, one would expect malonyl-CoA to be more labeled than the acetyl moiety of citrate. This is because a cytosolic acetyl-CoA synthetase would convert plasma [U-13C]acetate to [U-13C]acetyl-CoA used for malonyl-CoA synthesis. However, our data argue against a cytosolic activation of acetate. In the presence of [U-13C]acetate (this study, Figs 1A and 2A) or [U-13C]glucose (Table I of Ref. [17]), the enrichment of malonyl-CoA was about 40% that of the acetyl moiety of citrate (Figs 1A and 2A). Labeling of extramitochondrial acetyl-CoA from [U-13C]glucose can only occur via export of mitochondrial acetyl-CoA. Thus, the similar dilutions (malonyl-CoA)/(acetyl of citrate) with [U-13C]acetate and [U-13C]glucose show that no substantial activation of acetate occurs in heart cytosol, in agreement with the report from Fujino et al.
In summary, our data demonstrate that acetate strongly inhibits long-chain fatty acid oxidation in the heart. We therefore hypothesize that acetate may be a useful adjunctive therapy in myocardial reperfusion which is characterized by an abnormally high contribution of long-chain fatty acid to energy metabolism, coupled with the excessive production of reactive oxygen species [44]. Also, acetate might be useful for the treatment of acute decompensation of patients with long-chain fatty acid oxidation disorders [36].
Acknowledgments
This work was supported by grant from the NIH (5R01DK069752), and the Cleveland Mt. Sinai Health Care Foundation.
Abbreviations
- AceCS
acetyl-CoA synthetase
- CPT
carnitine palmitoyltransferase
- GC-MS
gas chromatography-mass spectrometry
- LC-MS
liquid chromatography-mass spectrometry
- MPE
molar percent enrichment
Footnotes
In most US centers, the use of high-acetate dialysis solution is being phased out in favor of low acetate solution (6–7 mM).
Mass isotopomers are defined as Mn, where n is the number of heavy atoms in the molecule.
Kasumov, T., Cendrowski, A.V., David, F., Jobbins, K.A., Anderson, V.E., and Brunengraber, H. “High efficiency of anaplerosis from propionate in the perfused rat heart”. Biochemical Journal (submitted).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Gavino VC, Somma J, Philbert L, David F, Garneau M, Belair J, et al. Production of acetone and conversion of acetone to acetate in the perfused rat liver. J Biol Chem. 1987;262:6735–40. [PubMed] [Google Scholar]
- 2.Constantopoulos G, Barranger JA. Nonenzymatic decarboxylation of pyruvate. Anal Biochem. 1984;139:353–8. doi: 10.1016/0003-2697(84)90016-2. [DOI] [PubMed] [Google Scholar]
- 3.Buckley BM, Williamson DH. Origins of blood acetate in the rat. Biochem J. 1977;166:539–45. doi: 10.1042/bj1660539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akanji AO, Humphreys S, Thursfield V, Hockaday TD. The relationship of plasma acetate with glucose and other blood intermediary metabolites in non-diabetic and diabetic subjects. Clin Chim Acta. 1989;185:25–34. doi: 10.1016/0009-8981(89)90127-7. [DOI] [PubMed] [Google Scholar]
- 5.Tolchin N, Roberts JL, Hayashi J, Lewis EJ. Metabolic consequences of high mass-transfer hemodialysis. Kidney Int. 1977;11:366–78. doi: 10.1038/ki.1977.54. [DOI] [PubMed] [Google Scholar]
- 6.Blaise G, Noel J, Vinay P, Cardoso M, Vinet B, Boulanger Y, et al. Metabolic effects of acetate on the heart. Clin Invest Med. 1989;12:254–61. [PubMed] [Google Scholar]
- 7.Siler SQ, Neese RA, Hellerstein MK. De novo lipogenesis, lipid kinetics, and whole-body lipid balances in humans after acute alcohol consumption. Am J Clin Nutr. 1999;70:928–36. doi: 10.1093/ajcn/70.5.928. [DOI] [PubMed] [Google Scholar]
- 8.Freinkel N, Singer DL, Arky RA, Bleicher SJ, Anderson JB, Silbert CK. Alcohol hypoglycemia. I. carbohydrate metabolism of patients with clinical alcohol hypoglycemia and the experimental reproduction of the syndrome with pure ethanol. J Clin Invest. 1963;42:1112–33. doi: 10.1172/JCI104797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodwin GW, Taegtmeyer H. Regulation of fatty acid oxidation of the heart by MCD and ACC during contractile stimulation. Am J Physiol. 1999;277:E772–E777. doi: 10.1152/ajpendo.1999.277.4.E772. [DOI] [PubMed] [Google Scholar]
- 10.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 11.Bian F, Kasumov T, Thomas KR, Jobbins KA, David F, Minkler PE, et al. Peroxisomal and mitochondrial oxidation of fatty acids in the heart, assessed from the 13C labeling of malonyl-CoA and the acetyl moiety of citrate. J Biol Chem. 2005;280:9265–71. doi: 10.1074/jbc.M412850200. [DOI] [PubMed] [Google Scholar]
- 12.Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev. 2002;7:115–30. doi: 10.1023/a:1015320423577. [DOI] [PubMed] [Google Scholar]
- 13.Taegtmeyer H, Hems R, Krebs HA. Utilization of energy-providing substrates in the isolated working rat heart. Biochem J. 1980;186:701–11. doi: 10.1042/bj1860701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randle PJ, England PJ, Denton RM. Control of the tricarboxylate cycle and its interactions with glycolysis during acetate utilization in rat heart. Biochem J. 1970;117:677–95. doi: 10.1042/bj1170677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bethencourt AV, Matos OE, Shipp JC. Acetate-1-C14 metabolism and the effect of acetate on glucose and palmitate metabolism in the perfused rat heart. Metabolism. 1966;15:847–55. doi: 10.1016/0026-0495(66)90177-6. [DOI] [PubMed] [Google Scholar]
- 16.O’donnell JM, Alpert NM, White LT, Lewandowski ED. Coupling of mitochondrial fatty acid uptake to oxidative flux in the intact heart. Biophys J. 2002;82:11–8. doi: 10.1016/S0006-3495(02)75369-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reszko AE, Kasumov T, David F, Jobbins KA, Thomas KR, Hoppel CL, et al. Peroxisomal fatty acid oxidation is a substantial source of the acetyl moiety of malonyl-CoA in rat heart. J Biol Chem. 2004;279:19574–9. doi: 10.1074/jbc.M400162200. [DOI] [PubMed] [Google Scholar]
- 18.Linn TC, Srere PA. Identification of ATP citrate lyase as a phosphoprotein. J Biol Chem. 1979;254:1691–8. [PubMed] [Google Scholar]
- 19.Reszko AE, Kasumov T, Comte B, Pierce BA, David F, Bederman IR, et al. Assay of the concentration and 13C-isotopic enrichment of malonyl-coenzyme A by gas chromatography-mass spectrometry. Anal Biochem. 2001;298:69–75. doi: 10.1006/abio.2001.5349. [DOI] [PubMed] [Google Scholar]
- 20.Ballard FJ. Supply and utilization of acetate in mammals. Am J Clin Nutr. 1972;25:773–9. doi: 10.1093/ajcn/25.8.773. [DOI] [PubMed] [Google Scholar]
- 21.Fujino T, Kondo J, Ishikawa M, Morikawa K, Yamamoto TT. Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J Biol Chem. 2001;276:11420–6. doi: 10.1074/jbc.M008782200. [DOI] [PubMed] [Google Scholar]
- 22.Abbas AS, Wu G, Schulz H. Carnitine acetyltransferase is not a cytosolic enzyme in rat heart and therefore cannot function in the energy-linked regulation of cardiac fatty acid oxidation. J Mol Cell Cardiol. 1998;30:1305–9. doi: 10.1006/jmcc.1998.0693. [DOI] [PubMed] [Google Scholar]
- 23.Ramsay RR, Zammit VA. Carnitine acyltransferases and their influence on CoA pools in health and disease. Mol Aspects Med. 2004;25:475–93. doi: 10.1016/j.mam.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Verleur N, Hettema EH, Van Roermund CW, Tabak HF, Wanders RJ. Transport of activated fatty acids by the peroxisomal ATP-binding-cassette transporter Pxa2 in a semi-intact yeast cell system. Eur J Biochem. 1997;249:657–61. doi: 10.1111/j.1432-1033.1997.00657.x. [DOI] [PubMed] [Google Scholar]
- 25.Groot PH. The activation of short-chain fatty acids by the soluble fraction of guinea-pig heart and liver mitochondria. The search for a distinct propionyl-CoA synthetase. Biochim Biophys Acta. 1975;380:12–20. doi: 10.1016/0005-2760(75)90040-5. [DOI] [PubMed] [Google Scholar]
- 26.Olowe Y, Schulz H. Regulation of thiolases from pig heart. Control of fatty acid oxidation in heart. Eur J Biochem. 1980;109:425–9. doi: 10.1111/j.1432-1033.1980.tb04811.x. [DOI] [PubMed] [Google Scholar]
- 27.Corkey BE, Brandt M, Williams RJ, Williamson JR. Assay of short-chain acyl coenzyme A intermediates in tissue extracts by high-pressure liquid chromatography. Anal Biochem. 1981;118:30–41. doi: 10.1016/0003-2697(81)90152-4. [DOI] [PubMed] [Google Scholar]
- 28.Sundqvist KE, Peuhkurinen KJ, Hiltunen JK, Hassinen IE. Effect of acetate and octanoate on tricarboxylic acid cycle metabolite disposal during propionate oxidation in the perfused rat heart. Biochim Biophys Acta. 1984;801:429–36. doi: 10.1016/0304-4165(84)90149-1. [DOI] [PubMed] [Google Scholar]
- 29.Liedtke AJ, Hacker T, Renstrom B, Nellis SH. Anaplerotic effects of propionate on oxidations of acetate and long-chain fatty acids. Am J Physiol. 1996;270:H2197–H2203. doi: 10.1152/ajpheart.1996.270.6.H2197. [DOI] [PubMed] [Google Scholar]
- 30.Oram JF, Wenger JI, Neely JR. Regulation of long chain fatty acid activation in heart muscle. J Biol Chem. 1975;250:73–8. [PubMed] [Google Scholar]
- 31.Yamashita H, Fukuura A, Nakamura T, Kaneyuki T, Kimoto M, Hiemori M, et al. Purification and partial characterization of acetyl-coA synthetase in rat liver mitochondria. J Nutr Sci Vitaminol (Tokyo) 2002;48:359–64. doi: 10.3177/jnsv.48.359. [DOI] [PubMed] [Google Scholar]
- 32.Bhaird NNA, Kumaravel G, Gandour RD, Krueger MJ, Ramsay RR. Comparison of the active sites of the purified carnitine acyltransferases from peroxisomes and mitochondria by using a reaction-intermediate analogue. Biochem J. 1993;294:645–51. doi: 10.1042/bj2940645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hexeberg S, Willumsen N, Berge RK. Eicosapentaenoic acid causes transient accumulation of lipids in rat myocardium. Biochim Biophys Acta. 1995;1256:341–5. doi: 10.1016/0005-2760(95)00043-c. [DOI] [PubMed] [Google Scholar]
- 34.Russell RR, III, Taegtmeyer H. Coenzyme A sequestration in rat hearts oxidizing ketone bodies. J Clin Invest. 1992;89:968–73. doi: 10.1172/JCI115679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russell RR, III, Taegtmeyer H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J Clin Invest. 1991;87:384–90. doi: 10.1172/JCI115008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roe CR, Ding JH. The metabolic and molecular bases of inherited diseases. New York: McGraw-Hill; 2001. Mitochondrial fatty acid oxidation disorders; pp. 2297–326. [Google Scholar]
- 37.Krahenbuhl S, Mang G, Kupferschmidt H, Meier PJ, Krause M. Plasma and hepatic carnitine and coenzyme A pools in a patient with fatal, valproate induced hepatotoxicity. Gut. 1995;37:140–3. doi: 10.1136/gut.37.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silva MF, Ruiter JP, Overmars H, Bootsma AH, van Gennip AH, Jakobs C, et al. Complete beta-oxidation of valproate: cleavage of 3-oxovalproyl-CoA by a mitochondrial 3-oxoacyl-CoA thiolase. Biochem J. 2002;362:755–60. doi: 10.1042/0264-6021:3620755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD. beta-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am J Physiol Heart Circ Physiol. 2003;285:H1626–H1631. doi: 10.1152/ajpheart.00332.2003. [DOI] [PubMed] [Google Scholar]
- 40.O'donnell JM, Alpert NM, White LT, Lewandowski ED. Coupling of mitochondrial fatty acid uptake to oxidative flux in the intact heart. Biophys J. 2002;82:11–8. doi: 10.1016/S0006-3495(02)75369-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barth C, Sladek M, Decker K. The subcellular distribution of short-chain fatty acyl-CoA synthetase activity in rat tissues. Biochim Biophys Acta. 1971;248:24–33. doi: 10.1016/0005-2760(71)90071-3. [DOI] [PubMed] [Google Scholar]
- 42.Barth C, Sladek M, Decker K. Dietary changes of cytoplasmic acetyl-CoA synthetase in different rat tissues. Biochim Biophys Acta. 1972;260:1–9. doi: 10.1016/0005-2760(72)90067-7. [DOI] [PubMed] [Google Scholar]
- 43.Scholte HR, Groot PH. Organ and intracellular localization of short-chain acyl-CoA synthetases in rat and guinea-pig. Biochim Biophys Acta. 1975;409:283–96. doi: 10.1016/0005-2760(75)90024-7. [DOI] [PubMed] [Google Scholar]
- 44.Gambert S, Vergely C, Filomenko R, Moreau D, Bettaieb A, Opie LH, et al. Adverse effects of free fatty acid associated with increased oxidative stress in postischemic isolated rat hearts. Mol Cell Biochem. 2006;283:147–52. doi: 10.1007/s11010-006-2518-9. [DOI] [PubMed] [Google Scholar]