

Summary
Bifunctional human PAPS synthetase (PAPSS) catalyzes, in a two-step process, the formation of the activated sulfate carrier 3′-phosphoadenosine 5′-phosphosulfate (PAPS). The first reaction involves the formation of the 5′-adenosine phosphosulfate (APS) intermediate from ATP and inorganic sulfate. APS is then further phosphorylated on its 3′-hydroxyl group by an additional ATP molecule to generate PAPS. The former reaction is catalyzed by the ATP-sulfurylase domain and the latter by the APS kinase domain. Here we report the structure of the APS kinase domain of PAPSS isoform 1 (PAPSS1) representing the Michaelis complex with the products ADP-Mg and PAPS. This structure provides a rare glimpse of the active conformation of an enzyme catalyzing phosphoryl transfer without resorting to substrate analogs, inactivating mutations, or catalytically non-competent conditions. Our structure shows the interactions involved in the binding of the magnesium ion and PAPS, thereby revealing residues critical for catalysis. The essential magnesium ion is observed bridging the phosphate groups of the products. This function of the metal ion is made possible by the DGDN-loop changing its conformation from that previously reported, and unambiguously identifies these loop residues as a Walker B motif. Furthermore, the second aspartate of this motif is the likely candidate for initiating nucleophilic attack on the ATP γ-phosphate by abstracting the proton from the 3′-hydroxyl of the substrate APS. We also report the structure of the APS kinase domain of human PAPSS1 in complex with two APS molecules, demonstrating the ability of the ATP/ADP binding site to bind APS. Both structures reveal extended N-termini that approach the active site of the neighboring monomer. Together, these results significantly increase our understandings of how catalysis is achieved by APS kinase.
Keywords: PAPS synthetase, APS kinase domain, phosphoryl transfer, crystal structure, magnesium binding, Michaelis complex
Introduction
Essential for all sulfonation reactions is the availability of a high-energy sulfuryl group donor molecule.1 In higher organisms, sulfate activation is catalyzed by the bifunctional PAPS synthetase that contains both an ATP sulfurylase (ATPS) domain and an APS kinase (APSK) domain. The former activates inorganic sulfate by a reaction with ATP to give adenosine-5′-phosphosulfate (APS) and pyrophosphate; APS is subsequently phosphorylated by the APSK domain to yield the universal high-energy sulfuryl donor 3′-phosphoadenosine-5′-phosphosulfate (PAPS).
In mammalian cells, PAPS is absolutely required for the sulfonation of proteins, hormones, neurotransmitters, carbohydrates, lipids, drugs and xenobiotics. All enzymes that catalyze sulfonation, called sulfotransferases, utilize PAPS as the sulfuryl donor.2 The fact that PAPS synthetase (PAPSS) is the sole source of PAPS makes this enzyme indispensable for all cells. Humans express two isoforms of PAPSS that are 78% identical.3 Isoform 1, PAPSS1, is highly expressed in brain and skin, while PAPSS2 is the predominant form in liver and cartilage.4,5 Mutations in PAPSS2 have been associated with several diseases in mice and humans (murine brachymorphism,6,7 human spondyloepimetaphyseal dysplasia - SEMD8 and some forms of osteoarthritis9,10).
In the majority of lower organisms and plants the ATP sulfurylase and APS kinase activities are present on two separate proteins. In contrast, these activities are combined to a single polypeptide in mammals. The biological implications for the fusion of the ATPS and APSK genes are still unclear. It had been proposed that a bifunctional enzyme would enable the intermediate APS to be channeled between the two active centers.11 However, recent extensive kinetic analysis12 and the structure of the bifunctional PAPSS113 reveal the absence of such a channel between the two active sites. Furthermore, experiments employing equimolar amounts of monofunctional proteins (ATPS and APSK domains cloned and expressed separately) showed that the mixture is equally efficient in catalyzing the overall reaction as is the bifunctional protein itself.14 Moreover, switching the order of the domains within a single polypeptide chain maintains a functional protein.14 These observations suggest that each domain functions independently, and support the proposal that fusion of the two activities onto a single polypeptide is mainly for attaining coordinated expression.15 However, one cannot discard the possibility that ligand-induced conformational changes allow for some measure of coordination between the two domains. As a first step towards increasing our understanding of these physiologically essential bifunctional enzymes we have undertaken experiments to structurally characterize the enzymatically active APSK domain of PAPSS1.
Kinetic studies done with the homologous monofunctional APS kinase from Penicillium chrysogenum16 have established an ordered reaction mechanism, in which the enzyme first binds ATP-Mg, and then binds the second substrate APS. Upon completion of phosphoryl transfer, PAPS leaves first, followed by ADP. The proposed kinetic mechanism is also supported by experiments based on the accessibility of the active site for chemical modifications17 or susceptibility to proteolysis when incubated with different sets of nucleotides,18 suggesting that substrate binding is accompanied by significant conformational changes. Crystal structures of P. chrysogenum APSK without any nucleotides in the active site,18 or those complexed with ADP, or the combination of ADP and APS,19 reveal the predicted conformational changes induced by nucleotide binding.
One of the intriguing hallmarks of this enzyme is the pronounced substrate inhibition with APS. Studies on APSK from P. chrysogenum,16 rat,20 and Arabidopsis thaliana21 have shown that this inhibition is uncompetitive in nature, suggesting the formation of the dead-end E-ADP-APS complex. This complex is predicted to form after completion of the enzymatic reaction, when PAPS leaves the active site and APS rebinds to the same site prior to ADP dissociation.
Our current structural information on the APSK domain of human PAPSS1 is limited to structures solved in the context of bifunctional enzyme.13 These PAPSS1 structures were obtained by soaking different ligands to the same crystal form, and unexpectedly all show an asymmetry between the two APSK domains that form the PAPSS1 dimer. These data were interpreted such that asymmetry in the PAPSS1 APSK domain is an intrinsic feature of the human enzyme.13 This asymmetry stands in contrast to structures of the P. chrysogenum APSK that show equivalent monomers.18,19 An alternative explanation for the observed asymmetry in the ASPK domain of PAPSS1 could be the presence of the ATP sulfurylase domain, or simply a manifestation of the crystallization conditions. Additional open questions regarding the APSK domain concern the binding mode of the essential magnesium ion,17 and of the transferred phosphoryl group. Finally, although several experiments suggest the formation of E-ADP-APS as the inhibitory complex, the question remains whether APS, due to its high structural resemblance to ADP, can also bind at the ATP/ADP binding site, thereby forming an inhibitory E-APS-APS complex. To address these issues we solved the crystal structures of the PAPSS1 APSK domain in complex with APS, and in complex with the products PAPS and ADP. Interestingly, both structures of isolated domain, obtained in different crystal forms, reveal symmetrical dimers. The structure of the Michaelis product complex (ADP plus PAPS), which includes the magnesium ion, reveals how the ion serves to organize the active site residues into a productive conformation and identifies residues involved in binding of the transferred phosphate. The structure with APS confirms that APS can indeed bind concomitantly to the APS and ATP/ADP sites. In addition, in both complexes we can model previously unseen N-terminal residues that are positioned close to the nucleotides of the neighboring monomer. Together, these results increase our structural and functional understanding of this important enzyme.
Results
Overall structure
The APS kinase (APSK) domain (residues 25–227) of human bifunctional PAPS synthetase 1 was crystallized in complex with APS, and with ADP plus PAPS. The APS complex crystallized in space group C2 with eight molecules in the asymmetric unit. Initial phases were obtained by molecular replacement using as the search model a truncated structure of the P. chrysogenum APS kinase solved in complex with ADP and APS.19 The model was refined at 2.05 Å resolution to a final R-factor of 22.3% and R-free of 28.2%. Two APS molecules per monomer, one at the APS binding site and one at the phosphoryl donor binding site, were clearly observed in the electron density maps. In six of the eight monomers electron density extending to the first residue present in our construct allowed visualization of the N-terminal region. In addition, a conformation not seen up to this work for a conserved region (DGDN-loop) revealed its role in magnesium binding (see below).
The APS kinase domain in complex with ADP and PAPS crystallized in space group P21 with two molecules in the asymmetric unit. Maximum resolution for this data set was 1.9 Å. The starting model used to solve the ADP-PAPS complex structure was the above APS complex. After alternating rounds of model building and refinement the structure converged to an R-factor of 18.7% and R-free of 23.6%. Excellent electron density for the two nucleotides, including the essential magnesium ion, was observed. The APS kinase dimer in complex with ADP and PAPS is shown in Figure 1(a). The N-terminal region and the magnesium binding competent conformation of the DGDN-loop resembled that present in the APS-APS complex. For both the APS-APS and the ADP-PAPS structures, all main chain dihedral angles are found in the allowed regions of Ramachandran plot. Data collection and refinement statistics are presented in Table 1.
Figure 1. The APSK domain of human bifunctional PAPSS1 forms a symmetrical dimer and adopts the same overall fold as observed in the context of the bifunctional enzyme.
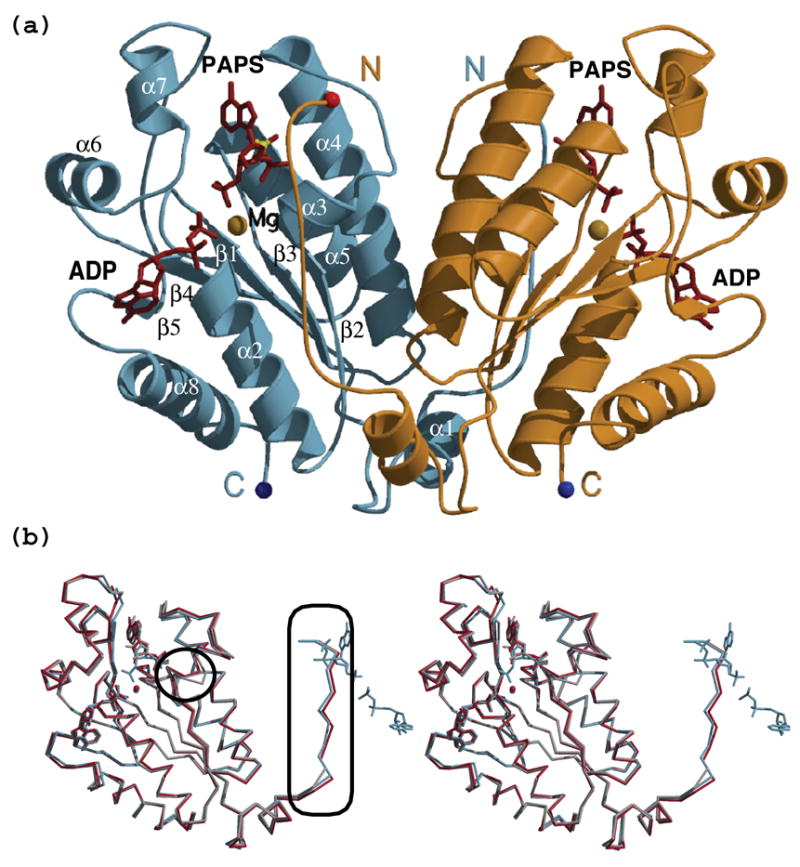
(a) Ribbon diagram of the APSK dimer (in light blue and orange) in complex with ADP and PAPS (maroon). The magnesium ion bridging the two nucleotides is depicted as a yellow sphere. Note that the N-terminal residues of one monomer approach the active site of the neighboring monomer. (b) Overlay of the APSK domain crystallized with ADP and PAPS (light blue), with APS (red), and with ADP and APS in the context of the full length PAPSS1 (gray - chain B in PDB entry 1XNJ). The rmsd for the two structures reported here is 0.44 Å over 201 common Cα atoms; between our structures and 1XNJ, it is 0.57–0.62 Å over 175–191 atoms. Important differences are: the conformation of the DGDN-loop (circle) and our ability to model 9 additional residues at the N-terminus (rectangle). These N-terminal residues approach the active site of the second monomer (the nucleotides of that monomer are included for orientation purposes).
Table 1.
Data collection and refinement statistics
| ADP-PAPS | APS-APS | |
|---|---|---|
| Data collection | ||
| Beamline | APS, SERCAT-BM | APS, SERCAT-BM |
| Wavelength (Å) | 1.000 | 0.9594 |
| a=51.40, b=63.32, c=61.69 | a=181.19 b=69.03 c=150.61 | |
| Unit cell (Å) | β=114.38 | β=116.65 |
| Space group | P21 | C2 |
| No. molecules/a.u. | 2 | 8 |
| Resolution limit (Å) | 20–1.9 | 10–2.05 |
| Measured reflections | 156,400 | 376,976 |
| Unique reflections | 27,832 | 101,066 |
| Completeness (%; overall/last shell) | 97.5/94.5b | 83.6/92.9b |
| I/σI (overall/last shell) | 18.83/9.30b | 12.4/4.36b |
| Rsyma (overall/last shell) | 7.1/14.8b | 8.2/24.0b |
| Refinement | ||
| Resolution limit (Å) | 20–1.9 | 10–2.05 |
| No. reflections (working/free) | 24,917/2,766 | 91,516/10,107 |
| Rcrystd (overall/last shell) | 0.187/0.205c | 0.223/0.234c |
| Rfree (overall/last shell) | 0.236/0.306c | 0.282/0.324c |
| No. protein/water/nucleotide/ion res. | 406/252/4/6 | 1,594/822/16/6 |
| rmsd from ideal geometry (Å) | ||
| Bond length | 0.012 | 0.011 |
| Angle distances | 1.571 | 1.729 |
| Estimated coordinate error (Å) | 0.178 | 0.233 |
| Ramachandran plot statistics | ||
| Residues in most favored regions | 90.4% | 90.6% |
| Residues in allowed regions | 8.4% | 8.9% |
| Residues in gen. allowed regions | 1.1% | 0.5% |
| Residues in disallowed regions | 0.0% | 0.0% |
| PDB-entry id | 2OFX | 2OFW |
Rsym = ∑|I-〈I〉|/∑ I
last shell (ADP/PAPS) =1.90–2.01 Å; (APS/APS) = 2.05–2.10 Å
last shell (ADP/PAPS) =1.90–1.95 Å; (APS/APS) = 2.05–2.10 Å
Rcryst = ∑||Fobs|−|Fcalc||/∑|Fobs|, 10% randomly omitted reflections were used for Rfree
The overall fold of both structures is the same as previously described for the APSK domain of the full length PAPSS113 (Figure 1(b)) and the APSK domain from PAPSS2 (PDB ID 2AX4). Interestingly, in the reported structures of the full length PAPSS1, the APS kinase domain exhibited differences between the molecules that form the physiological dimer: one molecule contained nucleotide(s) and the second was empty.13 In contrast, apart from small differences between monomers in the APS-APS complex, our structures reveal a symmetric dimer (rmsd between the two monomers of the ADP-PAPS structure is 0.19 Å, and range from 0.26 to 0.65 Å for the eight monomers of the APS-APS structure). Consistent with our results, the structure of the isolated APSK domain from PAPSS2 also contains a symmetrical dimer with one ADP molecule in each of the monomers’ active sites (PDB ID 2AX4). The presence of magnesium in our structures has ramifications not only for the nucleotides that it directly interacts with, but also for the conformation of the DGDN-loop and the N-terminus. The change in conformation of the DGDN-loop in comparison to that seen in the absence of magnesium (circle in Figure 1(b)) allows the N-terminus to adopt a stable conformation not previously seen (ellipse in Figure 1(b)). Note that the N-terminus of one monomer approaches the nucleotides bound to the counterpart monomer.
ADP binding
It has been observed in human bifunctional PAPSS1 that the base of the ADP (ADP was present in only one of the subunits) adopts the syn conformation. This is in contrast to the anti ADP conformation observed in structures of the P. chrysogenum APSK. The electron density for the ADP in our ADP-PAPS complex structure was unambiguous for the phosphate moieties but revealed two possible conformations for the sugar and the base. One conformation approximates the previously reported syn conformation and the other the anti conformation (Figure 2(a)).
Figure 2. Binding of nucleotides and magnesium in the ADP-PAPS complex.
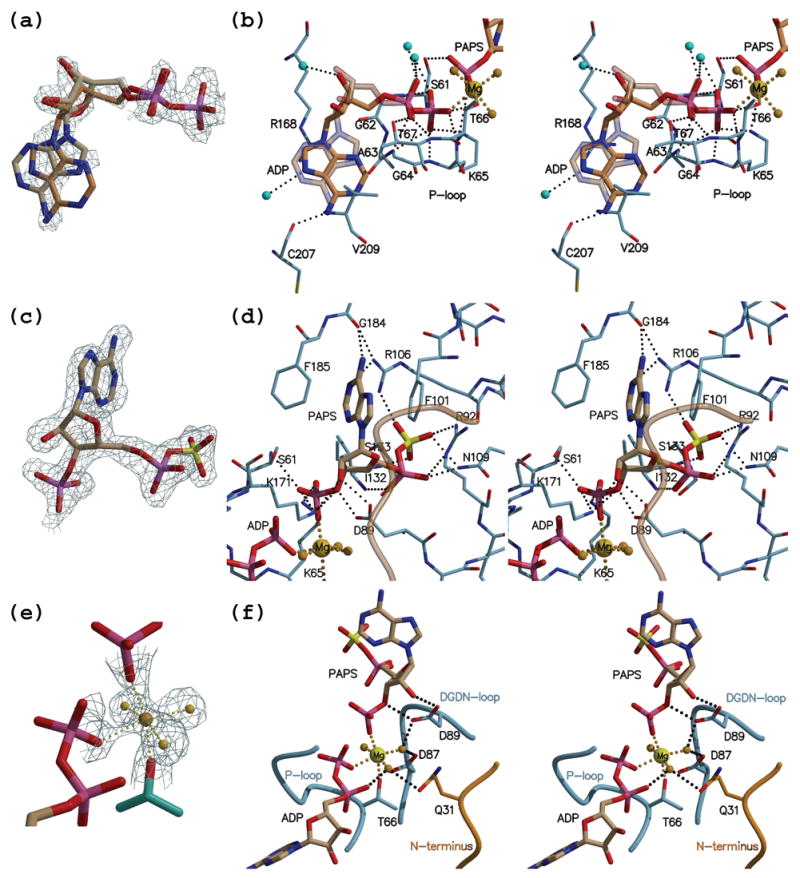
(a) Shown is ADP in its modeled syn and anti conformation with the simulated annealing composite omit map calculated with the coefficients 2Fobs-Fcalc (contoured at 1.3 σ). Invoking a mixture of the two base conformations accounts best the electron density maps. (b) Stereo view depicting the interactions made by ADP. Nitrogen atoms are rendered in blue, oxygen in red, sulfur in yellow and phosphorous in magenta. Protein carbon atoms are colored in light blue whereas nucleotide carbon atoms are in brown. Waters are depicted as cyan spheres, and the magnesium ion with it water ligands in yellow. The anti ADP conformation is shown as a semi-transparent model. (c) PAPS with its map as in (a) but contoured at 1.5 σ. (d) Stereo view of the interactions made by PAPS and colored as in (b). The N-terminus that originates from the neighboring monomer is rendered as a semi-transparent orange coil. (e) The octahedrally coordinated magnesium with the corresponding omit map as in (c). (f) Stereo view depicting magnesium coordination. The cation bridges the phosphates of ADP and PAPS, as well as interacting with Thr66 from the protein P-loop. The remaining three ligands are water molecules. One of the waters coordinated to magnesium is stabilized through interactions with Asp87 and Asp89 from the DGDN-loop. Another water ligand is at a hydrogen bond distance to Gln31 that originates from the N-terminus (orange) of the other monomer.
Interactions of the adenine base and the protein are limited only to π-stacking between Arg168 and Val209, and a sole hydrogen bond between main chain carbonyl of Cys207 and NH2 of the adenine. The interactions are maintained in both the syn and anti base conformations (Figure 2(b)). Therefore, these non-conformationally discriminating interactions and the observed electron density suggest that both the syn and anti ADP forms are present in our crystals at approximately an equal ratio. In contrast, the ADP phosphates are firmly positioned by main chain interactions with P-loop residues, and exhibit a single conformation. Interestingly, two P-loop side chains, Ser61 and Lys65, also interact with the 3′-phosphate of the PAPS molecule (Figure 2(b) and Figure 3). A paucity of interactions with the ribose hydroxyl groups suggests that both oxy and deoxy nucleotides can be equally efficient as phosphoryl donors.
Figure 3. Distance map of the active site of APSK domain in complex with ADP plus PAPS.
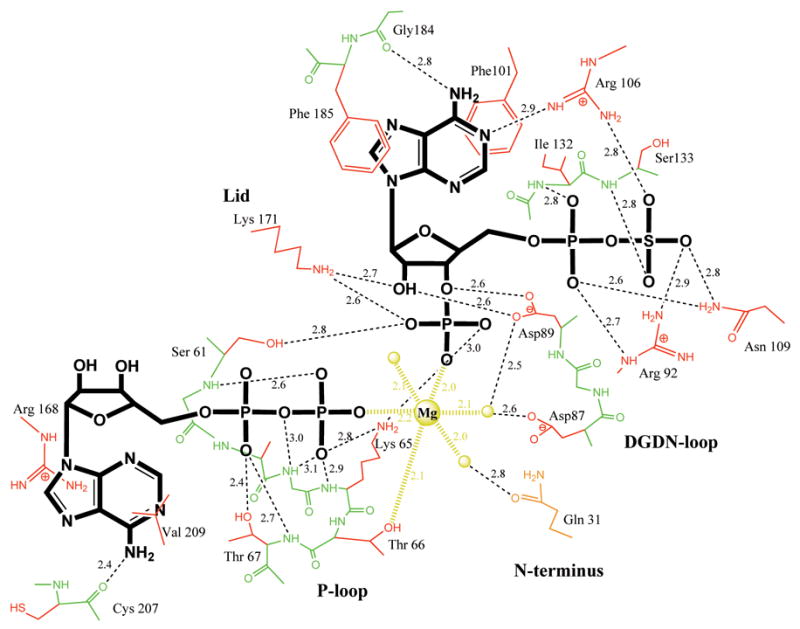
Schematic representation of the protein-nucleotides-magnesium interactions. Nucleotides are shown in thick bonds (ADP in anti conformation), main chain protein atoms in green, side chains in red, magnesium and its three water ligands in yellow. All distance are in Å.
PAPS binding
This is the first report of an APSK with PAPS bound in the active site. The electron density unequivocally shows a 3′-phosphate group, which differentiates PAPS from APS (Figure 2(c)). The portion of the PAPS molecule that is common to that of APS is observed to bind in a very similar fashion as previously described for APS.13,19 In short, the adenine base is in the syn conformation firmly stacked between two conserved phenylalanine residues (Phe185 and Phe101). The NH2 group, a hallmark of the adenine ring, is held in place by an interaction with the main chain carbonyl of Gly184. Arg106 contributes to the stabilization of the syn conformation by bridging the adenine ring N1 atom and an oxygen atom of the sulfate moiety. Oxygens of the 5′-phosphosulfate interact with the side chains of Arg92 and Asn109 (residues that are disordered prior to APS binding13) as well as the main chain nitrogen atoms of amino acids 132 and 133 (Figure 2(d) and Figure 3).
Notably, the side chain of Asn27 from the N-terminal region of the neighboring monomer is close (~3.5 Å) to the base of the phosphoryl acceptor. This N-terminal region, which in previous structures was not observed, approaches the APS/PAPS binding site in our structures (depicted as a transparent orange coil in Figure 2(d) and a solid orange coil in Figure 2(f)). While not directly interacting with PAPS, the Asn27 side chain (not shown) adopts a position only possible when the base is in the syn conformation. Since this is one of the few conserved residues in the N-terminal region we examined whether it has a role in facilitating APS/PAPS binding. Substituting Asn27 with Ala did not have any effect on the steady state activity of the protein (Table S1).
The PAPS ribose ring is held in position by Asp89 from the DGDN-loop through interactions with the 2′- and 3′-hydroxyl groups. These interactions ensure that the sugar ring adopts the 2′-endo, 3′-exo conformation. The 3′-phosphate of PAPS draws three different parts of the protein closer to the APS/PAPS binding region. This is accomplished by an interaction with Ser61 and Lys65 from the P-loop, which are situated in the core of the protein, with Asp89 from the DGDN-loop, and finally with Lys171 from the lid region. These interactions seem to provide extra rigidity to the structure. The latter interaction contributes to the final closure of the lid region and provides an additional basic residue that can participate in catalyzing phosphoryl transfer. Overlay of the APSK domain on guanylate kinase (a representative member of nucleoside monophosphate kinases) demonstrates that Lys171 is positioned similarly to the lid arginine (Arg137) of mouse guanylate kinase.22 Together, this suggests that the conserved Lys171 interacts with the γ-phosphate of ATP, and guides its transfer to the 3′-OH group of the acceptor APS.
Magnesium binding and DGDN-loop
Excellent electron density was observed for the octahedrally coordinated magnesium ion (Figure 2(e)). Thr66 of the P-loop (Ser in several other organisms) forms one of the metal ligands. Two additional ligands are provided by the phosphate groups of ADP (β-phosphate) and PAPS (3′-phosphate). The remaining three magnesium ion ligands are water molecules. Importantly, the observation of magnesium bridging the two substrates via what was the ATP γ-phosphate - now at 3′-position of PAPS- and the phosphate corresponding to the β-phosphate of ATP (Figure 2(f) and Figure 3), reveals the metal ion’s role in catalyzing phosphoryl transfer (see Discussion).
One of the magnesium ion water ligands is positioned through interactions with carboxyl groups of the highly conserved Asp87 and Asp89. This conformation of the loop holding Asp87 and Asp89 separated by a glycine residue (DGDN-loop) is first observed in the structure reported here (Figure 4(a)). The high conservation of the 86LDGDN90 sequence that is positioned in proximity to the active site was already noticed by MacRea et al.18 They suggested that this sequence is analogous to the Walker B motif (the motif that contributes a carboxylic acid required for magnesium-water-ligand coordination). This prediction was hard to reconcile with the available structures since the predicted aspartic acid residue was too distant from the P-loop (Walker A motif), and hence the ATP-Mg binding site. The authors suggested that a magnesium induced conformational change would bring the aspartic acid closer to the active site. Further support of the importance of the DGDN-loop comes from mutagenesis studies that demonstrated a requirement for strict conservation of this loop for enzyme activity.23 Our structures of the APSK domain, the first to include the magnesium ion, support the above predictions. Magnesium binding draws the DGDN-loop towards the nucleotides through the interactions with Asp87 and Asp89 (Figure 4(a)). The pronounced conformational change of the DGDN-loop upon magnesium binding requires the glycine residue for main chain flexibility. In fact, the mutation associated with brachymorphic mice, a phenotype characterized by short stature, has been traced to a change of this glycine residue to an arginine in PAPSS2. Therefore, our results supply a structural explanation for the loss of APS kinase activity that causes this disease, directly implicating the lost ability of such a mutant enzyme to bind magnesium.
Figure 4. The DGDN-loop changes conformation in the presence of magnesium.
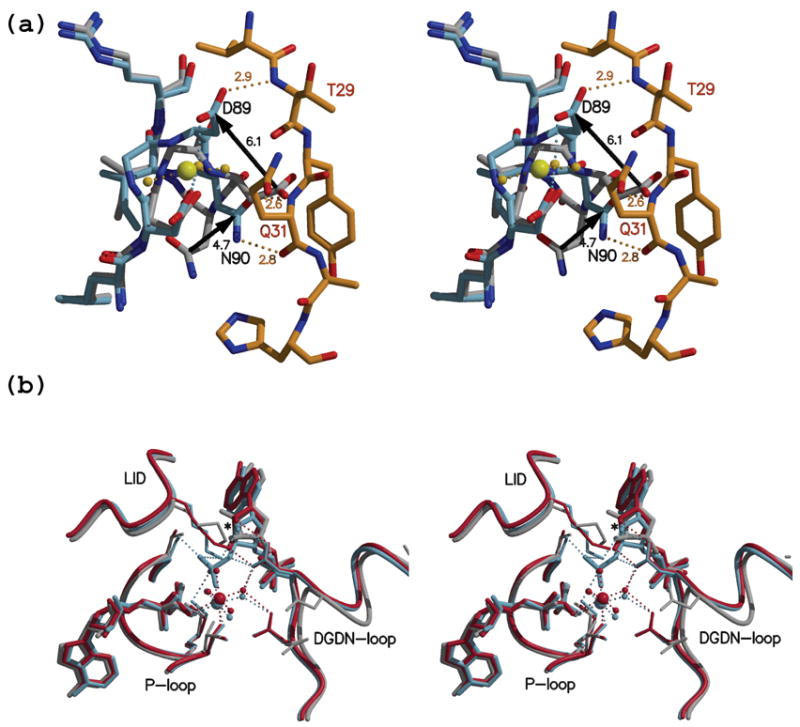
(a) Stereo view of an overlay of the DGDN-loop (light blue) from the ADP-PAPS complex with the N-terminus in orange and the previously reported ADP-APS complex (gray - PDB id 1XNJ). Due to the presence of magnesium in the ADP-PAPS complex, the DGDN-loop is stabilized at a conformation where Asp87 and Asp89 interact with a magnesium water ligand. This loop conformation, but not the DGDN-loop in the structure lacking magnesium, allows the N-terminus to adopt the position as seen in our structure. The three polar interactions between main chain atoms of the N-terminus and the DGDN-loop are shown as orange dashed lines with the corresponding distances in Å. Pronounced differences in side chain position are observed for Asp89 and Asn90 (black arrows). (b) Stereo view of the active site overlay between the APSK domain in complex with ADP plus PAPS (light blue), APS plus APS (red), and the monomer containing ADP plus APS in 1XNJ (gray). The presence of magnesium draws the DGDN-loop towards the active site, allowing Asp89 to interact with sugar hydroxyls (P)APS. In addition, the interaction between the PAPS 3′-phosphate group and the magnesium ion draws the nucleotide towards ADP.
An additional magnesium water ligand is observed to interact with the side chain of Gln31, which originates from the N-terminal sequence of the other molecule (orange Figure 2(f) and Figure 3). To test the importance of this interaction we mutated Gln31 to Ala. Surprisingly, this mutant was enzymatically indistinguishable from the wild type (Table S1).
Apart from drawing in the DGDN-loop, the magnesium ion also brings the PAPS molecule closer to ADP when compared to the position of the APS molecule in the APS complex structure. This shift, of ~0.7Å, is due to the aforementioned bridging character of the magnesium ion acting to bind both the β-phosphate of ADP and the 3′-phosphate of PAPS (Figure 4(b)).
N-terminal residues
We could model all of the APSK residues present in the construct (residues 25–227). The five most N-terminal residues have not been previously observed in any APS kinase structure. These residues show clear density in the omit maps (Figure 5(a)) and are in the extended conformation. This N-terminal region lies at the surface close to the active site of the neighboring monomer.
Figure 5. The extended conformation of the N-terminus propagates along the neighboring monomer and approaches the active site.
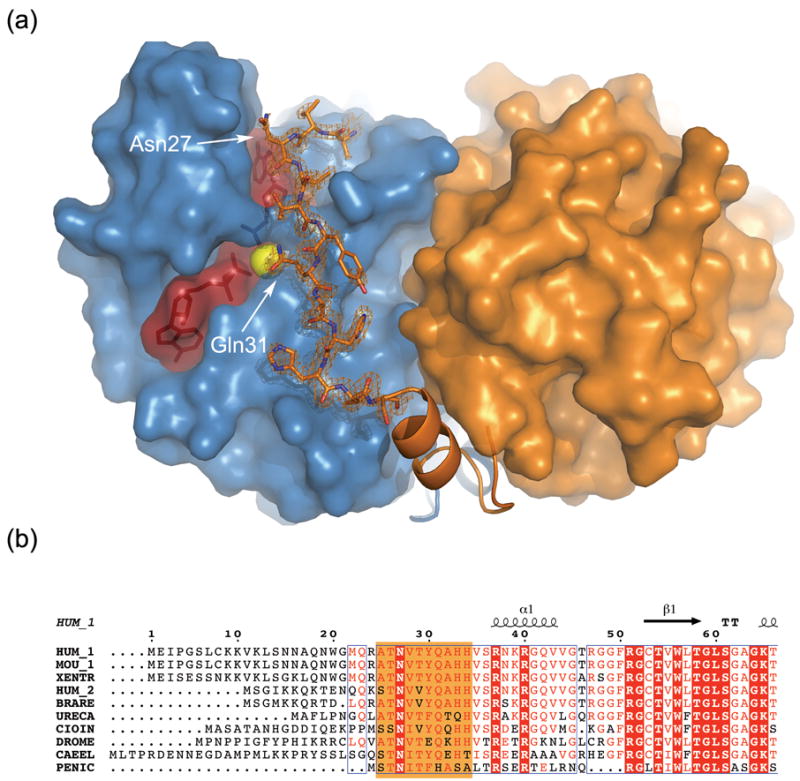
(a) Surface representation of an APSK dimer with one monomer in light blue and one in orange. Nucleotides, in maroon, are shown as semi-transparent space-filled and ball-and-stick objects, and the magnesium ion as a yellow sphere. The most N-terminal residues (25–36) are shown in a ball-and-stick representation surrounded by the corresponding simulated annealing omit map contoured at 1.5 σ. Residues 37–50 are represented as a ribbon (α-helix for residues 37–43). See text for details. (b) Sequence alignment of the N-terminus of several APSK domains from bifunctional enzymes (HUM_1 - human PAPSS1, MOU_1 - mouse PAPSS1, XENTR - Xenopus tropicalis PAPSS, HUM_2 - human PAPSS2, BRARE - Brachydanio rerio PAPSS, URECA - Urechis caupo PAPSS, CIOIN - Ciona intestinalis PAPSS, DROME - Drosophila melanogaster PAPSS, CAEEL - Caenorhabditis elegans PAPSS) and the monofunctional Penicillium chrysogenum (PENIC) APSK enzyme. Note the low sequence identity for the first 50 residues. Residues highlighted in orange correspond to those shown as ball-and-stick in (a).
All the interactions between the N-terminal residues and the neighboring monomer occur via main chain atoms. Specifically, the main chain amino group of Thr29 interacts with the side chain of Asp89, and the backbone NH and carbonyl moieties of Gln31 interact with the carbonyl and amino functionalities of the Asn90 side chain, respectively (Figure 4(a)). This is consistent with the low sequence conservation observed for the N-terminus (Figure 5(b)). The absence of interactions between the N-terminus and symmetry related molecules, along with very similar conformations for the N-terminal residues in both the ADP-PAPS and APS-APS structures (despite different crystallization conditions), suggests that the observed conformation is indeed relevant. By adopting this conformation, the N-terminus is able to interact with the critical DGDN-loop. However, we could not detect any appreciable change in the kinetic behavior of the point mutations N27A, Q31A, or in a truncation mutant lacking the first 34 N-terminal residues.
Structure with APS
Several monofunctional APS kinases24,25,26 and human bifunctional ATP sulfurylase/APS kinase12 have been reported to exhibit pronounced substrate inhibition with APS. Extensive kinetic studies performed with the P. chrysogenum enzyme16,25,27 indicate that this inhibition is uncompetitive with respect to ATP and that excess of substrate results in the formation of an abortive complex, where ADP and APS occupy the active site of the enzyme. Our kinetic analysis, performed on the APSK domain of human PAPSS1, also supports the formation of the abortive dead-end complex E-ADP-APS as a mechanism of substrate inhibition for the human enzyme (data not shown).
However, experiments done on P. chrysogenum APSK show that 0.4–1.2 mM APS alone provides significant protection against enzyme inactivation by 2,4,6-trinitrobenzene sulfonate (a compound which reacts with free amino groups),17 suggesting the ability of two APS molecules to bind to the enzyme and protect the active site from reacting with the chemical. Indeed, when cocrystallizing the APSK domain with 3 mM APS and 5 mM MgCl2, we obtained a structure in which two APS molecules are bound. Thus, APS occupies both nucleotide-binding sites of APSK simultaneously. One APS molecule binds to the ATP binding site in virtually the same manner as ADP (all of the interactions with the P-loop are maintained) and an additional one to the APS binding site. However, different from the observed mixture of syn and anti conformations of ADP in the ADP-PAPS structure, in this APS-APS structure, the APS binding in the ADP binding site is exclusively in the syn conformation (as is the APS in the APS binding site).
There are eight molecules in the asymmetric unit of the APS-APS complex forming four symmetrical dimers. Six of the eight monomers (chains B, C, D, E, F, and H) contain the magnesium ion and this correlates with the active conformation of the DGDN-loop. In fact, in those monomers of the APS-APS complex lacking magnesium ion, the inactive conformation of the DGDN-loop prevents the N-terminus of the neighboring molecule from adopting the extended conformation as described previously. The other two monomers (chains A and G) lack this magnesium, and they exhibit only partial (chain A) or no occupancy for the active conformation of the DGDN-loop. Therefore, the largest rmsd between monomers is observed for chains G and F (0.65 Å for 201 Cα atoms) and the smallest rmsd between chains C and D (0.26 Å for 203 Cα atoms). The overall structure resembles the previously discussed ADP-PAPS complex with highest similarity for chain C (0.45 Å for 204 atoms) and lowest for chain A (0.74 Å for 201 atom). Figures 1(b) and 4(b) demonstrate this similarity.
Bearing in mind that ADP and APS have very similar shapes (are isosteric) and chemical properties, it is not entirely surprising that APS can bind to the ADP binding site. The only difference between these nucleotides is due to the β-phosphate in ADP being replaced with sulfate in APS. Our results demonstrate that at high concentration APS can indeed bind in the ADP site. Moreover, the binding of APS to this site can mimic the ability of ATP-ADP to induce the conformational changes responsible for the formation of the APS binding site, enabling APS association. Although this complex can be seen as representing the APS inhibitory complex, it was formed using high APS concentration (3 mM). The much smaller value for inhibitory constants (about 50 μM) of APS12,16,26,28,29 argue that such an APS-APS complex is of lower importance in the physiological context in comparison to an ADP-APS complex.
Discussion
Here we report the crystal structure of the APS kinase domain of the human PAPSS1 enzyme in complex with the product nucleotides ADP and PAPS, or in complex with two substrate molecules of APS. Both structures are characterized by a symmetrical dimer, in contrast to the homodimeric, non-symmetrical complex reported in the context of the full-length enzyme.13 Harjes et al. observed an APSK domain with one monomer occupied by ADP (and APS after soak) while the other monomer is vacated. They assigned these differences to an intrinsic property of the human protein, specifically to a lack of one of the internal hydrogen bond within the P-loop due to the substitution of Ser (in P. chrysogenum) with Ala (in the human enzyme). However, the fact that we were able to observe a symmetrical dimer (obtained in different crystallization conditions that resulted in two crystal forms), analogous to the one observed in the monofunctional P. chrysogenum enzyme, demonstrates that both active sites can be identically occupied at the same time in the human APSK domain. Consistent with this result is the recent structure of the APSK domain of human PAPSS2, where also a symmetrical dimer was observed. The identical P-loop sequence between PAPSS1 and PAPSS2 argues against this loop as the cause of dimer asymmetry observed in the human bifunctional PAPSS1 structure.
Our structures highlight four major novel features: they provide information on (i) the binding of the reaction product PAPS, (ii) elucidate the active conformation of the Michaelis product complex (ADP-Mg plus PAPS), (iii) the binding of the magnesium ion that induces a conformational change in the DGDN-loop, and (iv) reveal the extended conformation of the N-terminal region of the protein. All of these findings are inter-related.
The ability to crystallize with PAPS in the active site allowed us to identify residues involved in the binding of the transferred phosphoryl group. Not surprisingly, interactions between the site harboring the 3′-phosphate of PAPS–previously occupied by the γ-phosphate of ATP - are supplied mainly by the P-loop (Ser61 and Lys65), but also by the highly conserved lid residue Lys171 (being analogous to the lid Arg in nucleoside monophosphate kinases22), exemplifying the importance of this residue in the phosphoryl transfer reaction. Together, the adenine stacking interaction of Arg168 and the aforementioned interaction of Lys171 make the sole connection between phosphoryl donor nucleotide and the mobile lid region of the protein. Although the electron density for the lid can be modeled in the structures with ADP and APS,13,19 as well as in our structure with two APS molecules (i.e. in structures missing the γ-phosphate, or 3′-phosphate), the conformation of the lid region in not completely rigid. This is demonstrated by the fact that the lid adopts several slightly different conformations in our structure with two APS molecules bound in the active site. This implicates Lys171 as the final interaction from the lid region needed for the formation of a functional active site.
The presence of the magnesium ion in the ADP-PAPS structure, as well as in several monomers of the APS-APS structure, unambiguously identifies Asp87 and Asp89 as residues of a Walker B motif sequence. The motif is required for magnesium binding by supplying a carboxylic acid residue that positions a water molecule that ligates the metal. This observation rationalizes mutational studies that identified the DGDN-loop as a structural motif indispensable for the activity of the enzyme,23 as well as the effect of the mutation G78R in PAPSS2 that results in the brachymorphic defect in mice. Based on our structures, mutation of the glycine will be detrimental to activity for two reasons: one, by decreasing the flexibility of the loop, the required conformational change will be hindered. This will prevent the binding of the essential magnesium ion. Two, any residue larger than an alanine will sterically clash with PAPS. The distance from the Cβ-atom of an alanine modeled at that position to the α-phosphate of PAPS is less than 2.8 Å. Also, interactions between the γ-phosphate, magnesium ion, and DGDN-loop may play a role in promoting the conformational changes that shape the APS binding site. This fact rationalizes the observed ordered binding mechanism.
In both the ADP-PAPS and the APS-APS complex structures, the carboxylic group of Asp89 interacts with the 2′- and 3′-hydroxyls of PAPS and APS, respectively. These interactions are required for the proper orientation (2′endo, 3′exo) of the sugar ring for the phosphorylation by ATP (Figure 4(b)). Moreover, this interaction reveals that Asp89 could function as the general base by abstracting a proton from the 3′-hydroxyl group of APS. This would increase the nucleophilicity of the 3′-group and promote the nucleophilic attack on the γ-phosphate of ATP. This proposed mechanism, involving Asp89, is more in agreement with the structural data in comparison to an alternative model, which proposed the P-loop Ser61 as the general base.19 Once Asp89 becomes protonated, its interactions with the sugar are weakened, promoting the relaxation of the DGDN-loop and subsequent dissociation of PAPS from the active site.
We were successful in trapping the complex of ADP-Mg and PAPS due to the extended conformation adopted by the N-terminus. This conformation sterically prevents the DGDN-loop from returning to its inactive position (Figure 4(a)). In fact, by interacting with DGDN-loop, the N-terminus seems to stabilize the products bound state in our structure. Surprisingly, mutational analysis of this region does not affect the steady state kinetic parameters of the enzyme. Neither the point mutations N27A and Q31A, nor the complete deletion of the first 34 N-terminal residues had any effect on the kinetics of the APSK domain (Table S1). A similar deletion mutant generated in the context of the full-length protein also reported no changes in the kinetic parameters.30 This suggests that, while positioned close to the active site, this N-terminal region of the APSK domain does not play a critical role in catalysis.
A significant aspect of the work is that we were successful in trapping the Michaelis complex of the APS kinase reaction. Notably, this was accomplished without resorting to substrate analogs (e.g. employing a non-hydrolyzable analog), removal of the essential magnesium ion (e.g. by addition of the chelator EDTA), or by enzyme inactivating mutations, three strategies that can be employed to allow the visualization of a Michaelis complex of an enzyme catalyzing phosphoryl transfer. Thermodynamic properties of PAPSS played an important part in our ability to solve the structure of the product complex composed of a molecule of ADP, a molecule of PAPS, and the magnesium ion. The equilibrium in solution of the APS kinase reaction favors the products by a factor of over 1000. This is due to the fact that during the reaction the higher energy phosphoanhydride bond of ATP (ΔG°′ = −32.2 kJ/mol) is broken and the lower energy phosphate ester (ΔG°′ = −13.8 kJ/mol) on the 3′-hydroxyl of APS is formed. We can conclude that the equilibrium on the enzyme mirrors that in solution and, thus, favors the products. In summary, we were able to observe the Michaelis products complex of the enzyme due to two facts. First, the thermodynamics of the system, which favors the products over the substrates. Second, the conformation of the N-terminus, which “locks” the protein in a conformation that prevents product release.
The Michaelis products structure exposes all of the amino acid side chains that are important in catalyzing phosphoryl transfer. In addition, the DGDN-loop is stabilized in its active conformation. Most importantly, the observation of the magnesium ion bridging the β-phosphate of ADP and the transferred γ-phosphate, now located at the 3′-position of PAPS, permits the assignment of at least two catalytic roles for the magnesium ion: one, stabilization of the additional negative charge that forms in the transition state of the transferred phosphoryl group, and two, guiding the phosphoryl group from the donor ATP to the acceptor APS. Together, these findings reveal the intricacies of phosphoryl transfer catalyzed by the APSK domain of human PAPSS1.
Materials and Methods
Protein expression and purification
In order to increase chances of crystallization, we have examined previously solved crystal structures and conservation of the N-terminal residues among different APS kinases (Figure 5(b)). We noticed that the first 25 residues are not conserved between APSK domains of bifunctional PAPSS and are not present in the monofunctional P. chrysogenum APSK. This observation prompted us to design an APSK domain construct lacking the first 25 residues. As the terminal residue for our construct we have chosen residue 227 of human PAPSS1 based on analysis of P. chrysogenum structures.
The APS kinase domain (25–227 amino acids) of the PAPSS1 coding region31 was cloned as a glutathione S-transferase fusion in the bacterial expression vector pGEX-RB32 in which the thrombin cleavage site was replaced by a TEV site. Expression was induced by 0.1 mM IPTG and E. coli BL-21 Codon-Plus (Stratagene) cells were grown overnight at 22 °C. After cell sonication, the soluble fraction was subjected to a Glutathione-Sepharose Fast Flow column (Pharmacia), washed with buffer consisting of 50 mM Tris/HCl pH 7.5, 200 mM KCl, 5 mM MgCl2 and 1 mM EDTA and eluted with the same buffer containing 10 mM glutathione. Eluted protein was cut overnight with TEV protease while glutathione was dialyzed out. Uncut protein was removed by reloading the protein on the Glutathione-Sepharose Fast Flow column and collecting the cut protein in the flow-through. As a final step of purification, APS kinase in the above buffer was subjected to gel filtration using Superdex 200 (Pharmacia). Almost all of the protein came out as a peak corresponding to MW of ~ 50kD, indicating that APSK forms a dimer in solution. During the purification procedure, the concentration of DTT was maintained at 5 mM. The total yield of the protein from 6 L bacterial culture was ~1.5 g. Purified protein was frozen at −80 °C. Prior to crystallization, the protein was dialyzed against a solution containing 25 mM Tris-HCl pH 7.5, 25 mM KCl, and 7 mM DTT.
Crystallization and X-ray data collection
Crystals were grown at room temperature using hanging drops containing equal volumes of protein and reservoir solution (2+2 μL). For generating the APS complex, the protein solution contained 5–8 mg/ml of 25N APSK, 3 mM APS and 5 mM MgCl2, while the reservoir contained 18–20% (w/v) PEG 3350 and 0.25 M calcium acetate. Rectangular, birefringent crystals appeared overnight. Crystals were very sensitive to manipulation so, as cryoprotectant, grains of glucose were added directly to the drop and let to dissolve for 5–10 min. Crystals were then frozen in liquid nitrogen.
For the ADP-PAPS complex, the protein solution contained 6 mg/ml of 25N APSK, 2 mM PAPS, 2 mM ADP and 5 mM MgCl2, while the reservoir contained only 0.2–0.4 mM ammonium dihydrogenphosphate. Crystals were transferred stepwise to a mother liquor solution containing 30% glucose, and then frozen in liquid nitrogen. The high resolution limit for the data set collected on these crystals was restricted by the crystal-to-detector distance. In addition to the nucleotides and magnesium ions, four phosphate molecules were also modeled bound at the surface of the protein. We could also model two extra N-terminal residues (Met and His) remaining after TEV cleavage.
Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory and processed with the XDS program package.33
Steady state kinetics
The APSK enzymatic assay in the forward reaction was performed by monitoring the rate of ADP formation using the pyruvate kinase/lactate dehydrogenase-coupled spectrophotometric assay that measures the decrease in absorbance at 340 nm due to the oxidation of NADH. The assay was performed with 200–400 nM enzyme in a buffer containing 50 mM Tris-HCl pH 7.5, 100 mM KCl, 2.5 mM MgCl2, 0.25 mM EDTA, 1.0 mM ATP, 0.4 mM phosphoenolpyruvate and 0.2 mM NADH. The concentration of APS was varied between 2 and 100 μM. The reaction mixture was left to equilibrate for 10 minutes at 37°C to allow for the consumption of trace ADP present in the ATP stock solution. APSK was added to trigger the reaction. All measurements were made in triplicate at 37°C using a Varian Cary 50 Bio UV-visible spectrophotometer.
Supplementary Material
Acknowledgments
We thank the SERCAT staff for assistance in data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. N.S. and A.L. were supported by NIH grant AI46943, and S.O., I.P., and M.K. were supported by the Max Planck Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Strott CA. Sulfonation and molecular action. Endocr Rev. 2002;23:703–32. doi: 10.1210/er.2001-0040. [DOI] [PubMed] [Google Scholar]
- 2.Klaassen CD, Boles JW. Sulfation and sulfotransferases 5: the importance of 3′-phosphoadenosine 5′-phosphosulfate (PAPS) in the regulation of sulfation. Faseb J. 1997;11:404–18. doi: 10.1096/fasebj.11.6.9194521. [DOI] [PubMed] [Google Scholar]
- 3.Franzon VL, Gibson MA, Hatzinikolas G, Woollatt E, Sutherland GR, Cleary EG. Molecular cloning of a novel human PAPS synthetase which is differentially expressed in metastatic and non-metastatic colon carcinoma cells. Int J Biochem Cell Biol. 1999;31:613–26. doi: 10.1016/s1357-2725(98)00155-1. [DOI] [PubMed] [Google Scholar]
- 4.Xu ZH, Otterness DM, Freimuth RR, Carlini EJ, Wood TC, Mitchell S, Moon E, Kim UJ, Xu JP, Siciliano MJ, Weinshilboum RM. Human 3′-phosphoadenosine 5′-phosphosulfate synthetase 1 (PAPSS1) and PAPSS2: gene cloning, characterization and chromosomal localization. Biochem Biophys Res Commun. 2000;268:437–44. doi: 10.1006/bbrc.2000.2123. [DOI] [PubMed] [Google Scholar]
- 5.Fuda H, Shimizu C, Lee YC, Akita H, Strott CA. Characterization and expression of human bifunctional 3′-phosphoadenosine 5′-phosphosulphate synthase isoforms. Biochem J. 2002;365:497–504. doi: 10.1042/BJ20020044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugahara K, Schwartz NB. Defect in 3′-phosphoadenosine 5′-phosphosulfate formation in brachymorphic mice. Proc Natl Acad Sci U S A. 1979;76:6615–8. doi: 10.1073/pnas.76.12.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurima K, Warman ML, Krishnan S, Domowicz M, Krueger RC, Jr, Deyrup A, Schwartz NB. A member of a family of sulfate-activating enzymes causes murine brachymorphism. Proc Natl Acad Sci U S A. 1998;95:8681–5. doi: 10.1073/pnas.95.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ul Haque MF, King LM, Krakow D, Cantor RM, Rusiniak ME, Swank RT, Superti-Furga A, Haque S, Abbas H, Ahmad W, Ahmad M, Cohn DH. Mutations in orthologous genes in human spondyloepimetaphyseal dysplasia and the brachymorphic mouse. Nat Genet. 1998;20:157–62. doi: 10.1038/2458. [DOI] [PubMed] [Google Scholar]
- 9.Ikeda T, Mabuchi A, Fukuda A, Hiraoka H, Kawakami A, Yamamoto S, Machida H, Takatori Y, Kawaguchi H, Nakamura K, Ikegawa S. Identification of sequence polymorphisms in two sulfation-related genes, PAPSS2 and SLC26A2, and an association analysis with knee osteoarthritis. J Hum Genet. 2001;46:538–43. doi: 10.1007/s100380170036. [DOI] [PubMed] [Google Scholar]
- 10.Ford-Hutchinson AF, Ali Z, Seerattan RA, Cooper DM, Hallgrimsson B, Salo PT, Jirik FR. Degenerative knee joint disease in mice lacking 3′-phosphoadenosine 5′-phosphosulfate synthetase 2 (Papss2) activity: a putative model of human PAPSS2 deficiency-associated arthrosis. Osteoarthritis Cartilage. 2005;13:418–25. doi: 10.1016/j.joca.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 11.Lyle S, Ozeran JD, Stanczak J, Westley J, Schwartz NB. Intermediate channeling between ATP sulfurylase and adenosine 5′-phosphosulfate kinase from rat chondrosarcoma. Biochemistry. 1994;33:6822–7. doi: 10.1021/bi00188a010. [DOI] [PubMed] [Google Scholar]
- 12.Lansdon EB, Fisher AJ, Segel IH. Human 3′-phosphoadenosine 5′-phosphosulfate synthetase (isoform 1, brain): kinetic properties of the adenosine triphosphate sulfurylase and adenosine 5′-phosphosulfate kinase domains. Biochemistry. 2004;43:4356–65. doi: 10.1021/bi049827m. [DOI] [PubMed] [Google Scholar]
- 13.Harjes S, Bayer P, Scheidig AJ. The crystal structure of human PAPS synthetase 1 reveals asymmetry in substrate binding. J Mol Biol. 2005;347:623–35. doi: 10.1016/j.jmb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Deyrup AT, Krishnan S, Singh B, Schwartz NB. Activity and stability of recombinant bifunctional rearranged and monofunctional domains of ATP-sulfurylase and adenosine 5′-phosphosulfate kinase. J Biol Chem. 1999;274:10751–7. doi: 10.1074/jbc.274.16.10751. [DOI] [PubMed] [Google Scholar]
- 15.Venkatachalam KV. Human 3′-phosphoadenosine 5′-phosphosulfate (PAPS) synthase: biochemistry, molecular biology and genetic deficiency. IUBMB Life. 2003;55:1–11. doi: 10.1080/1521654031000072148. [DOI] [PubMed] [Google Scholar]
- 16.Renosto F, Seubert PA, Segel IH. Adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Purification and kinetic characterization. J Biol Chem. 1984;259:2113–23. [PubMed] [Google Scholar]
- 17.Renosto F, Seubert PA, Knudson P, Segel IH. Adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Determining ligand dissociation constants of binary and ternary complexes from the kinetics of enzyme inactivation. J Biol Chem. 1985;260:11903–13. [PubMed] [Google Scholar]
- 18.MacRae IJ, Segel IH, Fisher AJ. Crystal structure of adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry. 2000;39:1613–21. doi: 10.1021/bi9924157. [DOI] [PubMed] [Google Scholar]
- 19.Lansdon EB, Segel IH, Fisher AJ. Ligand-induced structural changes in adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry. 2002;41:13672–80. doi: 10.1021/bi026556b. [DOI] [PubMed] [Google Scholar]
- 20.Lyle S, Geller DH, Ng K, Stanczak J, Westley J, Schwartz NB. Kinetic mechanism of adenosine 5′-phosphosulphate kinase from rat chondrosarcoma. Biochem J. 1994;301 ( Pt 2):355–9. doi: 10.1042/bj3010355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lillig CH, Schiffmann S, Berndt C, Berken A, Tischka R, Schwenn JD. Molecular and catalytic properties of Arabidopsis thaliana adenylyl sulfate (APS)-kinase. Arch Biochem Biophys. 2001;392:303–10. doi: 10.1006/abbi.2001.2453. [DOI] [PubMed] [Google Scholar]
- 22.Sekulic N, Shuvalova L, Spangenberg O, Konrad M, Lavie A. Structural characterization of the closed conformation of mouse guanylate kinase. J Biol Chem. 2002;277:30236–43. doi: 10.1074/jbc.M204668200. [DOI] [PubMed] [Google Scholar]
- 23.Singh B, Schwartz NB. Identification and functional characterization of the novel BM-motif in the murine phosphoadenosine phosphosulfate (PAPS) synthetase. J Biol Chem. 2003;278:71–5. doi: 10.1074/jbc.M206688200. [DOI] [PubMed] [Google Scholar]
- 24.Schriek U, Schwenn JD. Properties of the purified APS-kinase from Escherichia coli and Saccharomyces cerevisiae. Arch Microbiol. 1986;145:32–8. doi: 10.1007/BF00413024. [DOI] [PubMed] [Google Scholar]
- 25.MacRae IJ, Segel IH. Adenosine 5′-phosphosulfate (APS) kinase: diagnosing the mechanism of substrate inhibition. Arch Biochem Biophys. 1999;361:277–82. doi: 10.1006/abbi.1998.0993. [DOI] [PubMed] [Google Scholar]
- 26.Lee S, Leustek T. APS kinase from Arabidopsis thaliana: genomic organization, expression, and kinetic analysis of the recombinant enzyme. Biochem Biophys Res Commun. 1998;247:171–5. doi: 10.1006/bbrc.1998.8751. [DOI] [PubMed] [Google Scholar]
- 27.Renosto F, Martin RL, Segel IH. Adenosine-5′-phosphosulfate kinase from Penicillium chrysogenum: ligand binding properties and the mechanism of substrate inhibition. Arch Biochem Biophys. 1991;284:30–4. doi: 10.1016/0003-9861(91)90258-k. [DOI] [PubMed] [Google Scholar]
- 28.Hommes FA, Moss L, Touchton J. Purification and some properties of liver adenylylsulfate kinase. Biochim Biophys Acta. 1987;924:270–5. doi: 10.1016/0304-4165(87)90022-5. [DOI] [PubMed] [Google Scholar]
- 29.Satishchandran C, Markham GD. Adenosine-5′-phosphosulfate kinase from Escherichia coli K12. Purification, characterization, and identification of a phosphorylated enzyme intermediate. J Biol Chem. 1989;264:15012–21. [PubMed] [Google Scholar]
- 30.Deyrup AT, Krishnan S, Cockburn BN, Schwartz NB. Deletion and site-directed mutagenesis of the ATP-binding motif (P-loop) in the bifunctional murine ATP-sulfurylase/adenosine 5′-phosphosulfate kinase enzyme. J Biol Chem. 1998;273:9450–6. doi: 10.1074/jbc.273.16.9450. [DOI] [PubMed] [Google Scholar]
- 31.Venkatachalam KV, Akita H, Strott CA. Molecular cloning, expression, and characterization of human bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase and its functional domains. J Biol Chem. 1998;273:19311–20. doi: 10.1074/jbc.273.30.19311. [DOI] [PubMed] [Google Scholar]
- 32.Brundiers R, Lavie A, Veit T, Reinstein J, Schlichting I, Ostermann N, Goody RS, Konrad M. Modifying human thymidylate kinase to potentiate azidothymidine activation. J Biol Chem. 1999;274:35289–92. doi: 10.1074/jbc.274.50.35289. [DOI] [PubMed] [Google Scholar]
- 33.Kabash W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.