

Abstract
Phosphonatase functions in the 2-aminoethylphosphonate (AEP) degradation pathway of bacteria, catalyzing the hydrolysis of the C-P bond in phosphonoacetaldehyde (Pald) via formation of a bi-covalent Lys53ethylenamine/Asp12 aspartylphosphate intermediate. Because phosphonatase is a member of the haloacid dehalogenase superfamily, a family predominantly comprised of phosphatases, the question arises as to how this new catalytic activity evolved. The source of general acid-base catalysis for Schiff-base formation and aspartylphosphate hydrolysis was probed using pH-rate profile analysis of active-site mutants and X-ray crystallographic analysis of modified forms of the enzyme. The 2.9 Å X-ray crystal structure of the mutant Lys53Arg complexed with Mg+2 and phosphate shows that the equilibrium between the open and the closed conformation is disrupted, favoring the open conformation. Thus, proton dissociation from the cap domain Lys53 is required for cap domain-core domain closure. The likely recipient of the Lys53 proton is a water-His56 pair that serves to relay the proton to the carbonyl oxygen of the phosphonoacetaldehyde (Pald) substrate upon addition of the Lys53. The pH-rate profile analysis of active-site mutants was carried out to test this proposal. The proximal core domain residues Cys22 and Tyr128 were ruled out, and the role of cap domain His56 was supported by the results. The X-ray crystallographic structure of wild-type phosphonatase reduced with NaBH4 in the presence of Pald was determined at 2.4 Å resolution to reveal Nε-ethyl-Lys53 juxtaposed with a sulfate ligand bound in the phosphate site. The position of the C(2) of the N-ethyl group in this structure is consistent with the hypothesis that the cap domain Nε-ethylenamine-Lys53 functions as a general base in the hydrolysis of the aspartylphosphate bi-covalent enzyme intermediate. Because the enzyme residues proposed to play a key role in P-C bond cleavage are localized on the cap domain, this domain appears to have evolved to support the diversification of the HAD phosphatase core domain for catalysis of hydrolytic P-C bond cleavage.
Keywords: Schiff-base, phosphoryl transfer, phosphoenzyme, phosphoaspartate, structural enzymology, HAD superfamily, general acid catalysis, general base catalysis, enamine, phosphate ester hydrolysis, phosphonatase, phosphonate, cap domain, core domain, electrophilic catalysis
Introduction
Phosphonoacetaldehyde hydrolase (phosphonatase) catalyzes the hydrolysis of phosphonoacetaldehyde (Pald) to acetaldehyde and orthophosphate (Figure 1) [1]. Phosphonatase functions in the 2-aminoethylphosphonate (AEP) degradation pathway operative in strains of bacteria adapted for the use of the natural phosphonate AEP as a source of carbon, phosphorus, and nitrogen [2–4]. Phosphonates, which contain a P-C bond, differ in chemical stability from the more common phosphate monoesters, which possess a P-O-C bond linkage [5–7]. The catalytic pathways for their hydrolysis also differ [8, 9]. We were thus intrigued by the discovery that phosphonatase is a member of the haloacid dehalogenase superfamily (HADSF) [10], a family predominantly comprised of phosphatases.
Figure 1.
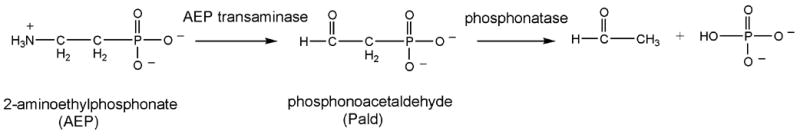
The bacterial AEP degradation pathway.
The vast majority of the enzymes in the HAD family are phosphatases[11, 12], followed by ATPases [13, 14] and phosphomutases [15]. The HAD phosphotransferases conserve a core catalytic domain that positions an Asp residue to function in nucleophilic catalysis[15, 16], and a second Asp residue to function in general acid/base catalysis. The second Asp protonates the leaving group oxygen in the first half of the reaction and deprotonates the water or alcohol nucleophile for reaction with the phosphoenzyme intermediate in the second half of the reaction. In addition, the catalytic domain provides a Lys/Arg residue to bind the substrate phosphoryl group and position the Asp nucleophile, and two carboxylate groups to assist in the binding of the Mg+2 cofactor (Figure 2). The Mg+2 cofactor forms a coordinate with the Asp nucleophile and the substrate phosphoryl group, thereby orienting and shielding the negative charges in the ground state and transition state [17]. The ATPase members of the HADSF, which do not require protonation of the ADP leaving group, and which require a comparatively slow rate of hydrolysis of the phosphoenzyme intermediate (allowing a hydrolysis coupled conformational change), have become specialized through the replacement of the Asp general acid/base by a Thr residue [18]. In this paper, we examine the extent to which the HAD phosphatase catalytic scaffold has diverged to support catalysis of hydrolytic cleavage of the phosphonate P-C bond by the enzyme phosphonatase.
Figure 2.
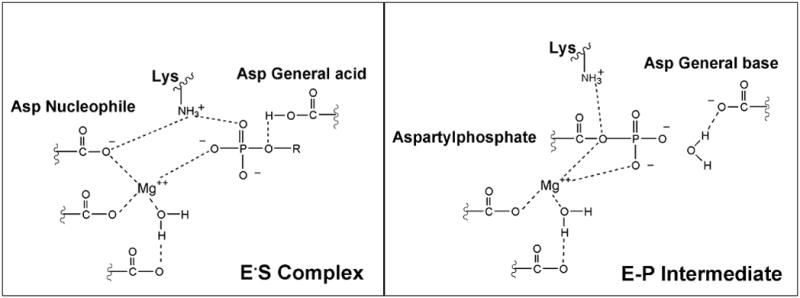
Schematic depicting the function of the conserved catalytic scaffold of the HAD phosphatases in phosphoryl transfer from substrate (left panel) and in the transfer of the phosphoryl group from the phosphoenzyme intermediate (E-P) to water (right panel).
Earlier work showed that catalysis of P-C bond cleavage of Pald by phosphonatase proceeds via an iminium ion (Schiff base) intermediate formed between the enzyme Lys53 and the substrate carbonyl group [8, 10, 19]. Thus, it was known that phosphonatase possesses the catalytic machinery for Schiff-base formation. The Schiff-base forming reaction between a substrate carbonyl and an enzyme active-site Lys residue is a common form of electrophilic catalysis employed by enzymes catalyzing aldol condensations or transketolization reactions [20]. The X-ray structure of phosphonatase bound with substrate or product analogs revealed that this HADSF member [17, 21], like the HADSF members phosphomutase β-PGM [22, 23] and phosphatase phosphoserine hydrolase [24–28], possesses a common phosphatase core domain. Moreover, the catalytic scaffold of the core domain is highly conserved among these enzymes. The active site is formed at the interface of the core domain and the cap domain, tethered by flexible linkers. The cap domain associates with the core domain to enclose the active site for catalysis and dissociates to allow product dissociation and substrate binding (Figure 3).
Figure 3.
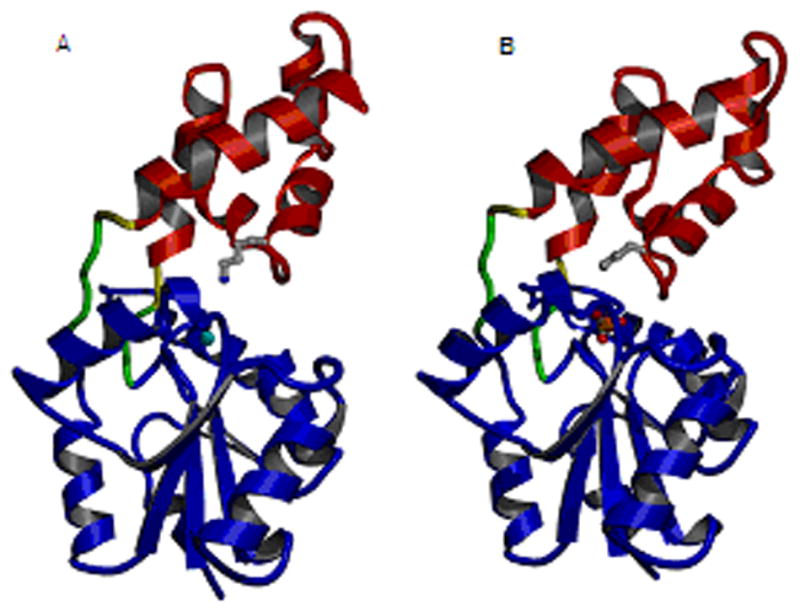
Ribbon diagram of Bacillus cereus phosphonatase [17] depicting a monomer from the (A) enzyme-Mg2+ complex in the “open conformation” and (B) the enzyme-Mg2+-tungstate complex in the “closed conformation”. The protein is differentially colored as follows: red, cap domain; blue, core domain; green, linkers; yellow, hinges; gray, Lys53; cyan, Mg2+; and orange, tungstate.
The Schiff-base forming Lys53 of phosphonatase is located on the surface of the cap domain, at the interface with the core domain. Inspection of the phosphonatase core domain confirmed that the Asp general acid/base, conserved among the HAD phosphatases, is replaced with Ala14. Thus, whereas the phosphonatase cap domain acquired the Lys for Schiff-base formation, the core domain lost the Asp for protonation of the leaving group, and deprotonation of the water nucleophile. Remarkably, the catalytic turnover rate for phosphonatase (15 s−1) [19] is comparable to that of the ATPases and the phosphatases reported to date (~10 s−1) [29–36].
Schiff-base formation, which proceeds via a carbinolamine intermediate, requires deprotonation of the Lys53 and protonation of the Pald C=O [37]. Based on inspection of the phosphonatase active site modeled with Pald (Figure 4, the Pald position is based on the position of the inhibitor vinyl sulfonate since the position of Pald in complex with the Asp12Ala mutant is slightly shifted [21]), a hydrogen bond network existing between Lys53, a water molecule, His56 and the Pald C=O was suggested as a possible means for proton transfer between enzyme and substrate. Attack of the Asp12 nucleophile at the phosphorus of the Schiff-base intermediate displaces Lys53N-ethyleneamine as the leaving group (Figure 5). Protonation of the leaving group is therefore not required in this step, consistent with presence of Ala in the general acid/base position of the HAD catalytic scaffold. However, general base catalysis of the hydrolysis of the aspartylphosphate intermediate must therefore be performed by the Lys53N-ethylenamine as there is no other residue properly positioned to fufill this function.
Figure 4.
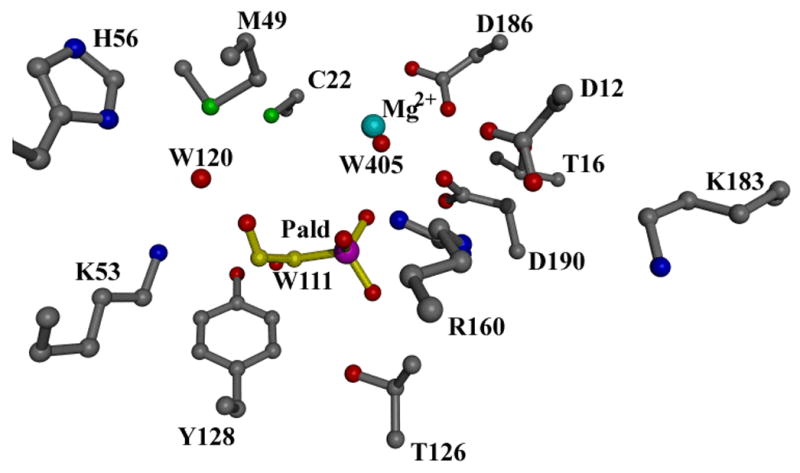
The active site model generated by replacing vinyl sulfonate with Pald in the structure of wild-type phosphonatase-Mg2+-vinyl sulfonate and then docking W111, W120 from the active site of the tungstate-bound wild type enzyme (W405 is a Mg2+ ligand).
Figure 5.
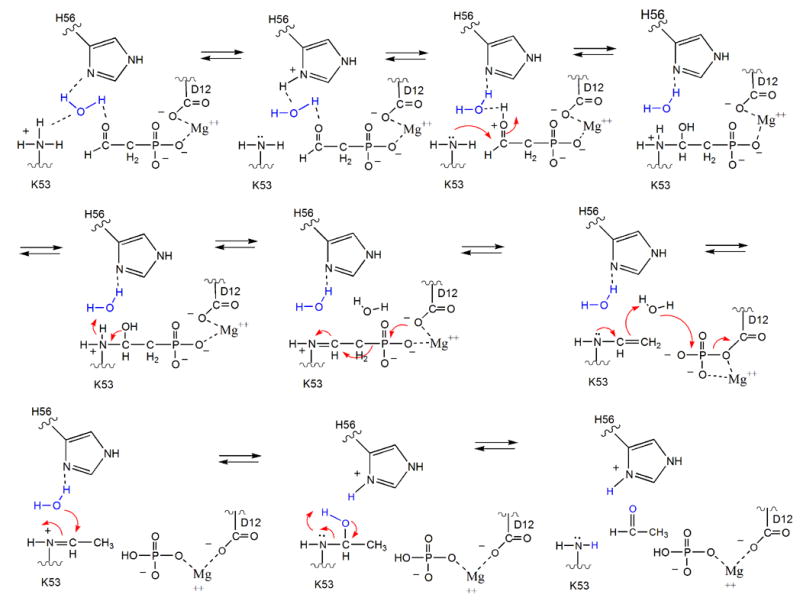
Schematic of the proposed phosphonatase catalytic mechanism.
Whereas the mechanism of proton shuttling outlined in Figure 5 is reasonable, it remained unsupported by experimentation. Herein, we report the results of pH-rate profile analysis of active-site mutants and the X-ray crystal structure of Lys53Arg phosphonatase complexed with Mg+2 and phosphate, which evidence Schiff-base formation via proton relay between the Lys53 and the Pald C=O. In addition, we report the structure of the borohydride-reduced product Schiff-base intermediate, which evidences the role of the Lys53N-ethyleneaimine in activation of the water nucleophile for aspartylphosphate hydrolysis. These results support the hypothesis that the phosphonatase cap domain is responsible for the successful adaptation of the HAD phosphatase core domain for catalysis of hydrolytic P-C bond cleavage in Pald.
Materials and Methods
Materials
E. coli JM109 competent cells were purchased from Promega and the expression vector pKK223-3 was purchased from Pharmacia Biotech. DNA manipulation enzymes were purchased from New England Biolabs. The phosphonatase mutants were generated using site-directed mutagenesis as previously described [21]. DNA sequencing was carried out at the Center for Genetics in Medicine, University of New Mexico School of Medicine. The wild-type and mutant phosphonatase proteins were purified as previously described [10]. Pald was synthesized according to the published procedure [38]. Yeast alcohol dehydrogenase, NADH, and all buffers were purchased from Sigma.
Enzyme Assay
Phosphonatase activity was measured at 25°C using the coupled, spectrophotometric assay described previously [21]. In this assay, acetaldehyde formation is coupled to NADH oxidation via the alcohol dehydrogenase (ADH) catalyzed reaction. Each 1 ml reaction mixture contained 50 mM K+HEPES (pH 7.5), 5 mM MgCl2, 0.15 mM NADH, 10 units ADH and various concentrations of Pald (0.5 to 5–10 Km). The reactions were monitored at 340 nm (Δε = 6.2 mM−1cm−1). The steady state kinetic parameters, kcat and Km, were obtained by analyzing the initial velocity data using equation 1 and the KinetAsystI computer program developed by Cleland [39]:
| (1) |
where V0 is the initial velocity, Km is the Michealis constant for the substrate, Vmax is the maximal velocity and [S] is the concentration of the substrate. The kcat was calculated from equation 2:
| (2) |
The enzyme concentration [E] was determined using the Bradford method [40].
pH Rate Profile Determinations
All reactions were carried out at 25°C using a universal buffer containing 20 mM MES, 30 mM HEPES, 30 mM CHES and 20 mM TAPS at a constant ionic strength attained using 0.2 M NaCl. The initial reaction rates were measured using the coupled assay as described above. The kcat and Km values were obtained using equations (1) and (2). At low pH (5.0~6.0) the background reaction of NADH was measured before initiating the phosphonatase reaction. The pH rate profile data were fitted to the equation 3.
| (3) |
where Y is the kcat/Km, H is the proton concentration of the reaction buffer, Ka and Kb are the apparent ionization constants of the acidic and basic groups, and C is the constant value of Y where it does not change with the solution pH.
Capture of the Phosphonatase-Schiff Base by Borohydride Reduction
Phosphonatase (25 μM) was incubated with 2 mM Pald in 100 mM Hepes (pH 8.0) containing 5 mM MgCl2 and 1 mM NaBH4 for 10 min at 0°C. The procedure was repeated to ensure complete inactivation of the enzyme (the first treatment caused 75% inactivation). The protein was concentrated in 1 mM Hepes pH 7.5, 0.1mM DTT and 10 mM MgCl2 in a Millipore Ultra-free concentrator to 15 mg/ml for crystallization.
Crystallization
Crystals of the reduced Schiff base-phosphonatase adduct were grown in solution containing 0.1 M NaCl, 0.1M Hepes pH 7.5 and 1.6 M ammonium sulfate by the hanging-drop vapor diffusion method. Crystals of the K53R mutant were grown in solution containing 50 mM KH2PO4 and 17% PEG 8000 by the hanging-drop vapor diffusion method.
The data sets were collected to 2.9 Å and 2.5 Å, respectively, using a Rigaku RU-300 X-ray generator equipped with an RAXIS-II area detector. Data collection for the K53R phosphonatase crystals was carried out at ambient temperature. Data collection for the reduced Schiff base-phosphonatase crystals was carried out at −180 ºC using 20% glycerol as a cryoprotectant. The data collection statistics are summarized in Table 1. The DENZO and SCALEPACK packages [41] were used for data indexing, reduction, and scaling. The K53R phosphonatase crystals are rhombohedral (R32 space group) with unit-cell dimensions a= 251.73 b= 251.73 c= 156.34. The reduced Schiff base-phosphonatase crystals are monoclinic belonging to space group C2 with unit-cell dimensions a=159.82 b=64.02 c=98.34 β=123.517°. A Matthews’s coefficient calculation suggested five and two monomers in the asymmetric units, respectively.
Table 1.
Crystallographic Data Collection and refinement statistics
| K53R | NaBH4 reduced protein | |
|---|---|---|
| Unit-cell dimensions (Å) | a = b= 251.73
c= 156.34 |
a = 159.82
b= 64.02 c= 98.34 β = 123.517 |
| Space group | R32 | C2 |
| Wavelength (Å) | 1.54 Å | 1.54 Å |
| Resolution range (Å) | 100-2.9 | 100-2.4 |
| Total/unique reflections | 368969/51994 | 238424/28942 |
| Completeness (%)* | 99.9 (99.5) | 92.8 (54) |
| I/σ (I) | 9 (2) | 8 (2) |
| Rmerge∞ (%) | 12 (30) | 19 (32) |
| Number of protein atoms/asymmetric unit | 10260 | 4082 |
| Number of reflections (working/free set) | 35598/4000 | 17839/1945 |
| Rwork1/Rfree 2 (%) | 25.27/26.6 | 23.0/27.2 |
| Average B-factors (Å2) | 38 | 27.9 |
| Amino acid residues | 36 | 21 |
| Mg2+ | 23 | 2 |
| Sulfate/phosphate | 30 | 31 |
| Water | 37 | 31 |
| Bond length (Å) | 0.007 | 0.008 |
| Angles (º) | 1.5 | 1.4 |
Statistics for the outermost resolution shell are shown in parentheses.
Rmerge = ∑hkl ∑i| Ihkl, i-<Ihkl> I | / ∑hkl∑i | Ihkl, i |, where <Ihkl> is the mean intensity of the multiple Ihkl, i observations for symmetry related reflections.
Rwork = ∑hkl|Fobs–Fcalc| / ∑hkl |Fobs|.
Rfree = ∑hkl ∑T|Fobs–Fcalc| / ∑hkl |Fobs|, where the test set T includes 10% of the data.
Structure Determination and Refinement
The molecular replacement method was used to determine the phases for both structures using wild-type phosphonatase as the model (1FEZ.pdb) in the program AMoRE [42]. The lowest R-factor and highest correlation coefficients were obtained using all “open” monomers to model the dimer of K53R phosphonatase and by using one “closed” monomer and one “open” monomer to model the dimer of the NaBH4 reduced Schiff base-phosphonatase. The resulting model solutions were subsequently subjected to rigid-body refinement in the CNS program suite [43], allowing independent movement of the cap and the core domains. Iterative rounds of minimization and simulated annealing using slow-cool torsional molecular dynamics followed by B-domain refinement and manual rebuilding were used to refine the structure in CNS. For statistical cross-validation purposes (calculation of Rfree, [44]) 10% of the data was excluded from refinement. Manual fitting was done using sigmaa weighted 2Fo - Fc and Fo - Fc electron density maps [45] in the graphics program O [46]. Refinement, using a MLF target was performed until Rfree ceased to decrease. Waters were added using the automated water-picking program in CNS. The model was checked using a 2fo-fc composite-omit map. The final model incorporated 257 out of 267 possible amino acids. Residues 1–4 at the N-terminus and residue 262–267 at the C-terminus were not visible in either structure in the electron-density map (also disordered in the wild-type model), and were omitted from the final model.
For the K53R phosphonatase structure refinement the Lys53 from the wild-type model was mutated to Ala for unbiased identification of the residue. In the later stages of refinement, based on the density, this residue was replaced with Arg53. The active site also contained strong density for one phosphate ion (from the crystallization conditions) and a Mg2+ cofactor from the protein solution. In the case of the NaBH4 reduced phosphonatase-Schiff base structure the electron density (contoured at 2 σ) connected to the Schiff-base Lys53 was observed in the active site of one monomer only. Additional densities corresponding to a sulfate ion (from the crystallization conditions) and a Mg2+ cofactor, respectively, were present in the active site of both monomers. The final Rfactor for the model was 27.5% (Rfree) and 25.9% (Rcryst) for the K53R structure and 26.8% (Rfree) and 22.7% (Rcryst) for the NaBH4 reduced structure. Analysis of the Ramachandran plot, as defined by PROCHECK [47], showed a good final model for both structures. Refinement and final model statistics are compiled in Table 1.
Results and Discussion
Schiff Base Formation: pH Rate Profile Analysis
The proposed model for Schiff base formation in phosphonatase catalysis is depicted in Figure 5. In this model the proton that is carried into the active site by Lys53 (pKa = 9.3; [48]) upon cap closure becomes delocalized by His56 and ultimately transferred to the Pald C=O via a hydrogen bond network that incorporates water120. The active configuration of the free enzyme should thus include the protonated form of Lys53 and the neutral (unprotonated) form of His56. A priori, it could be argued that the logkcat/Km versus pH profile for phosphonatase catalysis (first reported in reference (18); and again in Figure 6A) reflects loss of activity at low pH due to the protonation of His56 and loss of activity at high pH due to the deprotonation of the Lys53. However, the logkcat/Km versus pH profiles measured for phosphonatase active site mutants (described below) indicate that this is a simplistic interpretation.
Figure 6.
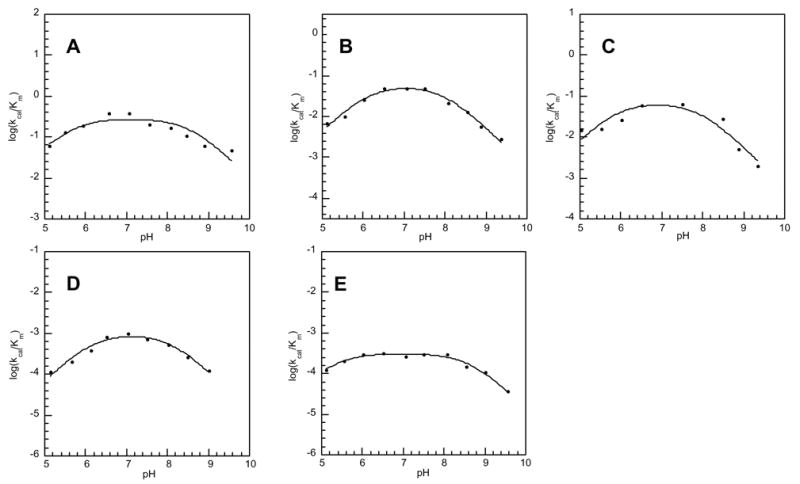
The kcat/Km pH profile for catalysis of Pald hydrolysis by wild-type (A) phosphonatase and the Cys22Ser (apparent pKa1 = 6.2 and apparent pKa2 = 8.1) (B), Cys22Ser/Tyr128Phe (apparent pKa1 = 6.4 and apparent pKa2 = 8.5) (C), His56Gln (apparent pKa1 = 6.6 and apparent pKa2 = 8.2) (D), and His56Ala (apparent pKa1 = 5.3 and apparent pKa2 = 8.7) (E) active-site mutants.
A more comprehensive view of the pH dependence of the kcat/Km is derived from a composite of the ionization of several groups (substrate and enzyme). Therefore, the apparent pKa values calculated from these data using equation 3 (reported in the legend to Figure 6) cannot be assigned to a specific acid group. On the other hand, we might expect that the replacement of an essential enzyme acid or base residue by site-directed mutagenesis would noticeably perturb the logkcat/Km versus pH profile. Whereas Table 2 provides the kcat/Km values for wild-type and mutant phosphonatase measured at a single pH value, the pH profiles shown in Figure 6, provide a comparison over the entire pH range. The kinetic constants measured for the Cys22Ser, Cys22Ser/Tyr128Phe, His56Gln and His56Ala active-site mutants at pH 7.5 indicate that the core domain active site residues Cys22 and Tyr128 contribute to the function of the catalytic site but do not appear, based on only a 10-fold reduction in kcat and kcat/Km values to participate in acid-base catalysis (Table 2). The log kcat/Km versus pH profiles of the Cys22Ser and Cys22Ser/Tyr128Phe mutants shown in Figures 6B and 6C, respectively are similar, but not identical, to that of the wild-type enzyme.
Table 2.
The Steady–State Kinetic Constants for Wild-type and Mutant Phosphonatase Measured at pH 7.5 and 25 °C.a
| Enzyme | kcat (s−1) | Km (μM) | kcat/Km (s−1 μM−1) |
|---|---|---|---|
| Wild-typeb | 1.5 ± 0.1 x 101 | 33 ± 2 | 5.0 x 10−1 |
| C22Sb | 2.26 ± 0.09 | 33 ± 5 | 6.9 x 10−2 |
| Y128F/C22Sb | 2.11 ± 0.03 | 33 ± 2 | 6.4 x 10−2 |
| H56Ab | 7.5 ± 0.1 x 10−2 | 145 ± 6 | 5.2 x 10−4 |
| H56Q | 3.6 ± 0.1 x 10−1 | 170 ± 10 | 2.0 x 10−3 |
Reactions contained Pald, 10 mM MgCl2, 0.15 mM NADH and 5 units alcohol dehydrogenase in 50 mM K+Hepes (pH 7.5, 25 °C). The concentration of phosphonatase used was in the range of 0.02-2 μM, depending on the mutant studied.
Reported in Morais et al [21].
The replacement of the cap domain His56 with Ala or Gln impairs catalytic efficiency to a greater extent than does the replacement of the core-domain active site residues Cys22 and Tyr128 (Table 2). The kcat of the His56Ala mutant is 200-fold less than that of the wild-type enzyme and the kcat/Km is 1000-fold less. The His56Gln mutant is also less active than the wild-type phosphonatase: the kcat is decreased 42-fold and the kcat/Km is decreased 250–fold. The log kcat/Km versus pH profiles of the His56Gln vs His56Ala mutants are shown in Figures 6D and 6E, respectively. The pH-rate profile of the His56Ala mutant differs to the greatest extent from that of the wild-type phosphonatase. In particular, the drop in activity below pH 7 observed in the wild-type enzyme and the Cys22Ser, Cys22SerTyr128Phe and His56Gln mutants, is minimal in the His56Ala mutant. The suppression of the response to increasing acidity might be attributed to the loss of that function carried out by His56, which can be maintained by Gln but not by Ala.
In Figure 7, we illustrate the hydrogen-bond network that surrounds the His56 imidazole side chain in the substrate bound, catalytically active form of the enzyme as suggested by the X-ray crystallographic data. Substitution of His56 with Gln might not necessarily interrupt this hydrogen bond network because the functions of the His imidazole nitrogens could conceivably be assumed by the Gln side chain carbonyl and amide nitrogen, respectively. On the other hand, the methyl side chain of the substituted Ala in the His56Ala mutant would certainly not support a hydrogen bond network. To the extent that this putative network is important for catalysis in the wild-type phosphonatase, we anticipate a loss in catalytic activity upon its disruption.
Figure 7.
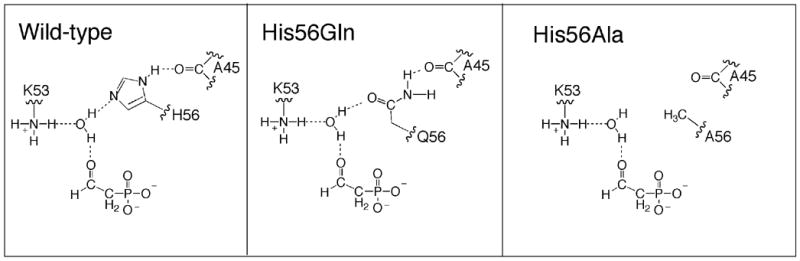
Schematic representation of the hydrogen-bond network in wild-type phosphonatase and in the His56Gln and His56Ala mutants.
Impact of Lys53 Substitution on Cap Closure: Structure of the Lys53Arg Mutant Complexed with Phosphate and Mg+2
The importance of delocalization of the proton on Lys53 in catalysis was probed by substituting Lys53 with Arg and determining whether or not the substitution destabilizes the cap closed conformation required for catalytic turnover. As illustrated in Figure 3 of the Introduction, phosphonatase cycles between an open conformation that allows substrate to bind and product to dissociate, and a closed conformation that is required for catalysis. When the cap closes over the active site, Lys53 enters the catalytic site and engages in the hydrogen bond network represented in Figures 5 and 7. Previous studies have shown that bound substrate or product ligands stabilize the cap closed conformation by an electrostatically driven induced-fit mechanism [48]. Crystallographic analyses of liganded and unliganded phosphonatase have captured the open conformation in the apoenzyme but only the closed conformation in the liganded enzyme [17, 21]. If the delocalization of the Lys53 proton is an integral part of the catalytically active conformation then we might expect that the substitution of Lys53 with Arg will disrupt the electrostatic balance and thus destabilize the cap closed conformation. Accordingly, we carried out the structure determination of the phosphonatase Lys53Arg mutant crystallized in the presence of the product phosphate and the Mg+2 cofactor. As might be expected, substitution of the Schiff-base forming Lys with Arg diminished catalytic activity to background levels in the enzymatic assay.
Crystals of Lys53Arg were non-isomorphous with the wild-type crystals. There are five monomers (2½ dimers) in the asymmetric unit and the physiological dimer interface is retained (one dimer is formed by crystallographic symmetry). The overall fold of the K53R mutant is no different from that of the wild-type enzyme, which is characterized by a distinct α-helical cap domain and α/β core domain. NCS restraints were applied during crystallographic refinement, thus all monomers are the same in their overall conformation (rmsd of 0.07Å over all Cα atoms). Figure 8A depicts three structures: wild-type open, wild-type closed and the K53R mutant wherein the core domains are superimposed to show the relative dispositions of the cap and core domains. The calculated values for the rmsd between the K53R monomer Cα positions, and those of the wild type open and closed conformation monomers, are 0.77Å and 1.02Å respectively.
Figure 8.
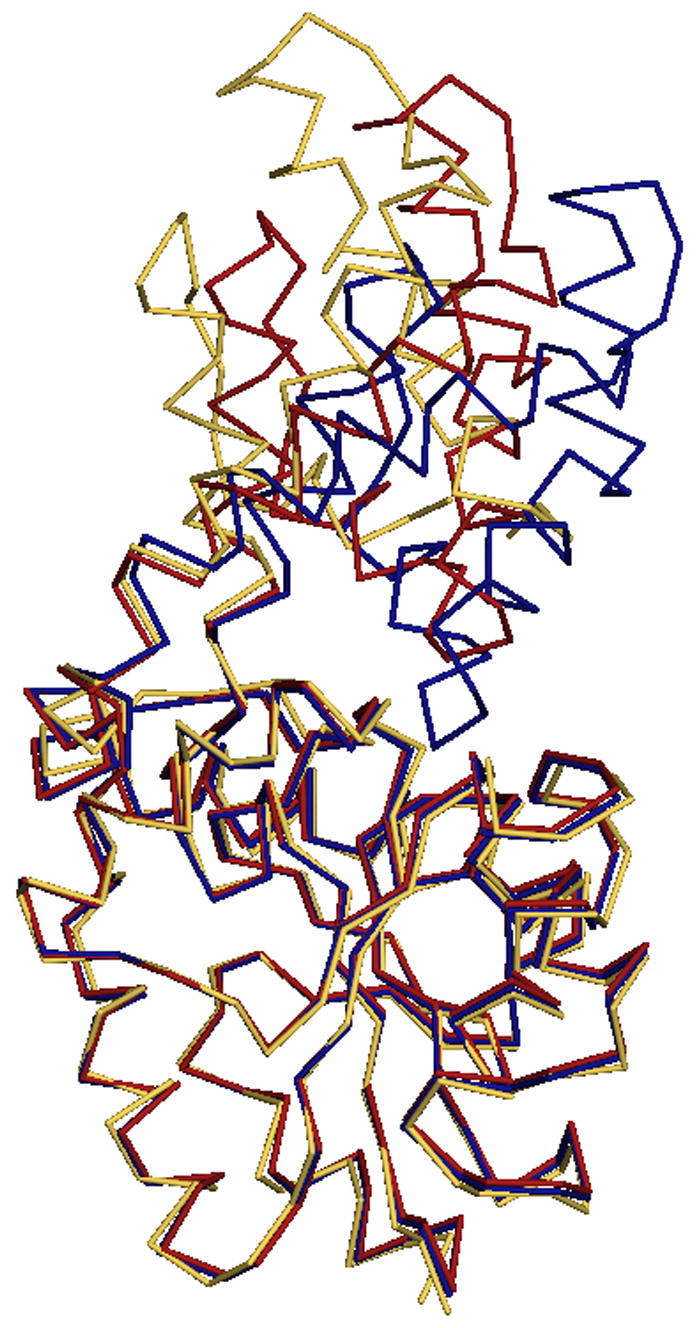
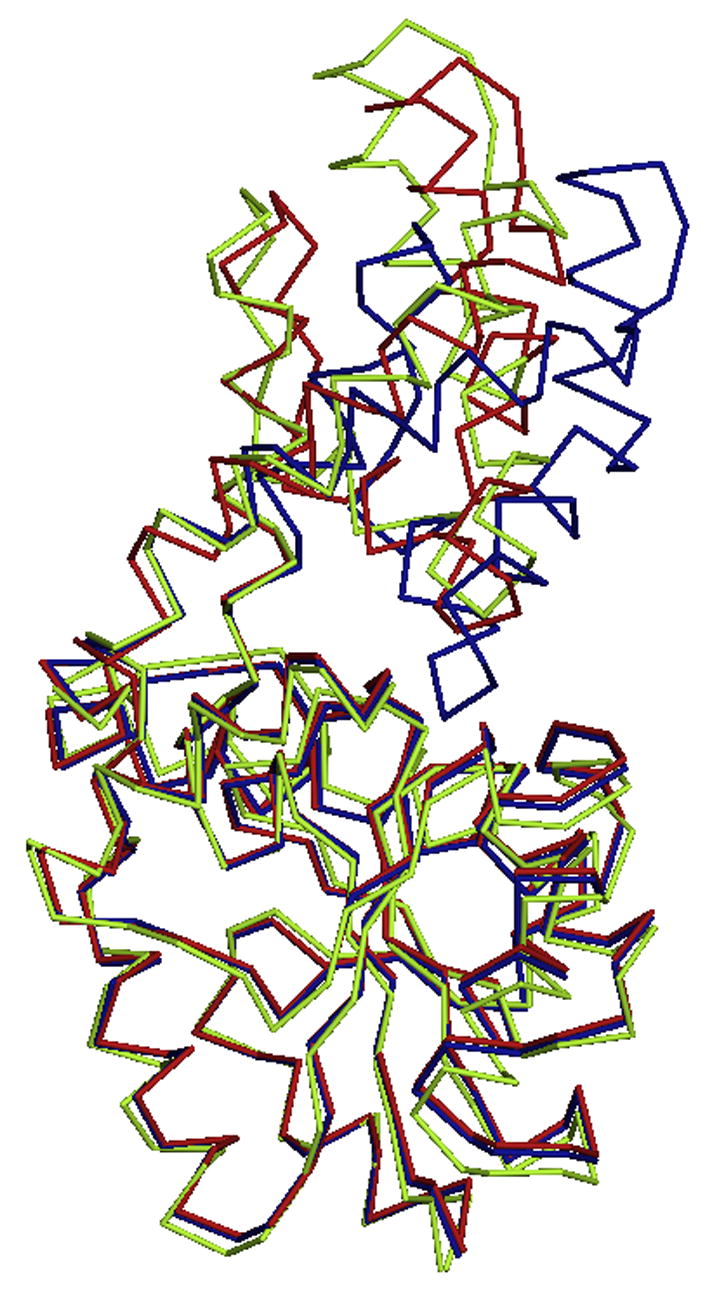
Superposition of the mutant and modified Cα traces of phosphonatase with wild type. (A) K53R Cα trace (yellow) with the open (red) and closed (blue) wild-type monomers. (B) The NaBH4 reduced phosphonatase backbone (green) with the open (red) and closed (blue) wild-type monomers.
The only significant difference in the wild-type and mutant structures is the angle by which the cap and core domains are disposed relative to one another via rigid-body movements. Specifically, the angle of rotation through the hinge axis (the linker connecting the cap and the core domain) is 22.35° between the wild-type open and closed monomers, while it is 42.88° between the K53R and wild type closed monomer (calculated with DYNDOM [49]). There is a slight change in the relative orientation of helices 2 and 3 of the cap domain. Because these two helices are connected by the “substrate-specificity loop” (residues 51–54) [50], the disposition of the nearby residues that contribute to the domain-domain interface, and that contribute functional groups to the catalytic site, were examined. Importantly, mutation of Lys53 to Arg results in only a slight structural rearrangement of the residues in the loop region, and all interactions between the protein and the ligands (Mg2+ and phosphate) are maintained in the K53R mutant as compared to the wild-type enzyme. Thus, although the open conformation observed with the K53R mutant is more open than that observed for the wild-type phosphonatase there is no evidence of a gross change in the active site structure. The loss of activity stems from the inability of the guanidinium group of the Arg residue in the K53R mutant to participate in Schiff-base formation, and not from large alterations in the enzyme conformation.
The fact that the phosphate bound K53R phosphonatase is observed in the cap-open conformation in the crystal structure is of special interest. In wild-type phosphonatase, the binding of anionic ligands (e.g. phosphate, sulphate, vinylsulphate or tungstate) which mimic the substrate cause the monomer to attain a closed conformation in both the crystal [11, 17, 21] and in solution [48]. Thus, the open conformation observed with the K53R mutant suggests that the replacement of the Lys53 primary ammonium group with the Arg guanidinium group interferes with the ligand induced domain closure. This finding is consistent with the predicted dependence of cap closure and catalytic turnover on the delocalization of the Lys53 charge via the hydrogen bond network depicted in Figure 5.
Structure of the Lys53ethylenamine adduct
The hydrolysis of the bi-covalent Lys53ethylenamine/Asp12 aspartylphosphate intermediate to product requires that the Lys53ethylenamine undergoes protonation at the C(2) position (Figure 5). In parallel, a water molecule must be activated by deprotonation for in-line attack at the phosphorus of the aspartylphosphate. The structure of the product Schiff-base was examined to identify the putative acid/base residues involved in these events. In the presence of Pald, NaBH4 inactivates phosphonatase by reducing the Lys53ethylenamine group of the reaction intermediate accumulated at steady-state (the product Schiff-base intermediate), thereby preventing its hydrolysis [7]. Structure determination of crystals of the NaBH4 reduced phosphonatase indeed revealed the Lys53ethyl residue. Fortuitously, the crystallization medium (which contained 1.6 M ammonium sulfate but no added phosphate) allowed the binding of a sulfate ion to the phosphate-binding subsite of the active site, affording a view of an analogue of the bi-covalent Lys53ethylenamine/Asp12aspartylphosphate intermediate. This provided for the assessment of the overall conformational state of this intermediate complex as well as identification of the active site residues proximal to the Lys53ethylenamine and Asp12aspartylphosphate groups. In addition, the structure revealed ordered solvent molecules positioned to participate in the hydrolysis of the Lys53ethylenamine and Asp12aspartylphosphate groups, and thus an overall picture of the active site poised for the conversion of the bi-covalent intermediate to the acetaldehyde and phosphate products.
The 2.5 Å structure of the Lys53ethylphosphonatase-sulfate-Mg+2 complex revealed a single physiological dimer in each asymmetric unit, with one subunit assuming the cap-open conformation and the other subunit the cap-closed conformation. The closed conformation does not differ significantly from that previously determined for the unliganded closed enzyme, with an rmsd of 0.16 Å. The open monomer, on the other hand, is more open than previously observed for the open conformation of unliganded phosphonatase (Figure 8B), with an overall rmsd of 1.24 Å. The interactions at the dimer interface are unchanged from those of the wild-type mixed dimer.
The active site of the open monomer contained the N-ethylLys53, the Mg2+ cofactor and sulphate ion (from the crystallization conditions) (Figure 8B). The position of the sulfate (which mimics phosphate) is identical to that of the tungstate bound in wild-type phosphonatase (1FEZ.pdb) and the phosphate bound in the K53R phosphonatase (vide supra). Thus, the sulfate interactions can be interpreted as mimicking the interaction with the product phosphate in the reaction pathway. In addition to the sulfate electron density, there is a strong electron density connected to the K53 (Figure 9A) that can be fitted well to N-ethyl lysine. The residues forming ligands to the Mg2+ are the same as in the wild type phosphonatase and as described for the K53R mutant structure.
Figure 9.
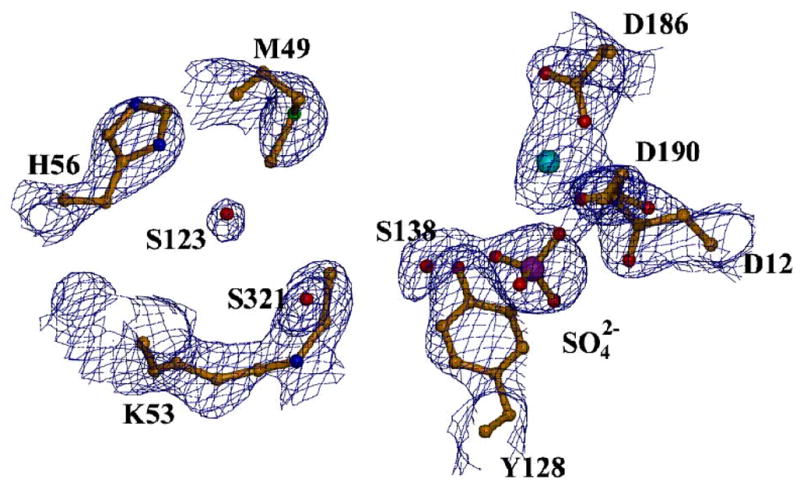
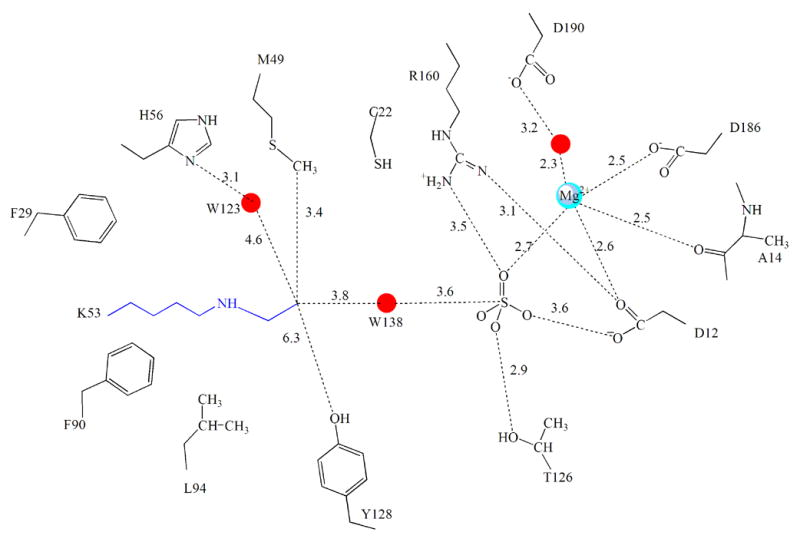
The active site of NaBH4 reduced phosphonatase. (A) Depicted with the electron density surrounding the modified Lys53. The active site residues (yellow) and ligands sulphate (orange) and cofactor Mg2+ (cyan) are depicted as ball-and -stick. The 2Fo-Fc electron density map (contoured at 1.5 σ) is shown as blue cages. (B). Chemdraw depiction of Ne-ethy-Lys53 and surrounding active-site residues (distances in Å).
The ethyl moiety of the N-ethyl-Lys53 is surrounded by a hydrophobic environment provided by the active-site residues Tyr128, Cys22 and Met49. It is interesting to note that in the open conformation of the unliganded enzyme, Met49 projects away from the active site but in the closed monomer it is positioned inside, to shield the catalytic site from solvent. In the cap-open structure of the N-ethyl-Lys53 phosphonatase, the Met49 is also positioned inside the active site where it forms hydrophobic contacts with the ethyl moiety. In addition, His56 (positioned within 6.8 Å of the ethyl-lysine group, see Figure 9B) forms a hydrogen bond to a water molecule, W123, near the C(2) of the ethyl group. This water molecule is also within hydrogen bonding distance of the Met49 sulfur atom and the backbone carbonyl of Leu51 (one of the residues of the linker connecting cap to core). Notably, positioned in-line to the bound sulfate and in between the sulfate and C(1) of the ethyl group of ethyl-Lys53 is the water molecule, W138. This water molecule is within hydrogen bonding distance of the sulfate and close to the terminal ethyl carbon (3.8 Å).
The structure of the N-ethyl-Lys53 phosphonatase sheds light on the mode of hydrolysis of the product Schiff-base generated from the second partial reaction of Pald hydrolysis. After C-P bond cleavage the C(2) of the displaced Lys53ethlyenamine must be protonated to form the Lys53ethyliminium ion prior to hydrolysis to acetaldehyde (Figure 5). Therefore, this step requires two catalysts: a general acid to protonate the C(2) carbon and a general base to deprotonate a water for attack. Within the active-site architecture there are two possibilities. Tyr 128 can act as a general acid to protonate at C(2) and the same Tyr 128, now ionized, may act as a general base and activate a water for addition to the C(1) of the Lys53ethyliminium ion. Alternatively, the Lys53ethylenamine itself removes a proton from water activating it for attack at the phosphoaspartate. The resulting Lys53ethyliminium might be hydrolyzed by the nucleophilic attack of a water molecule activated by the His56 nearby.
Through examination of the neighboring interactions of the N-ethyl-Lys53 adduct in the reduced structure, it can be seen that the position of Tyr128 is nearly 7Å away from the ethyl group of the Lys53N-ethyl moiety making it impossible for this residue to participate in the Lys53ethyliminium hydrolysis. Substitution of Tyr128 with Phe does not significantly alter catalytic efficiency (Table 2) nor the pH dependence of catalytic efficiency (Figure 6C). In fact, there is no enzymic residue in the correct position to deprotonate water 138 for hydrolysis of the phosphoenzyme.
Thus, it is most likely that the Lys53ethylenamine plays the key role in proton transfer required for the hydrolysis steps. Accordingly, the well-positioned enamine would abstract a proton from the water molecule oriented for in-line attack on the phosphorus of the phosphoaspartate. In the subsequent step of the reaction, water123, activated by His56, attacks the iminum carbon, thus initiating the release of acetaldehyde.
Conclusions
The presented structural results, together with pH-rate profile analysis, evidence the dependence of cap closure and catalytic turnover on the delocalization of the Lys53 charge via a hydrogen bond network between Lys53 and the substrate carbonyl, via a water molecule and His56. Additionally, based on both biochemical and structural data, our working hypothesis is that the Lys53ethlyenamine acts as the general base for Asp12aspartylphosphate hydrolysis. Such a mechanism would connect the reactivity of the Schiff-base subsite with that of the phosphoenzyme and thereby render the core domain Asp general acid/base of the HAD phosphotransferases obsolete. Indeed, in phosphonatase Ala replaces the Asp. We suggest that it is the specialization observed in the cap domain that is responsible for the successful adaptation of the HAD phosphatase core domain for catalysis of hydrolytic P-C bond cleavage.
Footnotes
This work was supported by NIH Grant GM61099 to K.N.A. and D.D-M.
The coordinates of the refined structures have been deposited with the Protein Data Bank, accession codes 2IOH and 2IOF for K53R and NaBH4 reduced phosphonatase, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.La Nauze JM, Rosenberg H, Shaw DC. Biochim Biophys Acta. 1970;212:332–50. doi: 10.1016/0005-2744(70)90214-7. [DOI] [PubMed] [Google Scholar]
- 2.La Nauze JM, Rosenberg H. Biochim Biophys Acta. 1968;165:438–47. doi: 10.1016/0304-4165(68)90223-7. [DOI] [PubMed] [Google Scholar]
- 3.Dumora C, Lacoste AM, Cassaigne A. Biochim Biophys Acta. 1989;997:193–8. doi: 10.1016/0167-4838(89)90186-6. [DOI] [PubMed] [Google Scholar]
- 4.Parker GF, Higgins TP, Hawkes T, Robson RL. J Bacteriol. 1999;181:389–95. doi: 10.1128/jb.181.2.389-395.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lad C, Williams NH, Wolfenden R. PNAS. 2003;100:5607–5610. doi: 10.1073/pnas.0631607100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Range K, McGrath MJ, Lopez X, York DM. J Am Chem Soc. 2004;126:1654–65. doi: 10.1021/ja0356277. [DOI] [PubMed] [Google Scholar]
- 7.Hildebrand RL. The Role of Phosphonates in Living Systems. CRC Press, Inc; Boca Raton, FL: 1983. [Google Scholar]
- 8.Olsen DB, Hepburn TW, Moos M, Mariano PS, Dunaway-Mariano D. Biochemistry. 1988;27:2229–34. doi: 10.1021/bi00406a063. [DOI] [PubMed] [Google Scholar]
- 9.Lee SL, Hepburn TW, Swartz WH, Ammon HL, Mariano PS, Dunaway-Mariano D. J Am Chem Soc. 1992;114:7346–7354. [Google Scholar]
- 10.Baker AS, Ciocci MJ, Metcalf WW, Kim J, Babbitt PC, Wanner BL, Martin BM, Dunaway-Mariano D. Biochemistry. 1998;37:9305–15. doi: 10.1021/bi972677d. [DOI] [PubMed] [Google Scholar]
- 11.Zhang G, Morais MC, Dai J, Zhang W, Dunaway-Mariano D, Allen KN. Biochemistry. 2004;43:4990–7. doi: 10.1021/bi036309n. [DOI] [PubMed] [Google Scholar]
- 12.Allen KN, Dunaway-Mariano D. Trends Biochem Sci. 2004;29:495–503. doi: 10.1016/j.tibs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Aravind L, Galperin MY, Koonin EV. Trends Biochem Sci. 1998;23:127–9. doi: 10.1016/s0968-0004(98)01189-x. [DOI] [PubMed] [Google Scholar]
- 14.Collet JF, van Schaftingen E, Stroobant V. Trends Biochem Sci. 1998;23:284. doi: 10.1016/s0968-0004(98)01252-3. [DOI] [PubMed] [Google Scholar]
- 15.Collet JF, Stroobant V, Pirard M, Delpierre G, Van Schaftingen E. J Biol Chem. 1998;273:14107–12. doi: 10.1074/jbc.273.23.14107. [DOI] [PubMed] [Google Scholar]
- 16.Collet JF, Stroobant V, Van Schaftingen E. Methods Enzymol. 2002;354:177–88. doi: 10.1016/s0076-6879(02)54014-3. [DOI] [PubMed] [Google Scholar]
- 17.Morais MC, Zhang W, Baker AS, Zhang G, Dunaway-Mariano D, Allen KN. Biochemistry. 2000;39:10385–96. doi: 10.1021/bi001171j. [DOI] [PubMed] [Google Scholar]
- 18.Lahiri SD, Zhang G, Radstrom P, Dunaway-Mariano D, Allen KN. Acta Crystallogr D Biol Crystallogr. 2002;58:324–6. doi: 10.1107/s0907444901019989. [DOI] [PubMed] [Google Scholar]
- 19.Olsen DB, Hepburn TW, Lee SL, Martin BM, Mariano PS, Dunaway-Mariano D. Arch Biochem Biophys. 1992;296:144–51. doi: 10.1016/0003-9861(92)90556-c. [DOI] [PubMed] [Google Scholar]
- 20.Voet D, Voet JG. Biochemistry. John Wiley & Sons; 1995. [DOI] [PubMed] [Google Scholar]
- 21.Morais MC, Zhang G, Zhang W, Olsen DB, Dunaway-Mariano D, Allen KN. J Biol Chem. 2004;279:9353–61. doi: 10.1074/jbc.M312345200. [DOI] [PubMed] [Google Scholar]
- 22.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. Biochemistry. 2002;41:8351–9. doi: 10.1021/bi0202373. [DOI] [PubMed] [Google Scholar]
- 23.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. Science. 2003:1082710. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- 24.Cho H, Wang W, Kim R, Yokota H, Damo S, Kim SH, Wemmer D, Kustu S, Yan D. Proc Natl Acad Sci U S A. 2001;98:8525–30. doi: 10.1073/pnas.131213698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim HY, Heo YS, Kim JH, Park MH, Moon J, Kim E, Kwon D, Yoon J, Shin D, Jeong EJ, Park SY, Lee TG, Jeon YH, Ro S, Cho JM, Hwang KY. J Biol Chem. 2002;277:46651–8. doi: 10.1074/jbc.M204866200. [DOI] [PubMed] [Google Scholar]
- 26.Peeraer Y, Rabijns A, Verboven C, Collet JF, Van Schaftingen E, De Ranter C. Acta Crystallogr D Biol Crystallogr. 2003;59:971–7. doi: 10.1107/s0907444903005407. [DOI] [PubMed] [Google Scholar]
- 27.Wang W, Kim R, Jancarik J, Yokota H, Kim SH. Structure (Camb) 2001;9:65–71. doi: 10.1016/s0969-2126(00)00558-x. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Cho HS, Kim R, Jancarik J, Yokota H, Nguyen HH, Grigoriev IV, Wemmer DE, Kim SH. J Mol Biol. 2002;319:421–31. doi: 10.1016/S0022-2836(02)00324-8. [DOI] [PubMed] [Google Scholar]
- 29.Clausen JD, McIntosh DB, Woolley DG, Andersen JP. J Biol Chem. 2001;276:35741–50. doi: 10.1074/jbc.M105434200. [DOI] [PubMed] [Google Scholar]
- 30.Sorensen TLM, Dupont Y, Vilsen B, Andersen JP. J Biol Chem. 2000;275:5400–5408. doi: 10.1074/jbc.275.8.5400. [DOI] [PubMed] [Google Scholar]
- 31.Toyoshima C, Nomura H, Sugita Y. FEBS Lett. 2003;555:106–10. doi: 10.1016/s0014-5793(03)01086-x. [DOI] [PubMed] [Google Scholar]
- 32.Kim Y, Yakunin AF, Kuznetsova E, Xu X, Pennycooke M, Gu J, Cheung F, Proudfoot M, Arrowsmith CH, Joachimiak A, Edwards A, Christendat D. J Biol Chem. 2004;279:517–526. doi: 10.1074/jbc.M306054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tremblay L, Zhang G, Dai J, Dunaway-Mariano D, Allen KN. Biochemistry. 2006;45:1183–93. doi: 10.1021/bi051842j. [DOI] [PubMed] [Google Scholar]
- 34.Lu Z, Dunaway-Mariano D, Allen KN. Biochemistry. 2005;44:8684–96. doi: 10.1021/bi050009j. [DOI] [PubMed] [Google Scholar]
- 35.Selengut JD, Levine RL. Biochemistry. 2000;39:8315–24. doi: 10.1021/bi0005052. [DOI] [PubMed] [Google Scholar]
- 36.Collet JF, Gerin I, Rider MH, Veigada-Cunha M, Van Schaftingen E. FEBS Letters. 1997;408:281–284. doi: 10.1016/s0014-5793(97)00438-9. [DOI] [PubMed] [Google Scholar]
- 37.Allen KN. In: Comprehensive Biological Catalysis. Sinnot M, editor. Academic press; New York: 1998. pp. 135–172. [Google Scholar]
- 38.Isbell AF, Englert LF, Rosenberg H. J Org Chem. 1969;34:755–756. [Google Scholar]
- 39.Cleland WW. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 40.Bradford MM. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Navaza J. Acta Crystallogr D Biol Crystallogr. 2001;57:1367–72. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- 43.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54 :905–21. doi: 10.1107/s0907444998003254. Pt 5. [DOI] [PubMed] [Google Scholar]
- 44.Brünger AT. Nature. 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 45.Read RJ. Methods Enzymol. 1997;278:110–128. doi: 10.1016/s0076-6879(97)77009-5. [DOI] [PubMed] [Google Scholar]
- 46.Jones TA, Zou JY, Cowan SW, Kjeldgaard GJ. Acta Crystallogr A. 1991;47 :110–9. doi: 10.1107/s0108767390010224. Pt 2. [DOI] [PubMed] [Google Scholar]
- 47.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 48.Zhang G, Mazurkie AS, Dunaway-Mariano D, Allen KN. Biochemistry. 2002;41:13370–13377. doi: 10.1021/bi026388n. [DOI] [PubMed] [Google Scholar]
- 49.Hayward S, Lee RA. J Mol Graph Model. 2002;21:181–3. doi: 10.1016/s1093-3263(02)00140-7. [DOI] [PubMed] [Google Scholar]
- 50.Lahiri SD, Zhang G, Dai J, Dunaway-Mariano D, Allen KN. Biochemistry. 2004;43:2812–20. doi: 10.1021/bi0356810. [DOI] [PubMed] [Google Scholar]