

Abstract
The binding of leptin to hypothalamic neurons elicits inhibition of orexigenic NPY/AgRP neurons and stimulation of anorexigenic POMC/CART neurons. Projections of serotonergic neurons onto POMC neurons suggest that leptin and serotonin converge onto POMC neurons to regulate body weight. We probed the interaction of these pathways by generating transgenic mice overexpressing leptin (LepTg) without 5HT2c receptors. On a chow-diet, the lean phenotype of LepTg mice was unaffected by the absence of 5HT2c receptors, whereas on a high fat diet, LepTg/5HT2c receptors knockout mice showed an exacerbation of diet-induced obesity. POMC mRNA levels were low in LepTg, 5HT2c receptors knockout and LepTg/5HT2c receptors knockout mice, demonstrating that perturbations of the 5HT2c receptor and leptin pathways either alone or in combination, negatively impact on POMC expression. Thus, on a chow diet, leptin action is independent of 5HT2c receptors whereas on a high fat diet 5HT2c receptors are required for the attenuation of obesity.
INTRODUCTION
Leptin activates neurons located in the mediobasal hypothalamus as well as in the brainstem [1,2]. The abundance of leptin receptors in the hypothalamus suggests that these regions are intimately associated with the regulation of food intake. Leptin receptors have been identified in orexigenic neurons that express neuropeptide Y (NPY), agouti-related protein (AgRP) and anorexigenic proopiomelanocortin (POMC), cocaine-and-amphetamine-regulated transcript (CART) peptides that are located in the ventromedial and ventrolateral arcuate nucleus, respectively, [3,4]. Leptin affects these two populations of neurons in opposite directions resulting in inhibition of NPY/AgRP neurons and stimulation of POMC/CART neurons [5–7].
Thus, in a physiological milieu of low leptin levels (as in fasting), the orexigenic pathway defined by NPY and AgRP is activated whereas the anorexigenic pathway mediated by POMC and CART neurons is inhibited. The location of NPY/AgRP and POMC/CART neurons in the arcuate nucleus (ARC) region of the hypothalamus define some the first-order of neurons directly responsive to leptin whereas other neurons located in the lateral hypothalamic area and in the perifornical area receive axonal projections from the ARC and constitute a second order set of neurons [5]. While serotonin has been implicated in the control of eating behavior and body weight [8], serotonergic neurons in Raphe nuclei of the brainstem contain leptin receptor mRNA and may be targets for leptin’s action [9]. Conversely, serotonin agonists activate POMC neurons [10], an effect that is contributed by a projection of serotonergic neurons to the arcuate nucleus [11]. Although serotonin binds to a variety of 5-hydroxytryptamine (5HT) receptors, the 5HT2c receptor (5HT2c-R) has gained particular interest in the regulation of food intake, namely because 5HT2c-R deficient mice exhibit late-onset obesity caused by abnormal feeding behavior [12]. Thus, mechanisms involving leptin-mediated melanocortin pathways as well as serotonin pathways appear to converge onto POMC neurons.
Transgenic mice overexpressing leptin (LepTg) fed a chow diet, have a reduced food intake, are lean and exhibit a severe lipodystrophy [13], which are altogether presumably owed to an overstimulation of the leptin-melanocortin axis. Paradoxically, these LepTg mice develop precipitous diet-induced obesity [14], thus serving as a switching animal model of leptin sensitivity and resistance states. In this study, we set out to determine whether 5HT2c receptors are required for the in the dual phenotypes of the LepTg mice and what would be the impact of their absence to the LepTg phenotypes. First, we asked whether inactivation of POMC signaling from the 5HT2c receptors, would revert or attenuate the effects of leptin overexpression by reverting the lean phenotype of the LepTg mice. Second, we asked to what extent would a leptin resistant state, which results in decreased leptin signaling to POMC neurons, be affected by inactivation of the leptin-independent anorectic arm of the POMC pathway via deletion of 5HT2c receptors. To address these two questions, we crossed the LepTg mice with the 5HT2c-R null mice to generate LepTg mice with and without 5HT2c receptors. Briefly, our results demonstrate that the absence of 5HT2c receptors from LepTg mice fails to affect their lipodystrophy on the chow diet, but exacerbates their obesity on the high fat diet. These findings demonstrate the dispensability of 5HT2c receptors in leptin action during a chow diet and their indispensability for the attenuation of DIO. We also show that the latter effect is partly mediated by a disturbance in the expression of the neuropeptides NPY, AgRP and POMC.
MATERIALS AND METHODS
Generation of mice overexpressing leptin in the absence of 5HT2c receptors
The UCSF Committee on Animal Research approved all experimental procedures in this study. 5HT2c receptor knockout (5HT2c-R-KO) and LepTg mice were previously described [12,13] and backcrossed each for over 15 generations onto the C57BL/6J genetic background. We crossed LepTg males with heterozygous 5HT2c-R-KO females and generated 4 groups of male mice consisting of normal, heterozygous LepTg, hemizygous 5HT2c-R-KO (The 5HT2c receptor is on the X chromosome) and heterozygous/hemizygous LepTg/5HT2c-R-KO mice. All mice were genotyped by PCR as previously described [12–13]. All mice in this study were males.
Body weights, food intake and fat pad weights
The LepTg mice were weaned at 6 weeks of age due to their cold susceptibility [13]. Body weights were monitored from 6–8 weeks of age to the nearest 0.1 g with an Ohaus precision scale from normal (n=8), LepTg (n=4), 5HT2c-R-KO (n=11) and LepTg/5HT2c KO (n=6) mice. At 8 weeks of age, four mice from each group were housed individually and monitored for food consumption over 10 days by periodically weighing the chow pellets on the wiretop of the cage, then killed to determine epidydimal white adipose tissue mass (WAT) since epididymal fat is the only fat depot that is present in the LepTg mice.
High fat diet treatment and metabolic assays
At 8 weeks of age, mice from the 4 groups (normal, n=7; LepTg, n=4; 5HT2c-R-KO, n=7; 5HT2c-R-KO/LepTg, n=5) were fed a high fat diet (Research Diets, Formula D12079B containing 40% kcal as fat, 43% kcal as carbohydrates and 17% kcal as protein with a total energy density of 4.7 kcal/g) for 12 weeks. Six weeks after the initiation of the high fat diet, food consumption of the individually housed mice was determined over 7 days as described above. At the end of the high fat diet treatment, the mice were brought to the laboratory from the mouse room in the afternoon, anesthetized with 2% Avertin, bled through the retrorbital and the resulting plasma frozen at −80°C. Glucose levels were determined at the time of killing using a Glucometer. Mouse leptin and insulin levels were quantitated with radioimmunoassay kits from Linco (St. Charles, MO).
Quantitation of neuropeptide mRNA expression
At the end of the high fat diet treatment, all mice were killed and their hypothalami removed, immediately frozen in liquid nitrogen and stored at −80°C. RNA was extracted from each hypothalamus with the Trizol reagent (Invitrogen, CA) according to the manufacturer’s recommendations. Approximately 10 μg of RNA were reverse transcribed into cDNA using MMLV reverse transcriptase and random hexamers according to standard procedures. The resulting cDNAs were then used in subsequent amplification reactions. Briefly, cDNAs were amplified in either duplicates or triplicates in microtiter plates on the MJ Research Opticon realtime PCR cycler using a sybergreen dNTP mix (Qiagen, CA). All PCR primers yielded single PCR products as visualized by gel electrophoresis. The PCR primers for NPY (111 bp) were 5′ CTCGTGTGTTTGGGCATTCT3′ and 5′GTAGTGTCGCAGAGCGGAGT3′, AgRP (115bp) 5′TTCCCAGGTCTAAGTCTGAATGGC3′ and 5′ACTCGCGGTTCTGTGGATCTA3′, POMC (116 bp) 5′ TTG CAT CCG GGC TTG CAA A 3′ and 5′ AGCGGAAGTGACCCATGACGTA 3′, CART (107 bp) 5′ GCTATGTTGCAGATCGAAGCGT 3′ and 5′AGCGTCACACATGGGGACTT 3′ and cyclophilin (162 bp) 5′ ATAATGGCACTGGCGGCAGG 3′ and 5′CCATCCAGCCATTCAGTCTTGG 3′. Reactions were denatured for 5 minutes at 95°C followed by 45 cycles at 95°C, 54.8°C, 72°C for 30 seconds each. Relative expression levels were calculated by the software Q-GENE [15, 16] after selecting Ct values for each target and reference genes in the linear portion of the amplification plot and taking into account the slope m of each the target and reference genes standard curves according to the formula:
All realtime amplifications reactions were carried out on the same cDNAs such that relative expression levels could be compared across the 4 groups. Relative quantities of cDNA obtained by realtime PCR for each amplicon were normalized to the means of the normal group in order to compare the ratios of orexigenic and anorexigenic peptides in the experimental groups.
Statistical analysis
Data are expressed as means and s.e.m. and were evaluated for statistical significance by student’s t-test for a two groups comparison or for multiple groups with analysis of variance (ANOVA) followed by post-hoc tests.
RESULTS
LepTg mice without 5HT2c receptors remain lipodystrophic
Four groups of mice resulted from the cross between heterozygous LepTg males and heterozygous 5HT2c-R-KO female mice. These were normal, hemizygous 5HT2c-R-KO, heterozygous LepTg and heterozygous/hemizygous LepTg/5HT2c-R-KO mice. On a chow diet and from 6–8 weeks of age, the body weights of the 4 groups of mice fell in two distinct groups consisting of high and low body weights (Fig. 1A). In the high body weight group, were normal and 5HT2c-R-KO mice and in the lower body weight group were the LepTg and LepTg/5HT2c-R-KO mice. The body weights of normal vs. 5HT2c-R-KO mice and LepTg vs. LepTg 5HT2c KO mice did not significantly differ from each other. However, the body weights of both LepTg mice with or without 5HT2c receptors were significantly lower than those of normal and 5HT2c-R-KO mice (p<0.01 in each comparison) by approximately 5g. Whereas the non-significant differences between normal and 5HT2c-R-KO mice and the significant differences between normal and LepTg mice are consistent with previous observations [12,13], the lack of significant differences in body weights between LepTg and LepTg/5HT2c-R-KO mice is novel and bears important implications for the downstream mechanisms of leptin action. Consistent with the body weights of mice in the four groups, the WAT mass of the LepTg/5HT2c-R-KO mice was significantly less than either normal or 5HT2c-R-KO mice (p<0.01 in each comparison) and essentially indistinguishable from the minute WAT mass of LepTg mice (Fig. 1B). This new finding demonstrates that the phenotype of LepTg/5HT2c-R-KO mice defaults back to that of LepTg mice, revealing that mechanisms underlying the leptin-mediated lipodystrophy of LepTg mice are independent of 5HT2c receptors. Broadly, our findings suggest the absence of a critical link between leptin action and the serotonergic system mediated by 5HT2c receptors on a chow diet. Furthermore, a 15–16% reduction in food intake was found in LepTg and LepTg/5HT2c-R-KO mice compared to their respective controls (p<0.05) (Fig. 1B).
Figure 1.
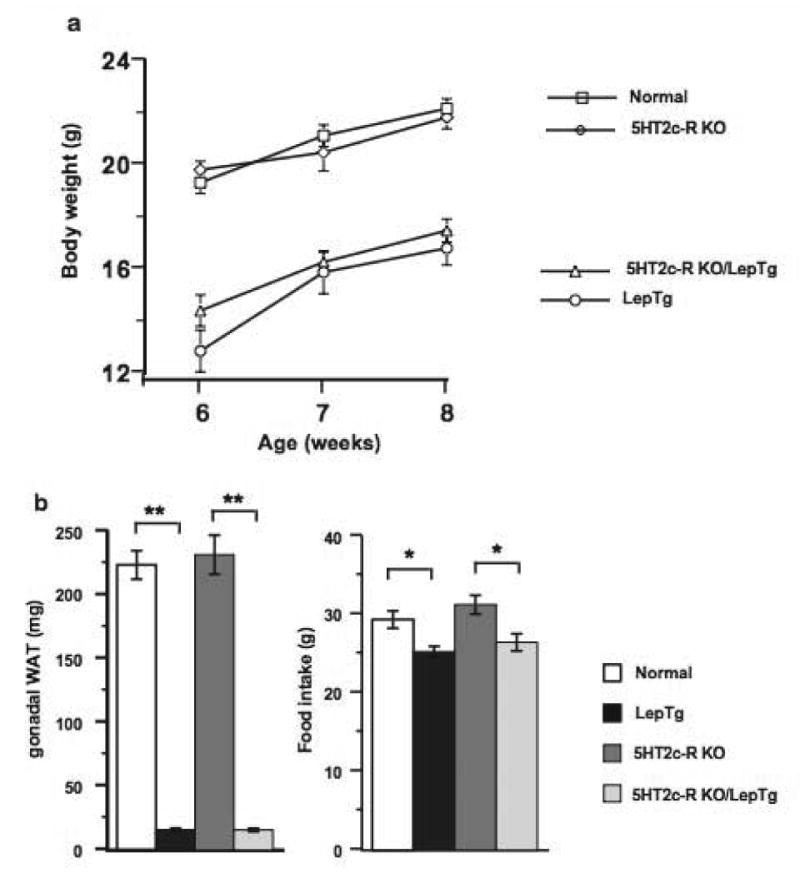
Interaction of leptin overexpression with absence of 5HT2c receptors during a chow diet. (a) Body weights of normal, LepTg, 5HT2c-R-KO and LepTg/5HT2c-R-KO mice from 6–8 weeks of age. Statistical significance among the four groups is discussed in the text. (b) Epidydimal white adipose tissue mass (WAT) and 10-day cumulative food intake of 4 mice from each of group. ** and * denote p<0.01 and p<0.05, respectively.
Exacerbation of DIO in LepTg mice without 5HT2c receptors
Fig. 2A shows the changes in body weight among the 4 groups as a result of the HFD treatment. The increase in body weights of the LepTg/5HT2c-R-KO mice at the end of the HFD treatment consisted of 100.5±14.9% compared to 63± 7.5%, 48±7.6% and 40.5±5.3% in LepTg, 5HT2c-R-KO and normal mice, respectively. Although the body weights of the normal and 5HT2cR KO mice fed the HFD followed a similar trend to that previously observed [17], it is likely that the lack of statistical significance between these two groups may have been caused by differences in the types of high fat diet, which in our study contained 5% less fat and 8% more carbohydrates than the original study [17].
Figure 2.
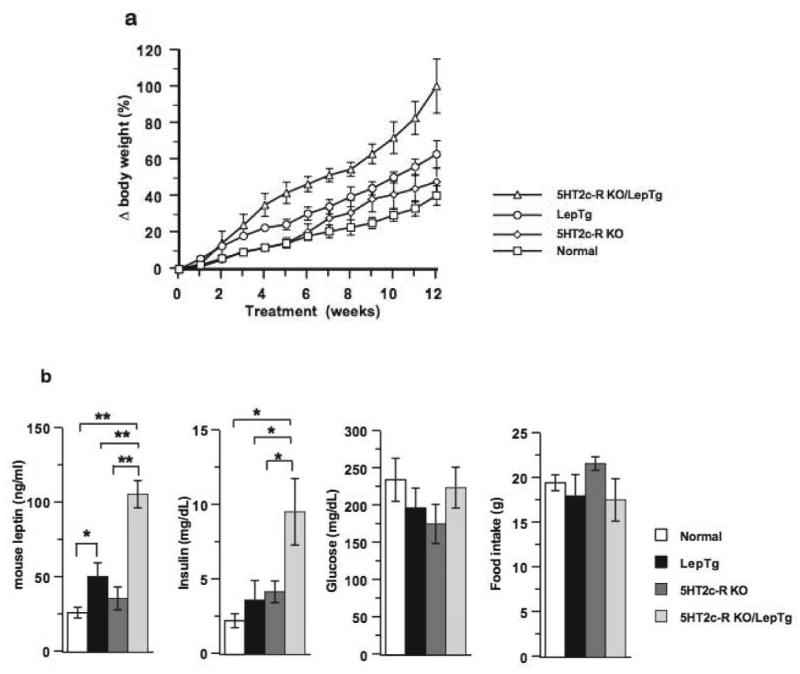
Interaction of leptin overexpression with absence of 5HT2c receptors during a HFD. (a) Changes in body weights of mice from the 4 groups during the course of the HFD. (b) Circulatory levels of endogenous leptin, insulin, and glucose at the end of the HFD. 7-day food consumption in mice from the 4 groups 6 weeks after the initiation of the HFD. ** and * denote p<0.01 and p<0.05, respectively.
The nature of the interaction between the leptin transgenic and 5HT2c-R-KO loci was determined by subtracting the mean body weights of LepTg, 5HT2c-R-KO and LepTg/5HT2c-R-KO mice from those of normal mice and then comparing the resulting values with each other, thus asking whether such interaction was suppressive, additive or synergistic. Using this analysis, we found that at the end of the HFD, the LepTg/5HT2c-R-KO mice had 60.1±9.5% more elevated body weight than normal mice, whereas 5HT2c-R-KO and LepTg mice weighed 7.5±2.3% and 22.6±2.2%, more than normal mice, respectively. Thus, statistical comparison of the normalized weights of the LepTg/5HT2c-R-KO mice vs. 5HT2c-R-KO and LepTg mice revealed that the effect produced by the presence of these two loci in the same mouse (LepTg/5HT2c-R-KO) was greater than the sum of the effects produced by these two loci separately (LepTg+5HT2c-R-KO) (p<0.01), thereby revealing a positive epistatic interaction. To determine adiposity in all mice, we measured at the end of the HFD treatment and in the afternoon when leptin levels are the highest [18], endogenous circulatory mouse leptin levels, which are proportional to fat mass and have been used as surrogate markers of adiposity [19–20]. LepTg mice overexpress human leptin, which is not detected by the mouse RIA. We found that leptin levels consisted in LepTg/5HT2c-R-KO mice of 106±35 ng/ml compared to 51±9 ng/ml, 36±8 ng/ml and 27± 4 ng/ml in LepTg, 5HT2c-R-KO and normal mice, respectively (p<0.01 in each comparison). These values translate for the LepTg/5HT2c-R-KO mice into increases of 4.1, 2.9 and 2.1 fold above those of normal, 5HT2c-R-KO, and LepTg mice, respectively (p<0.01 in each comparison). Similarly, the circulating insulin levels in LepTg/5HT2c-R-KO mice reached 9.6±2.2 ng/ml and were 2.9, 2.3 and 2.2 fold over those of normal, 5HT2c-R-KO and LepTg mice, respectively (p<0.05 in each comparison). Glucose levels and food consumption were not significantly different in mice across the 4 groups. (Fig. 2B).
Neuropeptide gene expression in DIO
Quantitation of the relative mRNA expression levels of the orexigenic peptides NPY, AgRP and anorexigenic peptides POMC, CART in the hypothalamus of mice fed the HFD (Fig. 3) revealed that CART was the highest expressed, but nevertheless not significantly different across the 4 groups. However compared to normal mice, NPY and AgRP levels, were significantly lower in the 5HT2c-R-KO mice, but not in LepTg and LepTg/5HT2c-R-KO mice. In contrast compared to normal mice, POMC levels were significantly decreased in all experimental groups by approximately 60% (p<0.01 in each group). Altogether, these determinations show that relative to normal mice, the 5HT2c-R-KO mice display low AgRP, NPY and POMC levels whereas the LepTg and LepTg/5HT2c-R-KO mice have low POMC and elevated NPY/AgRP levels.
Figure 3.
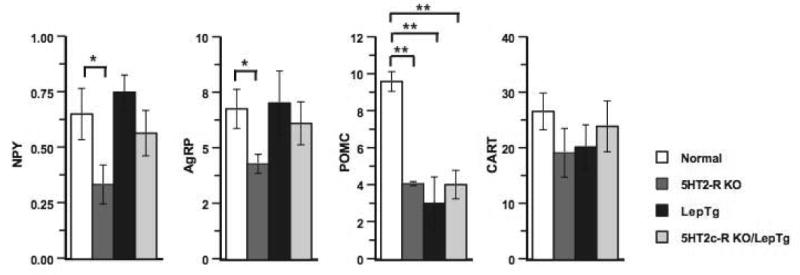
Relative mRNA expression levels of the neuropeptides NPY, AgRP, POMC and CART in the hypothalamus of normal, 5HT2c-R-KO, LepTg and LepTg/5HT2c-R-KO mice at the end of the HFD. * p<0.05; ** p<0.01.
DISCUSSION
Antagonism at the melanocortin 4 receptor by agouti related protein (AgRP) and α-MSH is a well-established downstream effector of leptin signaling, however, other factors, which feed into these pathways, may contribute to the cascade of events initiated by leptin. Of these, the serotonergic system has been implicated in the regulation of appetite and body weight [8, 21, 22]. A supporting a role for 5HT2c receptors in energy metabolism was the finding that 5HT2c-R-KO mice are hyperphagic, obese and under specific dietary manipulations predisposed to diet-induced obesity [17]. Furthermore, stimulation of the 5HT2c/1B receptor in rats resulted in decreased food intake and activation of neuronal activity in a manner similar to that induced by the anorectic drug D-fenfluramine, which was previously used to treat obesity [11]. Thus, the question arises whether a significant crosstalk exists between the 5HT2c receptor pathway and the leptin pathway such that it would impact on the phenotypes of the LepTg mice.
Our findings that the LepTg mice are lean and almost fatless on a chow diet (13) but gain substantial amounts of fat when fed a high fat diet (HFD) (14) provides an experimental model to study the interaction of the 5HT2c receptor and leptin pathways. Thus, we investigated via a genetic strategy, the role of 5HT2c receptors in LepTg mice. Our findings that the LepTg/5HT2c-R-KO mice are similar in weight and adiposity to LepTg mice with intact 5HT2c receptors demonstrate that leptin has a greater effect on body weight than 5HT2c-R signaling.
Thus, 5HT2c receptors may not constitute a critical component of the overall leptin pathway with respect to body weight and adiposity. Rather, our data complement previous findings [17] by strongly supporting the concept that the serotonergic system mediated by 5HT2c receptors regulates body weight and food intake via a neuronal circuitry different from that of leptin.
The cross between the LepTg and 5HT2c-R-KO mice followed by a HFD addressed the question whether these two predispositions to DIO [12, 14] would attenuate or exacerbate the DIO of either strain, thus defining the roles of their individual or combined pathways. Our findings indicate that these two predispositions synergistically interact to exacerbate DIO. Thus, the coupling of a hyperactive leptin pathway at the beginning of the HFD coupled with loss of 5HT2c-R function results in severe DIO. A corollary of this finding would suggest that presence of 5HT2c receptor signaling might actually attenuate DIO.
The nature of the mechanisms that underlie these differential effects on body weight and adiposity in LepTg mice with and without 5HT2c receptors was addressed by measuring the relative expression levels of the orexigenic neuropeptides NPY, AgRP and anorexigenic neuropeptides POMC and CART, which are respectively downregulated and upregulated by leptin [3–7, 23–26].
Our findings that the LepTg, 5HT2c-R-KO and LepTg/5HT2c-R-KO mice have relatively low and similar POMC levels suggest that perturbations in the 5HT2c-R and leptin pathways downregulate POMC expression to the same extent, precluding their differential phenotypes based on POMC expression alone.
Although, the elevated AgRP and NPY levels in LepTg and LepTg/5HT2c-R-KO but not 5HT2c-R-KO mice may account at least partly, for the shift of the leptin pathway from catabolic on the chow diet to anabolic on the high fat diet, their differential phenotypes is likely to be accounted for by the dysregulation of additional neuropathways regulating energy balance, such as leptin signaling defects [27] in intra and extra hypothalamic brain regions [28, 29].
In summary, our findings demonstrate that during low fat feeding states, mechanisms downstream or independent of leptin signaling do not appear to require the presence of 5HT2c receptors as determined by the continuous lipodystrophy of LepTg/5HT2c-R-KO mice.
Conversely, the exacerbation of DIO inflicted upon HFD fed mice with two genetic susceptibilities to DIO indicates that the presence of 5HT2c receptors actually attenuates DIO, which may be caused at least in part by a disturbance in the relative ratios of POMC, AgRP and NPY levels. The extension of our findings suggest that although obesity treatment modalities aimed at 5HT2c receptors or the leptin/melanocortin pathway might be beneficial in DIO, a greater benefit could be derived from simultaneous pharmacological targeting of both pathways resulting possibly in synergistic therapeutic effects.
Acknowledgments
We thank L. Tecott and E. Goulding for the generous gift of 5HT2c-R-KO mice and genotyping protocols, L. Tecott, E. Goulding and Streamson Chua for critical reading of the manuscript and Stuart Beale for advise on statistical analysis. This work was supported by NIH grants HD35142, T32DK 07636 and a UCSF grant from the Academic Senate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elmquist JK, Ahima RS, Maratos-Flier E, Flier JS, Saper CB. Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology. 1997;138:839–842. doi: 10.1210/endo.138.2.5033. [DOI] [PubMed] [Google Scholar]
- 2.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci USA. 1998;95:741–746. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baskin DG, Breininger JF, Schwartz MW. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes. 1999;48:828–833. doi: 10.2337/diabetes.48.4.828. [DOI] [PubMed] [Google Scholar]
- 4.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 1997;134:4489–4492. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 5.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 6.Stephens TW, Basinski M, Bristow PK, Bue-Valleskey JM, Burgett SG, Craft L, Hale J, Hoffmann J, Hsiung HM, Kriauciunas A, MacKellar W, Rosteck PR, Schoner B, Smith D, Tinsley FC, Zhang XY, Heiman M. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. 1995;377:530–532. doi: 10.1038/377530a0. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz MW, Baskin DG, Bukowski TR, Kuijper JL, Foster D, Lasser G, Prunkard DE, Porte D, Jr, Woods SC, Seeley RJ, Weigle DS. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;45:531–535. doi: 10.2337/diab.45.4.531. [DOI] [PubMed] [Google Scholar]
- 8.Leibowitz SF, Alexander JT. Hypothalamic serotonin in control of eating behavior, meal size, and body weight. Biol Psychiatry. 1998;44:851–864. doi: 10.1016/s0006-3223(98)00186-3. [DOI] [PubMed] [Google Scholar]
- 9.Finn PD, Cunningham MJ, Rickard DG, Clifton DK, Steiner RA. Serotonergic neurons are targets for leptin in the monkey. J Clin Endocrinol Metab. 2001;86:422–426. doi: 10.1210/jcem.86.1.7128. [DOI] [PubMed] [Google Scholar]
- 10.Heisler LK, Cowley MA, Tecott LA, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Marcus JN, Holstege H, Lee CE, Cone RD, Elmquist JK. Activation of central melanocortin pathways by fenfluramine. Science. 2002;297:609–611. doi: 10.1126/science.1072327. [DOI] [PubMed] [Google Scholar]
- 11.Heisler LK, Cowley MA, Kishi T, Tecott LH, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Zigman JM, Cone RD, Elmquist JK. Central serotonin and melanocortin pathways regulating energy homeostasis. Ann N Y Acad Sci. 2003;994:169–174. doi: 10.1111/j.1749-6632.2003.tb03177.x. [DOI] [PubMed] [Google Scholar]
- 12.Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, Julius D. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- 13.Qiu J, Ogus S, Lu R, Chehab FF. Transgenic mice overexpressing leptin accumulate adipose mass at an older, but not younger age. Endocrinology. 2001;142:348–358. doi: 10.1210/endo.142.1.7909. [DOI] [PubMed] [Google Scholar]
- 14.Ogus S, Ke Y, Qiu J, Wang B, Chehab FF. Hyperleptinemia precipitates diet-induced obesity in transgenic mice overexpressing leptin. Endocrinology. 2003;144:2865–2869. doi: 10.1210/en.2002-0178. [DOI] [PubMed] [Google Scholar]
- 15.Muller PY, Janovjak H, Miserez AR, Zuzana Dobbi Z. Processing of gene expression data generated by quantitative real-time RT-PCR. BioTechniques. 2002;32:1372–1379. [PubMed] [Google Scholar]
- 16.Simon P. Q-Gene:processing quantitative real-time RT-PCR data. Bioinformatics. 2003;19:1439–1440. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- 17.Nonogaki K, Strack AM, Dallman MF, Tecott LH. Leptin-independent hyperphagia and type 2 diabetes in mice with a mutated serotonin 5-HT2c receptor gene. Nature Med. 1998;4:1152–1156. doi: 10.1038/2647. [DOI] [PubMed] [Google Scholar]
- 18.Cha MC, Chou CJ, Boozer CN. High-fat diet feeding reduces the diurnal variation of plasma leptin concentration in rats. Metabolism. 2000;49:503–507. doi: 10.1016/s0026-0495(00)80016-5. [DOI] [PubMed] [Google Scholar]
- 19.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 20.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern PA, Friedman JM. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 21.Blundell JE, Lawton CL, Hallford JC. Serotonin, eating behavior and fat intake. Obes Res. 1995;3:471S–476S. doi: 10.1002/j.1550-8528.1995.tb00214.x. [DOI] [PubMed] [Google Scholar]
- 22.Simansky KJ. Serotonergic control of the organization of feeding and satiety. Behav Brain Res. 1996;73:37–42. doi: 10.1016/0166-4328(96)00066-6. [DOI] [PubMed] [Google Scholar]
- 23.Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, Clausen JT, Jensen PB, Madsen OD, Vrang N, Larsen PJ, Hastrup S. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature. 1998;393:72–76. doi: 10.1038/29993. [DOI] [PubMed] [Google Scholar]
- 24.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–2123. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 26.Thornton JE, Cheung CC, Clifton DK, Steiner RA. Regulation of hypothalamic proopiomelanocortin mRNA by leptin in ob/ob mice. Endocrinology. 1997;138:5063–5066. doi: 10.1210/endo.138.11.5651. [DOI] [PubMed] [Google Scholar]
- 27.El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–1832. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 29.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;23:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]