

Abstract
Ultrasound and poly(ethylenimine) (PEI) have each separately been shown to increase DNA transfection efficiency. This study tested the hypothesis that the combination of ultrasound and PEI can have a synergistic effect to increase DNA transfection. This in vitro study assessed transfection efficiency of two different DNA plasmids encoding green fluorescent protein and firefly luciferase in two different cells types, a primary culture of human aortic smooth muscle cells and an immortal line of human prostrate cancer cells. We found that ultrasound sonication increased transfection up to 18-fold, DNA complexation with PEI increased transfection up to 90-fold, and the combination of ultrasound and PEI synergistically increased transfection up to 200-fold, which resulted in reporter gene expression by 34% of cells. Kinetic measurements found that the effects of ultrasound alone acted quickly, whereas increased transfection by PEI either alone or in combination with ultrasound strongly benefited from a 4-h incubation with the DNA plasmid after sonication. Although serum reduced absolute expression levels, it did not affect the relative increase in transfection when ultrasound was added to PEI enhancement. Flow cytometry measurements showed that sonication increased intracellular uptake of labelled DNA complexed to PEI by 55% relative to PEI complexation alone. Electrophoresis assay showed no damage to DNA or PEI-DNA complexes after sonication. Overall, these results suggest that the combination of ultrasound and PEI can have a synergistic effect to increase DNA transfection.
Keywords: DNA transfection, gene delivery, nonviral vector, PEI, ultrasound
1. Introduction
Pharmacological treatment of many diseases only alleviates symptoms, often without addressing the root cause. In contrast, gene therapy offers the promise of a cure by correcting the root cause at a genetic level. Ideal targets for gene therapy are diseases caused by a single gene defect, of which there are more than 6,000 already known [1]. However, progress in gene therapy has been delayed by limited ability to deliver appropriate amounts of DNA to required sites of action and thereby achieve therapeutic gene expression levels [2]. Viral and non-viral vectors have been used for DNA delivery with varying levels of success, but always accompanied by disadvantages, such as insufficient expression levels or safety concerns [3,4].
One of the most promising non-viral vectors is poly(ethylenimine) (PEI), which is the most widely used gene delivery vector among cationic polymers [5–7]. PEI has been shown to be a relatively efficient gene transfer agent without the need for additional endosomolytic or lysosomotropic agents [8]. Notably, PEI:DNA complexes have been used successfully for in vivo applications, including direct application to various anatomical sites [9,10].
Despite success with PEI, it would be advantageous to further increase transfection efficiency. In this study, we considered the combination of ultrasound with PEI to achieve greater transfection levels. We are guided by the hypothesis that ultrasound and PEI can have a synergistic effect to increase DNA transfection. This hypothesis was motivated by previous studies addressing the use of ultrasound for increased gene transfection in other contexts [11,12].
A number of studies have shown that exposure of cells to ultrasound can increase DNA transfection by up to orders of magnitude through both in vitro and in vivo studies [13–16]. The mechanism remains under investigation, but is believed to involve increased uptake of DNA molecules into the cell and, possibly, upregulation of gene expression by an undetermined pathway. Another advantage of ultrasound-mediated transfection is that ultrasound can be non-invasively focused on almost any location in the body, which provides significant clinical advantages [17].
Although most studies have addressed transfection with naked DNA using ultrasound, there have been reports showing that ultrasound can act synergistically with other gene delivery vectors, such as viral vectors and cationic lipids [18]. For example, the application of ultrasound was shown to increase the transfection efficiency in rat myocardium in vivo using ultrasound-targeted destruction of microbubbles containing an adenovirus encoding a beta-galactosidase reporter gene [19,20]. Other studies demonstrated that ultrasound increased transfection of a variety of different cell types in vitro in combination with liposome-complexed reporter plasmids [21–23]. However, the effects of ultrasound on transfection by cationic polymers, such as PEI, have not been studied before.
Given the promising results for PEI and ultrasound each used independently, this study sought to investigate whether the combination of PEI and ultrasound could provide still greater transfection efficiency, possibly by a synergistic interaction. To address this idea, we examined the effects of ultrasound on the intracellular delivery and transfection efficiency of DNA both with and without PEI. The effects of ultrasound pressure and exposure time were studied using two different cell lines: human aortic smooth muscle cells and prostate cancer cells. The stability of naked DNA and PEI:DNA complexes was also assessed after ultrasound exposure.
2. Materials and Methods
2.1 Materials
2.1.1 DNA and other reagents
Two different DNA plasmids were used. The 5.75-kb eukaryotic expression plasmid gWIZ GFP, containing the green fluorescent protein gene, was purchased from Aldevron (Fargo, ND, USA) and used as received. The 4.7-kb PGL3-Control Vector plasmid, containing the firefly luciferase gene, was obtained from Promega (Madison, WI, USA). This plasmid was transformed into MAX Efficiency DH5α Competent Cells (Invitrogen, Carlsbad, CA, USA) using the heat shock method (according to the protocol from Invitrogen, catalogue no.18258–012). The amplification of the plasmid was done using a QIAGEN EndoFree Plasmid Giga Kit (Valencia, CA, USA) [24].
Additional reagents were prepared as follows. RPMI-1640 and MCDB-131 tissue culture media (Sigma-Aldrich, St. Louis, MO, USA) were supplemented with 10% (v/v) heat-inactivated, fetal bovine serum (FBS; Cellgro, Mediatech, Herndon, VA, USA). A Luciferase assay kit was obtained from Promega. Bradford assay reagent was used as obtained (Sigma-Aldrich). The Bradford assay was calibrated using solutions of human serum albumin (Sigma-Aldrich) in water with concentrations ranging from 1 to 1400 μg/ml. The fluorescent dye YOYO-1 iodide (Molecular Probes, Eugene, OR, USA) was used as a 1 mM stock solution in DMSO. Branched poly(ethylenimine) (Sigma-Aldrich) had a molecular weight of 25 kDa. The marker for agarose gel electrophoresis was a 1-kb DNA ladder (New England Biolabs, Beverly, MA, USA).
2.1.2. Cell lines
DU 145, human prostrate cancer cells (lot no. 1145858; American Type Culture Collection, Manassas, VA, USA) were cultured in RPMI-1640 media supplemented with 10% FBS and 1% penicillin-streptomycin solution at 37oC and 5% CO2. The cells were subcultured once they reached 70–80% monolayer confluence using Trypsin-EDTA to detach adherent cells from the tissue culture substrate.
Human aortic smooth muscle cells (AoSMC; catalogue no. CC-2571, lot no. 7F0787, Clonetics, San Diego, CA, USA) were initiated from frozen stock and harvested at passage 7 prior to each experiment. They were cultured in MCDB-131 media supplemented with 10% FBS, 1% penicillin-streptomycin, and 2 mM L-glutamine (Cellgro).
2.1.3. Ultrasound equipment
As previously described in detail [25], the ultrasound apparatus consisted of a focused, 500-kHz, piezoelectric transducer with an 8.9-cm focal length and a beam width of 3 mm (−6 db intensity area) at the focal beam point (Fig. 1). A sinusoidal voltage was produced by a waveform generator (model no. DS345, Stanford Research Instruments, Sunnyvale, CA, USA) and amplified by a custom tone-burst amplifier (Techno Scientific, Concord, ON, Canada) that powered and controlled the response of the transducer. The transducer was placed in a polycarbonate tank (30 × 29 × 37 cm) containing 26 L of deionised, distilled and partially degassed water. Spatial-peak-temporal-peak negative pressure (P) was measured at the focal beam point using a 0.2-mm aperture PVDF membrane hydrophone (model no. MHA200A, NTR Systems, Seattle, WA, USA) in the absence of a sample container. The spatial-peak acoustic energy (E) was calculated using the following equation:
Figure 1.
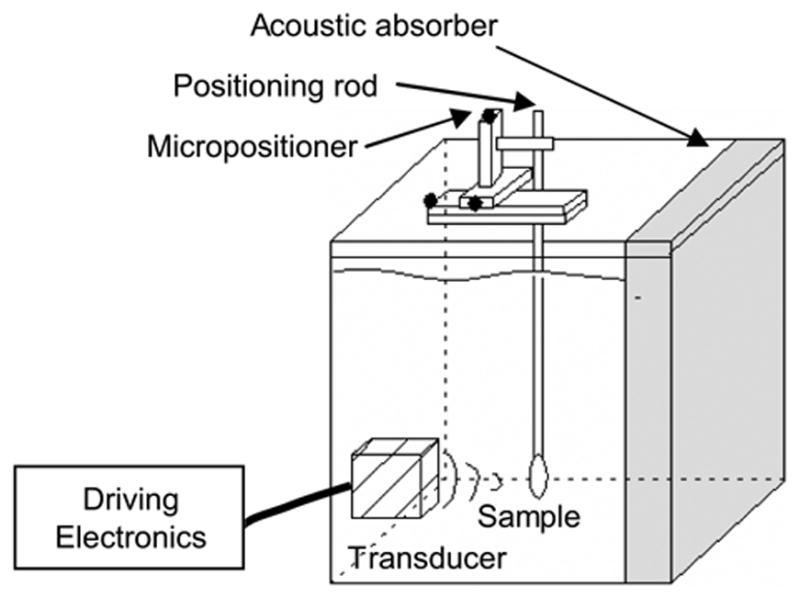
Schematic of the ultrasound apparatus. A transducer focused ultrasound energy on a cell sample positioned using a micropositioner. Acoustic reflections and standing waves were minimized by an acoustic absorber.
where P is the rms pressure, ρ the density of water (0.9982 g/cm3), and c the speed of sound in water (1482 m/s).
2.2 Methods
2.2.1. Preparation of PEI:DNA complexes
An aqueous stock solution of PEI (10 mM monomer content) was prepared by diluting 9 mg of the 50% (w/v) commercial solution in 10 ml DI water, bringing the solution to neutral pH with HCl, and filtering at 0.2 μm (Millipore, Bedford, MA, USA). Then, 42 μl of the PEI stock solution was added to 100 μL of a solution of 0.2 mg/ml DNA in RPMI-1640 in a 1.5 ml microcentrifuge tube (Eppendorf, Brinkmann Instruments, Westbury, NY, USA) and incubated for 20 min at room temperature. This produced PEI:DNA complexes with the desired PEI nitrogen:DNA phosphate ratio of 7:1, based on the recognition that 1 μl of PEI stock solution contains 10 nmol of amine nitrogen and 1 μg of DNA contains 3 nmol of phosphate [26]. In some cases, FBS was added at a final concentration of 10%.
2.2.2. Transfection protocol using ultrasound
Cells were trypsinized from culture flasks and resuspended in RPMI-1640 (with or without 10% FBS). Cell suspensions were added to the solutions containing naked DNA or PEI:DNA complexes immediately prior to ultrasound. Unless otherwise specified, cells were suspended in RPMI-1640 at a concentration of 2.5×106 DU 145 cells/ml or 7.5×105 AoSMC cells/ml, as determined with the aid of a Coulter counter (Multisizer II, Beckman Coulter). After briefly vortexing the cell suspensions, albumin-stabilized gas bubbles (17 ± 0.05 μl/ml, ~1.1×107 bubbles/ml, bubble diameter = 2.0–4.5 μm; Optison, Mallinckrodt, St. Louis, MO, USA) stored in a locking, airtight syringe (Sample Lock Syringe Hamilton, VWR, San Francisco, CA, USA) were slowly added to the cell suspension using a 22-gauge flat needle (400 μm i.d.) to serve as nucleation sites for acoustic cavitation.
After adjusting the volume to 400 μl with RPMI-1640, the suspension was slowly drawn into a 3-ml syringe (Becton Dickinson, Franklin Lakes, NJ, USA) with a 22-gauge needle (Perkin Elmer, Foster City, CA, USA) and transferred into 400-μl polyethylene transfer pipettes (6.1 mm internal diameter, 0.1 mm wall thickness, and 3.7 mm height; catalogue no. 292, Samco, San Fernando, CA, USA). An aluminum metal rod was then inserted into the tube of the transfer pipette, which sealed it, and was attached to a three way micropositioner (1-mm resolution, Velmex, Bloomfield, NY, USA), which positioned it in the focal beam point of the transducer.
Ultrasound was applied using 1–8 pulses each of 60 ms duration for a total exposure time between 60–480 ms. The duty cycle was 6%, which means that the duration of each experiment was 1–8 s. Spatial-peak-temporal-peak negative pressure was varied between 0.25 and 3.0 MPa. Control or “sham” samples were treated in the same way as other samples, but were not exposed to ultrasound. Unless otherwise specified, sonicated cells were then placed into the wells of a 6-well plate and 1 ml of RPMI-1640 (without 10% FBS) was added to each well immediately. After incubating the cells for 4 h at 37oC and 5% CO2, the transfection media was removed and replaced with 3 ml RPMI-1640 with 10% FBS, pre-warmed to 37oC. Cells were then incubated for another 24 h, in the case of DU 145 cells involving plasmid gWIZ GFP, or for 48 h, in all other cases, before assessment of reporter gene expression.
For transfection experiments in presence of serum, 10% FBS was added to the samples immediately before sonication. These samples were then placed into the wells of a 6-well plate and 1 ml of RPMI-1640 (with 10% FBS) was added to each well and left for 4 h. After the 4 h incubation period, the cells were treated as described for those without serum.
2.2.3. Assessment of GFP expression and cell viability
To assess GFP reporter gene expression, cells were washed twice with PBS, trypsinized, and suspended in 1 mL PBS. Fluorescent calibration beads (2.4 × 105 beads/mL; Linear Flow Green Flow Cytometry Intensity Calibration Kit L-14821, Molecular Probes) were added to the cell suspension to act as an internal volumetric standard to help determine cell viability, as described previously [27]. Cell samples were placed on ice until analysis by flow cytometry (BD LSR Flow Cytometer, Becton Dickinson) [28]. Using excitation with a 488-nm laser, the scattered light signals (i.e., forward and side scatter) were used to identify and distinguish between cells, fluorescent beads, and debris. Among data collected from 20,000 cells per sample, cells exhibiting green fluorescence greater than that of 99.9% of cells in the control samples were considered as GFP-positive. Cell viability was calculated by comparing the ratio of the viable cells and the fluorescent beads in each sample to the ratio measured in control samples.
2.2.4. Detection of luciferase activity
To assess luciferase reporter gene expression, cells were washed twice with PBS, incubated in 300 μl cell lysis buffer from a luciferase detection kit (Promega), and scraped off the multi-well substrate. The resulting lysate was transferred to a microcentrifuge tube. Luciferase activity was measured by mixing 20 μl of lysate with 80 μl of luciferin reagent (Promega) and measuring luminescence (5 s delay, 10 s integration time; Sirius Luminometer, model D-75173, Berthold Detection Systems, Pforzheim, Germany). The instrument was calibrated for background noise first with water and then with luciferin reagent.
To help interpret luciferase luminescence measurements, recoverable cellular protein concentration was determined as a measure of cell concentration using the modified Bradford assay [29]. Aliquots containing 20 μl of luciferase detection lysate were diluted with 80 μl of water. After adding 3 ml of Bradford reagent and incubating for 5–6 min, absorption at 595 nm was measured (Lambda 950, Perkin Elmer, Wellesley, MA, USA). Recoverable cellular protein concentration was determined by calibration relative to a standard curve generated using known concentrations of aqueous bovine serum albumin between 0–1000 μg/ml. Finally, luminescence measurements (RLU/ml) were divided by protein concentration measurements (μg protein/ml) to normalize luciferase expression in terms of relative light units (RLU)/μg cellular protein,
2.2.5. Quantification of DNA uptake
To measure DNA uptake, as opposed to expression, plasmid-DNA PGL3 was labelled with YOYO-1 iodide (Molecular Probes) at a ratio of 1 dye molecule per 100 nucleotide bases for naked DNA and a ratio of 1 dye molecule per 300 nucleotide bases for PEI:DNA complexes [30]. After sonicating cell samples containing labelled DNA, intracellular uptake was measured by flow cytometry. Microspheres with calibrated fluorescence (Quantum 25 FITC high level, Bangs Laboratories, Fishers, IN, USA) were used to quantify the number of plasmid DNA molecules delivered per cell [31].
2.2.6. Confocal imaging of DNA uptake
To image cells taking up DNA after sonication, AoSMC cells were first trypsinized and resuspended in RPMI-1640 at a final concentration of 106 cells/mL. To label them for viewing by confocal microscopy, a solution of red-fluorescent, TRITC-labeled wheat germ Lectin (Sigma-Aldrich) was added at a final concentration of 2 μM to the cell suspension and mixed for 5 min at room temperature on a nutator. The suspension was then centrifuged at 1000×g (model GS-15R, Beckman Coulter), the supernatant was pipetted off, and the cells were resuspended in RPMI-1640 for sonication in the presence of naked DNA or PEI:DNA complexes labeled with YOYO-1. After sonication, the 400 μl cell samples were fixed at different time intervals by mixing with 500 μl of 2.5% glutaraldehyde (Sigma). Samples were then centrifuged at 735×g for 3 min, washed 5 times with PBS, and observed under a multiphoton confocal microscope (LSM 510 Meta, Zeiss, Thornwood, NY).
2.2.7. Stability of PEI:DNA complexes after sonication
To assess their stability after sonication, PEI:DNA complexes were prepared by incubation at a N/P ratio of 7:1 in 400 μl of buffer containing 40 mM Tris acetate and 1 mM EDTA for 20 min at room temperature and exposed to ultrasound in the presence of Optison as described above, but in the absence of cells. After sonication, the complexes were lyophilized completely for 1 h (Cambridge Scientific, Cambridge, MA, USA). After resuspending in 4 μl DNA loading buffer (0.25% w/v bromophenol blue, 40% (w/v) glycerol in water) and 16 μl Tris acetate-EDTA buffer, samples were loaded into the wells of a 0.8% (w/v) agarose gel containing 0.5 μg/mL ethidium bromide. Electrophoresis was carried out for 1 h in Tris acetate-EDTA buffer (pH 7.4) at 100 V. DNA was visualized by fluorescence under UV irradiation. To visualize PEI, the gel was immersed in a PEI-staining solution (0.1% w/v Coomassie blue, 50% v/v methanol and 10% v/v glacial acetic acid) for 4 h, after which it was destained using 10% methanol and 10% glacial acetic acid for a further 4 h.
2.2.8. Statistical analysis
Statistical analyses were performed using Statview software (Version 3; SAS, Cary, NC, USA). Significance of variation between sets of data was determined using the one-way analysis of variance (ANOVA) test and Student’s t-test with P<0.05 being significant. In all the experiments, the standard error of the mean (SEM) was determined between each of the replicate experiments.
3. Results
3.1. Effect of ultrasound on transfection by naked DNA
Our initial studies assessed the effect of ultrasound on the transfection efficiency of AoSMC by naked DNA encoding GFP. Cells incubated with naked DNA were exposed to ultrasound at pressures between 0.5 MPa (corresponding to a mechanical index of 0.7, which is near the expected threshold for inertial cavitation in the presence of a cavitation agent [32]) and 2.0 MPa (which was the maximum pressure of the transducer used). Over this range of pressures, ultrasound increased transfection with increasing pressure in comparison to the sham (Fig. 2A, Student’s t-test, p < 0.0001). At the highest pressure used (which corresponds to an acoustic energy of 32 J), transfection reached its maximum value, which corresponded on a relative basis to an 18-fold increase in expression over the sham and on an absolute basis to transfection of 3.0% of exposed cells. “Sham” samples were treated identically to sonicated samples, except the ultrasound was not turned on. Increased pressure also decreased cell viability to 70–95% in most cases and to 43 ± 8.3% at the highest pressure used (Fig. 2B). We therefore conclude that ultrasound significantly enhanced the transfection efficiency of naked DNA, but also lead to a loss in cell viability. These finding are similar to previous results concerning transfection efficiency of naked DNA [31,33,34,35].
Figure 2.
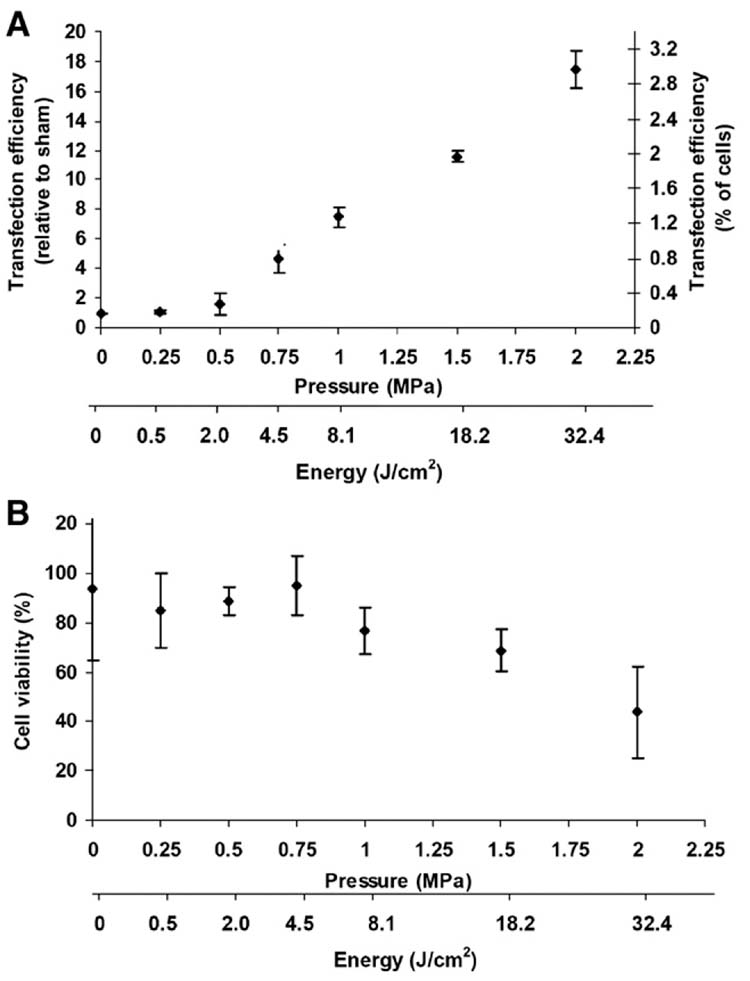
Effect of ultrasound on transfection and viability of AoSMC by naked DNA expressing GFP. (A) Transfection efficiency, expressed on a relative basis by normalizing to sham-exposed cells and on an absolute basis as the percent of cells transfected, is shown as a function of acoustic pressure at a constant exposure time of 240 ms. The corresponding acoustic energy is shown as well. (B) Cell viability is shown as a function of pressure for the same population of cells as in (A). Data are expressed as the average ± SEM for n ≥ 3 replicates.
3.2. Effect of ultrasound on transfection of AoSMC by PEI:DNA complexes expressing GFP
Independently, DNA transfection has been shown in this study to be increased by exposure to ultrasound (Fig. 2) and in the literature to be increased by complexation with PEI [5–7]. This study seeks to test the hypothesis that when used together, ultrasound and PEI can have a synergistic effect to increase DNA transfection. To test this hypothesis, cells were incubated with PEI:DNA complexes and exposed to ultrasound. During sham ultrasound exposure, PEI complexation increased DNA transfection to 15% of cells (Fig. 3A), which is a 90-fold increase over naked DNA. Exposure to ultrasound over a range of pressures increased transfection with increasing pressure in comparison to sham (Student’s t-test, p < 0.001). At the highest pressure used, transfection reached its maximum value, which corresponded on a relative basis to more than a doubling in expression over the PEI:DNA complex sham and a 200-fold increase over the naked DNA sham, which corresponds on an absolute basis to transfection of 34% of exposed cells. Increased pressure also decreased cell viability to approximately 50% in most cases (Fig. 3B).
Figure 3.
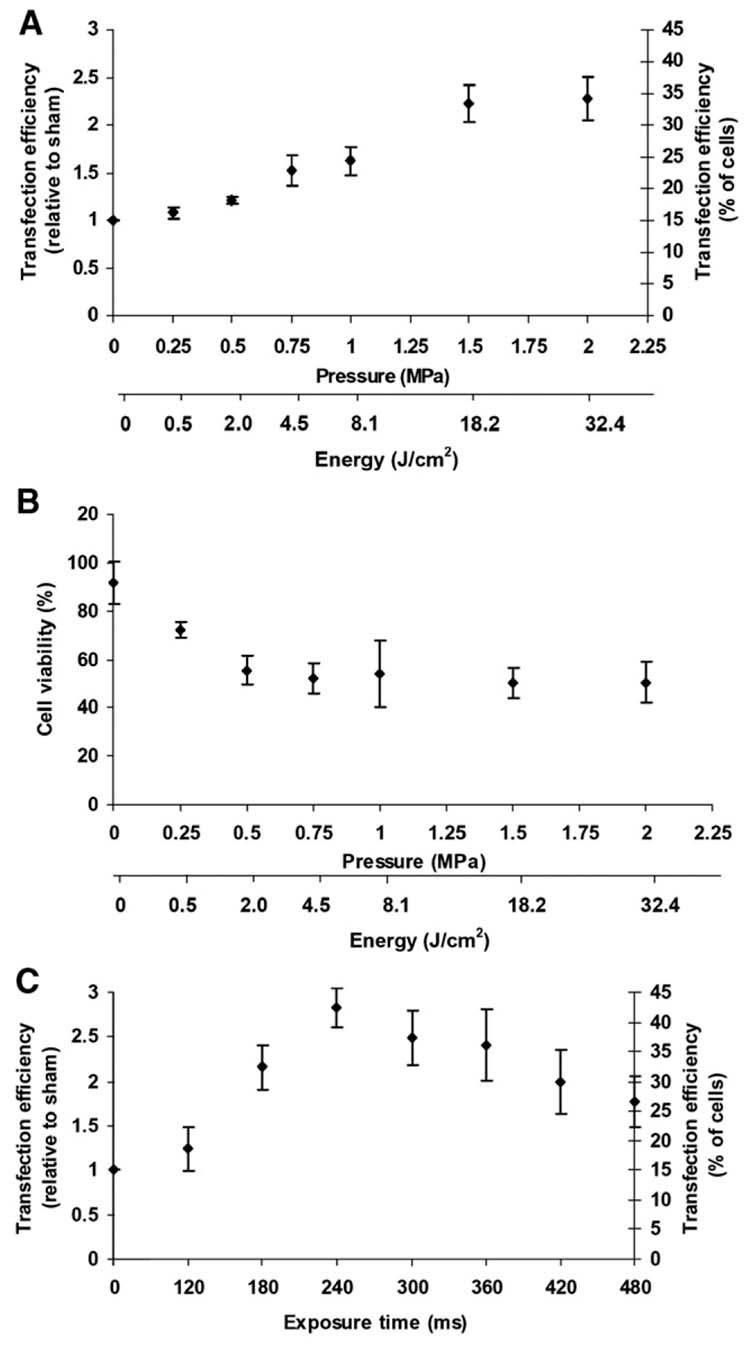
Effect of ultrasound on transfection and viability of AoSMC by PEI:DNA complexes expressing GFP. (A) Transfection efficiency, expressed on both a relative and absolute basis (see Fig. 2), is shown as a function of acoustic pressure at a constant exposure time of 240 ms. (B) Cell viability is shown as a function of pressure for the same populations of cells as in (A). (C) Transfection efficiency is shown as a function of acoustic exposure time at a constant pressure of 1.5 MPa. (average ± SEM, n ≥ 3).
In addition to the effects of pressure, we also measured the effect of ultrasound exposure time on transfection efficiency of PEI:DNA complexes (Fig. 3C). Initially, transfection efficiency increased with increasing exposure time (Student’s t-test, p < 0.0001) until a maximum value at 240 ms, above which transfection efficiency decreased. At the optimal conditions (i.e., 1.5 MPa, 240 ms), transfection efficiency was increased approximately 3-fold relative to the PEI:DNA complex sham.
These data show that ultrasound and PEI together increased DNA transfection more than either enhancement method alone. In addition, the data show that this enhancement was synergistic. These methods were not simply additive, because the sum of enhancement by PEI (15% transfection, Fig. 2A) and enhancement by ultrasound (3% transfection, Fig. 1A) is less than the enhancement by the PEI+ultrasound combination (34% enhancement, Fig. 2A).
3.3. Effect of ultrasound on transfection of DU 145 cells by PEI:DNA complexes expressing luciferase
To determine if the ability of ultrasound and PEI to increase transfection is more broadly applicable, we studied the effects of ultrasound on transfection by PEI complexed to a different DNA plasmid (encoding luciferase) in a different cell line (DU 145 cells). In this different system, transfection activity again increased with increasing pressure, the largest transfection value was again at 1.5 MPa (Student’s t-test, p < 0.01), and again corresponded to an increase close to two-fold relative to sham (Fig. 4). This demonstrates that ultrasound can increase transfection by PEI:DNA complexes using multiple DNA plasmid constructs and in multiple cell lines.
Figure 4.
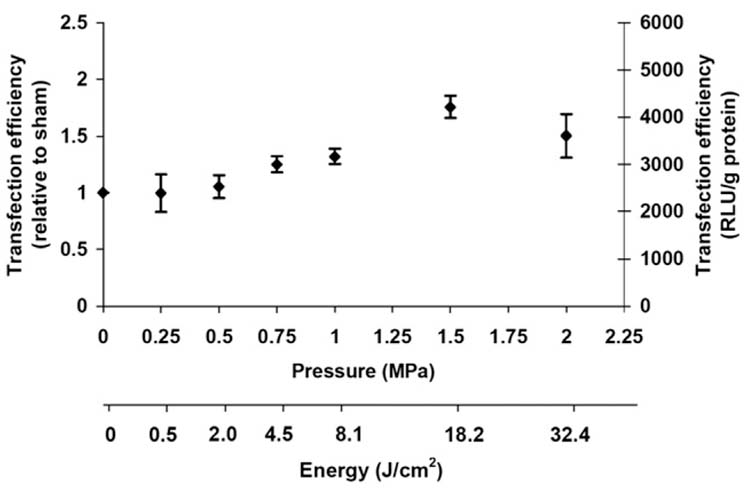
Effect of ultrasound on transfection of DU 145 cells by PEI:DNA complexes expressing luciferase. Transfection efficiency, expressed on both a relative and absolute basis (see Fig. 2), is shown as a function of pressure at a constant exposure time of 240 ms. (average ± SEM, n ≥ 3.
3.4. Effect of incubation time and serum on ultrasound-mediated transfection by DNA and PEI:DNA complexes
To better understand the mechanism by which ultrasound and PEI can synergistically increase transfection, we examined the effects of incubation time and serum on DNA expression. To first examine the kinetics of the naked DNA transfection process, cells were either immediately washed or allowed to incubate with DNA solution for 4 h after ultrasound treatment (Fig. 5). For both real and sham exposure to ultrasound, transfection efficiency was not significantly different in the presence or absence of the 4-h incubation (Student’s t-test, P > 0.05). This suggests that the initial processes related to association and uptake of naked DNA by cells is a rapid one that does not require extensive incubation.
Figure 5.
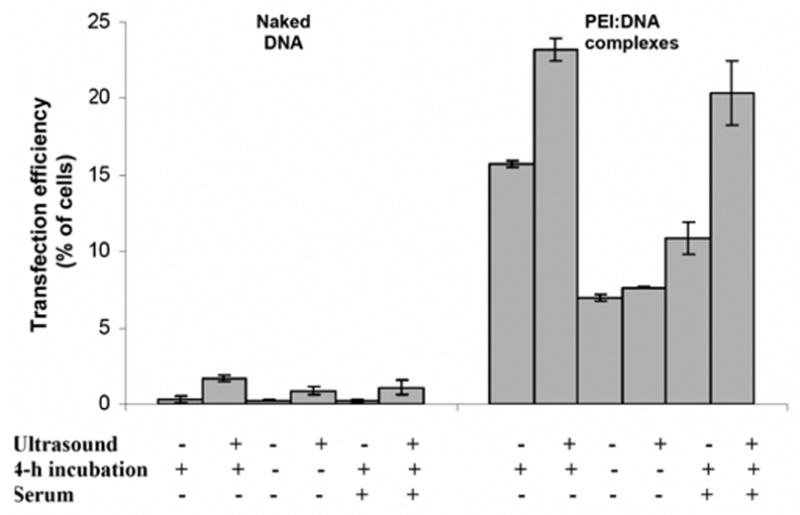
Effect of incubation time and serum on ultrasound-mediated transfection of AoSMC by naked DNA and PEI:DNA complexes expressing GFP. The effect of sonication is shown on cells (i) incubated for 4 h after sonication without serum, (ii) not incubated for 4 h after sonication without serum and (iii) incubated for 4 h after sonication with 10% fetal bovine serum. (average ± SEM, n ≥ 3).
In contrast, transfection efficiency by PEI:DNA complexes was significantly lower in the absence of a 4-h incubation following both real and sham exposure to ultrasound (Student’s t-test, P < 0.01). This suggests that the initial association and uptake of PEI:DNA complexes by cells is a slower one that requires extensive incubation. Most notably, the ability of ultrasound to increase transfection efficiency was lost in the absence of a 4-h incubation (Student’s t-test, P > 0.05). This suggests that the synergistic effect of ultrasound on transfection by PEI:DNA complexes acts primarily on this slow process that occurs during incubation.
To further examine mechanisms, the effect of serum on transfection efficiency was also examined. This scenario is of interest because the presence of serum better emulates in vivo conditions and has been previously shown to reduce transfection efficiency of PEI:DNA systems [36]. Consistent with previous findings, transfection efficiencies of both naked DNA and PEI:DNA complexes were reduced in presence of serum for both real and sham exposure to ultrasound (Student’s t test, p < 0.001). Although the absolute transfection values were reduced, the relative enhancement caused by ultrasound was unaffected. In the absence of serum, ultrasound increased transfection by naked DNA and PEI:DNA complexes by 4.8- and 1.5-fold, respectively. In the presence of serum, ultrasound similarly increased transfection by naked DNA and PEI:DNA complexes by 5.0- and 1.8-fold, respectively. This demonstrates that the ability of ultrasound to enhance transfection was unaffected by serum, which is a promising sign for future applications in vivo.
3.5. Effect of ultrasound on intracellular uptake of DNA and PEI:DNA complexes
The elevated transfection observed in this study might be explained by increased intracellular uptake of DNA by ultrasound and PEI complexation. To test this hypothesis, uptake of naked DNA was measured by flow cytometry using fluorescently tagged DNA and found to be increased by sonication (Fig. 6A). Similarly, uptake of DNA complexed with PEI was increased by sonication (Fig. 6B). Consistent with the proposed hypothesis, the relative levels of uptake (Fig. 6) scaled in rank order with the relative levels of transfection (Figs. 1–2), where (i) naked DNA with sham exposure was lowest, followed by (ii) naked DNA with sonication, then (iii) PEI:DNA complex with sham exposure and finally (iv) PEI:DNA complex with sonication as the optimal condition for both uptake and transfection. Quantitiative calibration of these results determined that an average of 6.0 × 104 DNA molecules/cell were delivered in the case of PEI complexation with sham exposure and an average of 9.3 × 104 DNA molecules/cell were delivered in the case of PEI complexation with sonication.
Figure 6.
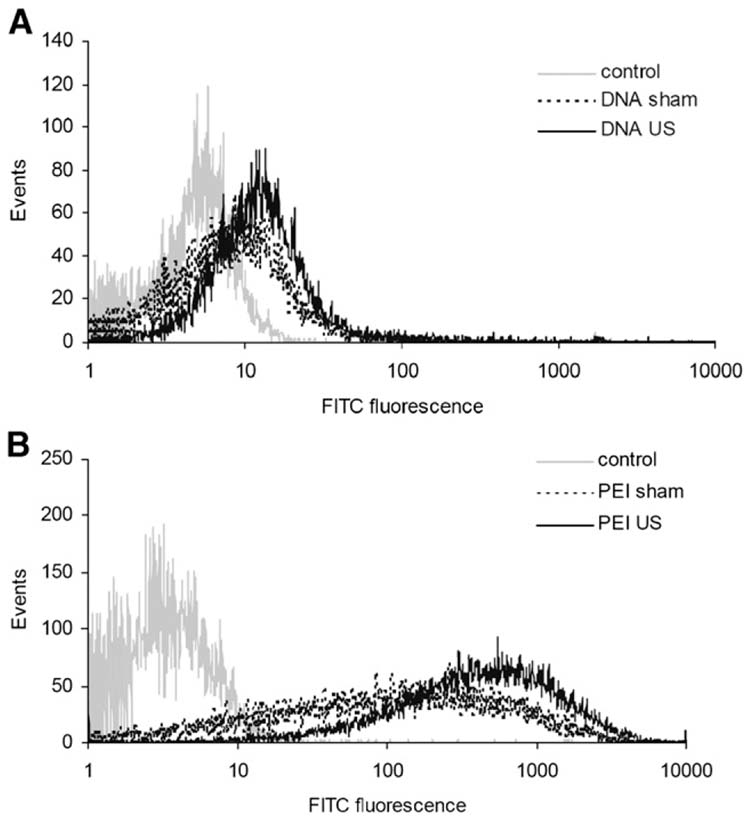
Effect of ultrasound on intracellular uptake of (A) naked DNA and (B) PEI:DNA complexes by AoSMC. Cellular fluorescence of DNA labeled with YOYO-1 dye was measured by flow cytometry for cells exposed to sham ultrasound or sonication at 1.5 MPa for 240 ms.
Confocal microscopy was employed to further study the intracellular uptake process associated with PEI:DNA complexes. First, control cells were labeled with red-fluorescent wheat germ lectin and exposed to ultrasound in the absence of DNA. A representative cell with a red-stained membrane is shown in Fig. 7A. Next, cells were either sonicated or sham sonicated in the presence of green-fluorescent DNA complexed to PEI. When these cells were fixed within 2 s after ultrasound exposure, sham-exposed cells contained negligible amounts of green-fluorescent DNA fluorescence (Fig. 7B, sham), whereas many sonicated cells displayed bright intracellular green fluorescence due to internalized DNA (Fig. 7B, sonicated). The green fluorescence appeared to be in the cytoplasm and excluded from the nucleus. Rapid uptake of naked DNA following sonication has been observed previously [37,38]. When examined after 10 min, sham samples exhibited fluorescence from DNA associated with the plasma membrane (Fig. 7C, sham), whereas many sonicated cells still displayed intracellular DNA fluorescence, often with greater intensity (Fig. 7C, sonicated). Finally, when viewed after 4 h, green fluorescent DNA was visible in the cytoplasm of sham cells (Fig. 7D, sham). DNA internalization after 4 h observed here is similar to previous findings showing intracellular trafficking of PEI:DNA complexes in the absence of ultrasound over a similar time scale [39]. After 4 h, intracellular DNA continued to be seen in sonicated cells, and at still greater intensity (Fig. 7D, sonicated). This increasing intensity over time after sonication suggests that uptake of PEI:DNA complexes may be rapidly initiated and continues to occur for hours after sonication.
Figure 7.
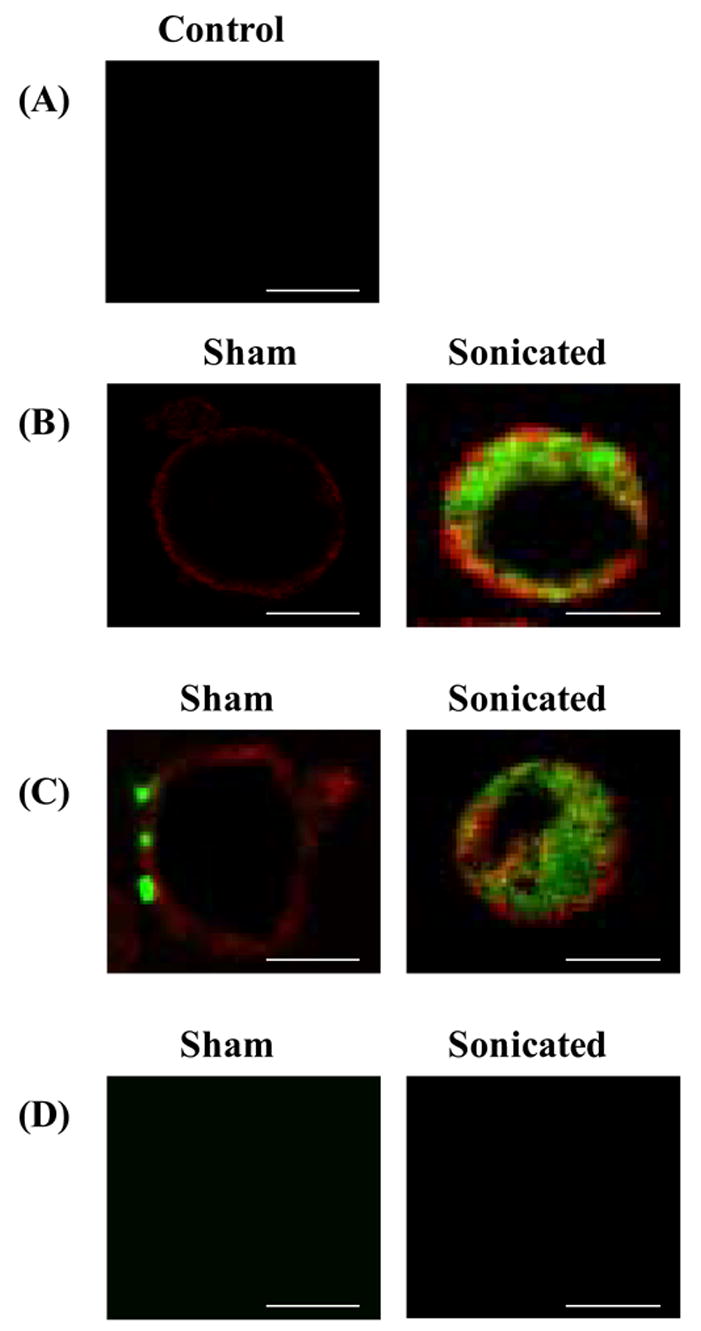
Confocal microscopy imaging of the effect of ultrasound on intracellular uptake of PEI:DNA complexes at (A) A control cell with its membrane labeled with red-fluorescent wheat germ lectin exposed to ultrasound in the absence of PEI:DNA complexes. Cells labeled with ref-fluorescent wheat germ lectin and exposed to sham ultrasound (left) or ultrasound (right) in the prescence of green-fluorescent PEI:DNA complexes imaged at (B) 2 s, (C) 10 min and (D) 4 h after sonication. Scale bar = 10 μm.
3.6. Stability of DNA and PEI:DNA complexes after sonication
Because ultrasound is known to damage DNA at extreme conditions, we assessed the stability of DNA and PEI:DNA complexes after sonication. Agarose gel electrophoresis showed no changes in the DNA after sonication over the range of conditions used in this study (Fig. 8A). A similar set of experiments showed no changes in the PEI:DNA complexes after sonication either (Fig. 8B). The lack of bands toward the positive electrode show that no negatively charged DNA was released from the PEI:DNA complexes. The presence of unchanged bands toward the negative electrode show that positively charged PEI:DNA complexes remained intact. To validate that the DNA within the PEI:DNA complexes remained intact, these complexes were dissociated using poly(aspartic acid) after sonication [40]. The liberated DNA was then analysed by agarose gel electrophoresis, which showed no changes in the DNA over the range of conditions used in this study (data not shown).
Figure 8.
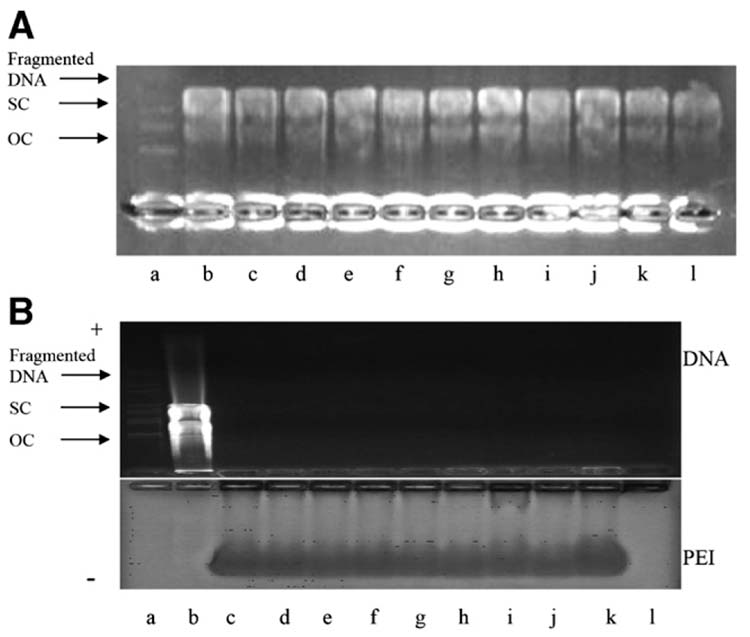
Stability of naked DNA and PEI:DNA complexes after sonication assessed by agarose gel electrophoresis. (A) Stablity of naked DNA after sonication: (a) molecular weight markers; (b) untreated DNA; (c) 0.5 MPa, 240 ms; (d) 0.75 MPa, 240 ms; (e) 1.0 MPa, 240 ms; (f) 1.5 MPa, 240 ms; (g) 2.0 MPa, 240 ms; (h) 1.5 MPa, 300 ms; (i) 1.5 MPa, 360 ms; (j) 0.5 MPa, 420 ms; (k) 0.5 MPa, 480 ms; (l) sham ultrasound. (B) Stability of PEI:DNA complexes after sonication: (a) molecular weight marker; (b) naked DNA, sham ultrasound; (c) PEI:DNA complex, sham ultrasound; (d) 0.5 MPa, 240 ms; (e) 0.75 MPa, 240 ms; (f) 1.0 MPa, 240 ms; (g) 1.5 MPa, 240 ms; (h) 2.0 MPa, 240 ms; (i) 1.5 MPa, 180 ms; (j) 1.5 MPa, 300 ms; (k) 1.5 MPa, 360 ms; (l) 1.5 MPa, 420 ms. OC = Open circular DNA, SC = supercoiled DNA.
4. Discussion
This study examined the effects of ultrasound and PEI:DNA complexation on transfection of AoSMC and DU 145 cells with GFP and luciferase reporter genes. Sonication of naked DNA increased transfection by up to 18-fold relative to sham. DNA complexation with PEI increased transfection by 90-fold relative to naked DNA. The combination of sonication with PEI complexation increased transfection up to 200-fold relative to naked DNA with sham ultrasound. Altogether, these data indicate that the combination of ultrasound with PEI:DNA complexation is an effective method to increase transfection.
This study also tested the hypothesis that ultrasound and PEI can have a synergistic effect to increase DNA transfection. The data from this study are consistent with this hypothesis. First, the combination of ultrasound and PEI:DNA complexation transfected cells with greater efficiency than either method alone. Moreover, this combination also transfected cells with greater efficiency than the sum of the transfection efficiencies of either method alone, i.e., sonication alone transfected up to 3% of cells (Fig. 2), PEI;DNA complexation with sham ultrasound transfected 15% of cells (Fig. 3), and PEI:DNA complexation with sonication transfected 34% of cells (Fig. 3), which is greater than the sum, 3% + 15% = 18%. This indicates a synergistic effect of sonication and PEI:DNA complexation on transfection.
The mechanism of how ultrasound synergistically increases the transfection efficiency of PEI:DNA complexes may involve a number of factors. First, the mechanism is believed to involve cavitation bubble activity stimulated by ultrasound. Indeed, cavitation activity was present in the sonicated samples in this study, as determined by hydrophone measurements showing increased broadband noise (data not shown). In addition, sonication in the absence of Optison contract agent, which serves to nucleate cavitation activity, had not significant effect on cell transfection or viability (data not shown).
Ultrasound is also known to increase intracellular uptake of large molecules [41], including enhanced delivery of artificial chromosomes complexed with cationic lipids, which are as large as 1–2 μm in size [42]. Increased intracellular delivery is believed to occur due to the formation of transient pores of nanometer to micron dimensions [25,43,44]. Consistent with this expectation, our flow cytometry results show that ultrasound increased intracellular DNA delivery for both naked DNA and PEI:DNA complexes (Fig. 6), which could at least partially explain the increased transfection efficiency. Confocal microscopy also suggested that more DNA might be delivered to cells after sonication (Fig. 7).
The total increase in intracellular DNA due to sonication, however, was relatively small. DNA delivery using PEI:DNA complexes was only increased relative to sham by 55%, while the increase of naked DNA delivery was even less (Fig. 6). These modest increases in DNA uptake may not be sufficient to explain the much larger increases in transfection, especially for naked DNA. Moreover, it is also not clear why just delivering more DNA by two independent methods should have a synergistic, rather than additive, effect. Thus, other effects of ultrasound on cell behaviour may be involved.
Additional mechanistic insight may come from the observation that (i) PEI-enhanced and ultrasound-enhanced uptake occurred both with and without a 4-h incubation, but enhancement of PEI-enhanced uptake was larger with the 4-h incubation and (ii) the synergistic effect of PEI complexation and sonication disappeared without the 4 h incubation. This suggests that while ultrasound can affect naked DNA at the time of sonication, its effect on PEI:DNA complexes is a long-lived effect that takes place with a time scale up to hours. In a related study, transfection efficiency of Lipofectamine:DNA complexes was similarly only increased by sonication when followed by a 3-h incubation [18].
A final mechanistic consideration is that sonication could alter the DNA or PEI:DNA complexes in a manner that affected transfection. However, the integrity of naked DNA, PEI:DNA complexes, and DNA isolated from PEI:DNA complexes was unchanged by sonication at the conditions used in this study. This suggests that sonication did not alter the DNA or PEI:DNA complexes in a manner that might explain the synergistic increase in transfection efficiency. These data also suggest that sonication did not damage the DNA, which can occur at conditions more extreme than those used in this study [40]. Although not observed here, previous work has shown that complexing DNA with cationic polymers and lipids can increase the stability of plasmid DNA during sonication [18,40,45,46].
Although sonication and PEI:DNA complexation can increase transfection efficiency, it can also be accompanied by cell death, as shown in this and previous studies [6,47,48]. Thus, practical applications require balancing increased transfection against losses of viability. For many in vitro and some in vivo applications, significant cell death may be acceptable and therefore stronger ultrasound conditions that lead to larger transfection efficiencies may be used. For other applications where only little or no cell death can be tolerated, weaker ultrasound conditions should be used to cause more modest increases in transfection efficiency at higher cell viability.
Conclusions
This study demonstrated that sonication of naked DNA increased transfection up to 18-fold, DNA complexation with PEI increased transfection 90-fold, and the combination of sonication with PEI:DNA complexation increased transfection up to 200-fold relative to naked DNA with sham ultrasound. This optimal condition transfected 34% of cells. Further analysis of these data supported the hypothesis that ultrasound and PEI can have a synergistic effect to increase DNA transfection. Increased transfection efficiency was at least partially explained by increased intracellular delivery of DNA due to sonication. The synergistic increase of transfection by combining ultrasound with PEI:DNA complexation was found to require a 4-h incubation after sonication and occurred even in the presence of serum. No effects of ultrasound on the integrity of either naked DNA or PEI:DNA complexes were found. Thus, ultrasound can be used to synergistically increase transfection efficiency of PEI:DNA complexes for gene delivery applications, possibly involving extracorporeal ultrasound focused non-invasively on tissues inside the body.
Acknowledgments
The authors would like to thank Esi Ghartey-Tagoe, Robyn Schlicher, Vladimir Zarnitsyn, Johnafel Crow, Pavel Kamaev and Daniel Hallow for helpful discussions. This work was supported in part by the National Institutes of Health. MRP is the Emerson-Lewis Faculty Fellow and is a member of the Institute for Bioengineering and Bioscience and the Center for Drug Design, Development and Delivery at Georgia Tech.
Footnotes
Submitted to the Journal of Controlled Release for consideration as a Research Paper in the Gene Delivery section.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McKusick VA. Mendelian inheritance in man: A catalogue of human genes and genetic disorders. 11. Baltimore: John Hopkins University press; 1994. [Google Scholar]
- 2.Park TG, Jeong JH, Kim SW. Current status of polymeric gene delivery systems. Adv Drug Deliv Rev. 2006;58:467–86. doi: 10.1016/j.addr.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Rolland AP. From genes to gene medicines: recent advances in nonviral gene delivery. Crit Rev Ther Drug Carrier Syst. 1998;15:143–198. doi: 10.1615/critrevtherdrugcarriersyst.v15.i2.20. [DOI] [PubMed] [Google Scholar]
- 4.Iwamoto HS, Trapnell BC, McConnell CJ, Daugherty C, Whitsett JA. Pulmonary inflammation associated with repeated, prenatal exposure to an E1, E3-deleted adenoviral vector in sheep. Gene Ther. 1999;6:98–106. doi: 10.1038/sj.gt.3300804. [DOI] [PubMed] [Google Scholar]
- 5.Lungwitz U, Breunig M, Blunk T, Gopferich A. Polyethylenimine-based non-viral gene delivery systems. Eur J Pharm Biopharm. 2005;60:247–66. doi: 10.1016/j.ejpb.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 6.Neu M, Fischer D, Kissel T. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J Gene Med. 2005;7:992–1009. doi: 10.1002/jgm.773. [DOI] [PubMed] [Google Scholar]
- 7.Godbey WT, Wu KK, Mikos AG. Poly(ethylenimine) and its role in gene delivery. J Control Release. 1999;60:149–160. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- 8.Kichler A, Leborgne C, Coeytaux E, Danos O. Polyethyleneimine-mediated gene delivery: a mechanistic study. J Gene Med. 2001;3:135–144. doi: 10.1002/jgm.173. [DOI] [PubMed] [Google Scholar]
- 9.Boletta A, Benigni A, Lutz J, Remuzzi G, Soria MR, Monaco L. Non-viral gene delivery to the rat kidney with poly(ethylenimine) Hum Gene Ther. 1997;8:1243–1251. doi: 10.1089/hum.1997.8.10-1243. [DOI] [PubMed] [Google Scholar]
- 10.Lemkine GF, Goula D, Becker N, Paleari L, Levi G, Demeneix BA. Optimisation of polyethylenimine based gene delivery to mouse brain. J Drug Target. 1999;7:305. doi: 10.3109/10611869909085513. [DOI] [PubMed] [Google Scholar]
- 11.Miller DL, Song J. Tumor growth reduction and DNA transfer by cavitation-enhanced high-intensity focused ultrasound in vivo. Ultrasound Med Biol. 2003;29:887–893. doi: 10.1016/s0301-5629(03)00031-0. [DOI] [PubMed] [Google Scholar]
- 12.Mehier-Humbert S, Bettingerb T, Yanb F, Guy RH. Ultrasound-mediated gene delivery: Kinetics of plasmid internalization and gene expression. J Control Release. 2005;104:203–211. doi: 10.1016/j.jconrel.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 13.Mitragotri S. Healing sound: the use of ultrasound in drug delivery and other therapeutic applications. Nat Rev Drug Discov. 2005;4:255–60. doi: 10.1038/nrd1662. [DOI] [PubMed] [Google Scholar]
- 14.Miller DL, Pislaru SV, Greenleaf JE. Sonoporation: mechanical DNA delivery by ultrasonic cavitation. Somat Cell Mol Genet. 2002;27:115–34. doi: 10.1023/a:1022983907223. [DOI] [PubMed] [Google Scholar]
- 15.Taniyama Y, Tachibana K, Hiraoka K, Aoki M, Yamamoto S, Matsumoto K, Nakamura T, Ogihara T, Kaneda Y, Morishita R. Development of safe and efficient novel nonviral gene transfer using ultrasound: Enhancement of transfection efficiency of naked plasmid DNA in skeletal muscle. Gene Ther. 2002;9:372–380. doi: 10.1038/sj.gt.3301678. [DOI] [PubMed] [Google Scholar]
- 16.Frenkel PA, Chen S, Thai T, Shohet RV, Grayburn PA. DNA-loaded albumin microbubbles enhance ultrasound-mediated transfection in vitro. Ultrasound Med Biol. 2002;28:817–822. doi: 10.1016/s0301-5629(02)00518-5. [DOI] [PubMed] [Google Scholar]
- 17.Rosenthal I, Sostaric JZ, Riesz P. Sonodynamic therapy – a review of the synergistic effects of drugs and ultrasound. Ultrason Sonochem. 2004;11:349–63. doi: 10.1016/j.ultsonch.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Lawrie A, Brisken AF, Francis SE, Tayler DI, Chamberlin J, Crossman DC, Cumberland DC, Newman CM. Ultrasound enhances reporter gene expression after transfection of vascular cells in vitro. Circulation. 1999;99:2617–2620. doi: 10.1161/01.cir.99.20.2617. [DOI] [PubMed] [Google Scholar]
- 19.Shohet RV, Chen S, Zhou YT, Wang Z, Meidell RS, Unger RH, Grayburn PA. Echocardiographic destruction of albumin microbubbles directs gene delivery to the myocardium. Circulation. 2000;101:2554–2556. doi: 10.1161/01.cir.101.22.2554. [DOI] [PubMed] [Google Scholar]
- 20.Chen S, Shohet RV, Bekeredjian R, Frenkel P. Optimization of ultrasound parameters for cardiac gene delivery of adenoviral or plasmid deoxyribonucleic acid by ultrasound-targeted microbubble destruction. J Am Coll Cardiol. 2003;42:301–308. doi: 10.1016/s0735-1097(03)00627-2. [DOI] [PubMed] [Google Scholar]
- 21.Koch S, Pohl P, Cobet U, Rainov NG. Ultrasound enhancement of liposome-mediated cell transfection is caused by cavitation effects. Ultrasound Med Biol. 2000;26:897–903. doi: 10.1016/s0301-5629(00)00200-3. [DOI] [PubMed] [Google Scholar]
- 22.Unger EC, McCreery T, Sweitzer RH. Ultrasound enhances gene expression of liposomal transfection. Invest Radiol. 1997;32:723–727. doi: 10.1097/00004424-199712000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Lawrie A, Brisken AF, Francis SE, Cumberland DC, Crossman DC, Newman CM. Microbubble-enhanced ultrasound for vascular gene delivery. Gene Ther. 2000;7:2023–2027. doi: 10.1038/sj.gt.3301339. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J, Russel DW. Molecular cloning: A laboratory manual. 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- 25.Guzman HR, Nguyen DX, Khan S, Prausnitz MR. Ultrasound-mediated disruption of cell membranes. I. Quantification of molecular uptake and cell viability. J Accoust Soc Am. 2001;110:588–596. doi: 10.1121/1.1376131. [DOI] [PubMed] [Google Scholar]
- 26.Boussif O, Lezoualc’H F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr J-P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prausnitz MR, Lau BS, Milano CD, Conner S, Langer R, Weaver JC. A quantitative study of electroporation showing a plateau in net molecular transport. Biophys J. 1993;65:414–422. doi: 10.1016/S0006-3495(93)81081-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guzman HR, Nguyen DX, Khan S, Prausnitz MR. Ultrasound-mediated disruption of cell membranes. II. Heterogenous effects on cells. J Accoust Soc Am. 2001;110:597–606. doi: 10.1121/1.1376130. [DOI] [PubMed] [Google Scholar]
- 29.Bradford MM. Rapid and sensitive method for quantification of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 30.Ogris M, Wagner E, Steinlein P. A versatile assay to study cellular uptake of gene transfer complexes by flow cytometry. Biochim Biophys Acta. 2000;1474:237–243. doi: 10.1016/s0304-4165(00)00018-0. [DOI] [PubMed] [Google Scholar]
- 31.Zarnitsyn VG, Prausnitz MR. Physical parameters influencing optimisation of ultrasound-mediated DNA transfection. Ultrasound Med Biol. 2004;30:527–538. doi: 10.1016/j.ultrasmedbio.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 32.AIUM (American Institute of Ultrasound in Medicine) Mechanical bioeffects from diagnostic ultrasound: AIUM consensus statements. Section 7. Discussion of the mechanical index and other exposure parameters. J Ultrasound Med. 2000;19:143–148. doi: 10.7863/jum.2000.19.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang HD, Lu QL, Xue SA, Halliwell M, Kodama T, Cosgrove DO, Stauss HJ, Partridge TA, Blomley MJ. Optimisation of ultrasound-mediated gene transfer (sonoporation) in skeletal muscle cells. Ultrasound Med Biol. 2004;30:1523–1529. doi: 10.1016/j.ultrasmedbio.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 34.Taniyama Y, Tachibana K, Hiraoka K, Namba T, Yamasaki K, Hashiya N, Aoki M, Ogihara T, Yasufumi K, Morishita R. Local delivery of plasmid DNA into rat carotid artery using ultrasound. Circulation. 2002;105:1233–1239. doi: 10.1161/hc1002.105228. [DOI] [PubMed] [Google Scholar]
- 35.Kim HJ, Greenleaf JF, Kinnick RR, Bronk JT, Bolander ME. Ultrasound-mediated transfection of mammalian cells. Hum Gene Ther. 1996;7:1339–1346. doi: 10.1089/hum.1996.7.11-1339. [DOI] [PubMed] [Google Scholar]
- 36.Tang GP, Zeng JM, Gao SJ, Ma YX, Shi L, Li Y, Too HP, Wang S. Polyethylene glycol modified polyethylenimine for improved CNS gene transfer: effects of PEGylation extent. Biomaterials. 2003;24:2351–2362. doi: 10.1016/s0142-9612(03)00029-2. [DOI] [PubMed] [Google Scholar]
- 37.Duvshani-Eshet M, Machluf M. Therapeutic ultrasound optimization for gene delivery: A key factor achieving nuclear DNA localization. J Control Release. 2005;108:513–528. doi: 10.1016/j.jconrel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 38.Mehier-Humberta S, Bettingerb T, Yanb F, Guy RH. Plasma membrane poration induced by ultrasound exposure: Implication for drug delivery. J Control Release. 2005;104:213–222. doi: 10.1016/j.jconrel.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 39.Godbey WT, Wu K, Mikos AG. Tracking the intracellular path of poly(ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci USA. 1999;96:5177–5181. doi: 10.1073/pnas.96.9.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuo JS, Jan M, Sung KC. Evaluation of the stability of polymer-based plasmid DNA delivery systems after Ultrasound exposure. Int J Pharm. 2003;257:75–84. doi: 10.1016/s0378-5173(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 41.Guzman HR, Nguyen DX, McNamara AJ, Prausnitz MR. Equilibrium loading of cells with macromolecules by ultrasound: effects of molecular size and acoustic energy. J Pharm Sci. 2002;91:1693–1701. doi: 10.1002/jps.10156. [DOI] [PubMed] [Google Scholar]
- 42.Oberle V, de Jong G, Drayer JI, Hoekstra D. Efficient transfer of chromosome-based DNA constructs into mammalian cells. Biochim Biophys Acta. 2004;1676:223–230. doi: 10.1016/j.bbaexp.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 43.Tachibana K, Uchida T, Ogawa K, Yamashita N, Tamura K. Induction of cell-membrane porosity by ultrasound. Lancet. 1999;353:1409. doi: 10.1016/S0140-6736(99)01244-1. [DOI] [PubMed] [Google Scholar]
- 44.Schlicher RK, Radhakrishna H, Tolentino TP, Apkarian RP, Zarnitsyn V, Prausnitz MR. Mechanism of intracellular delivery by acoustic cavitation. Ultrasound Med Biol. 2006;32:915–924. doi: 10.1016/j.ultrasmedbio.2006.02.1416. [DOI] [PubMed] [Google Scholar]
- 45.Kleemann E, Dailey LA, Abdelhady HG, Gessler T, Schmehl T, Roberts CJ, Davies MC, Seeger W, Kissel T. Modified polyethylenimines as non-viral gene delivery systems for aerosol gene therapy: investigations of the complex structure and stability during air-jet and ultrasonic nebulization. J Control Release. 2004;100:437–450. doi: 10.1016/j.jconrel.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 46.Wassan EK, Reimer DL, Bally MB. Plasmid DNA is protected against ultrasonic cavitation-induced damage when complexed to cationic liposomes. J Pharm Sci. 1996;85:427–433. doi: 10.1021/js9504752. [DOI] [PubMed] [Google Scholar]
- 47.Fischer D, Bieber T, Li Y, Elsasser HP, Kissel T. A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: effect of molecular weight on transfection efficiency and cytotoxicity. Pharm Res. 1999;16:1273–1279. doi: 10.1023/a:1014861900478. [DOI] [PubMed] [Google Scholar]
- 48.Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005;11:990–995. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]