

The transition metal-catalyzed allylic substitution with unstabilized carbon nucleophiles represents an important cross-coupling reaction for the construction of ternary carbon stereogenic centers.1 A key and significant limitation with this approach is the necessity to employ allylic alcohol derivatives that provide symmetrical π-allyl intermediates, which thereby circumvent problems associated with regiochemical infidelity.2 Another serious issue arises from the basic nature of the unstabilized nucleophile, which either promotes elimination of the metal-allyl intermediate or hydrolysis of the leaving group in the allylic alcohol fragment. Furthermore, the stereospecific alkylation is often subject to stereochemical inversion through the direct addition of the nucleophile to the metal center followed by concomitant reductive elimination.3
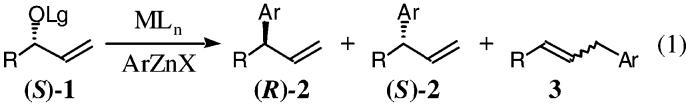
We envisioned that the rhodium-catalyzed allylic alkylation with an organozinc would facilitate the regio- and enantiospecific alkylation as a result of the propensity for the reaction to proceed through a configurationally stable π-allyl or enyl (σ + π) organometallic intermediate and the low basicity of the nucleophilic reagent.4,5 Herein, we now describe the first regio- and enantiospecific metal-catalyzed intermolecular allylic alkylation of unsymmetrical allylic alcohol derivatives (S)-1, using aryl organozinc halides, for the construction of 3-aryl propenyl derivatives (R)-2 with inversion of absolute configuration (eq 1).
Preliminary studies demonstrated that the trimethyl phosphite-modified Wilkinson's catalyst that had proven so general for the allylic alkylation using stabilized carbon and heteroatom nucleophiles was not an effective catalyst with organozinc reagents. Interestingly, although the application of a hydrotris(pyrazolyl)-borate rhodium complex had not been examined in the context of an allylic substitution reaction, this catalyst proved optimum for organozinc reagents as nucleophiles.6 While traditional leaving groups favored the formation of the primary allylic alkylation adducts, fluorinated leaving groups dramatically improved the specificity (2°:1° = (CF3)2CHOCO > CF3CO > MeOCO > MeCO). Table 1 outlines the optimization of the nucleophile and catalyst components of this challenging cross-coupling reaction with the optimal leaving group. Treatment of the allylic carbonate 1a with a catalytic amount of TpRh(C2H4)2 and the organozinc reagent derived from the transmetalation of phenyllithium with a zinc halide salt, furnished the alkylation products 2a/3a, demonstrating that zinc bromide was the optimum salt for the alkylation (entry 2 vs 1/3). Additional studies examined the effect of the organozinc reagent on selectivity (entries 2, 4, and 6). This study suggested that lithium bromide, generated as a consequence of the transmetalation step, might influence the regiospecificity through catalyst/nucleophile modification (entries 2/4 vs 6).7 Interestingly, the addition of lithium bromide to the catalyst prior to the introduction of the organozinc and allylic carbonate 1a, furnished the secondary allylic alkylation adduct 2a with significantly improved regiospecificity (entries 5 and 7). Finally, dibenzylidenacetone (dba) was added to improve the selectivity and reproducibility of the cross-coupling reaction, by serving as a surrogate ligand, given the volatility of the ethylene ligands (entry 8).
Table 1.
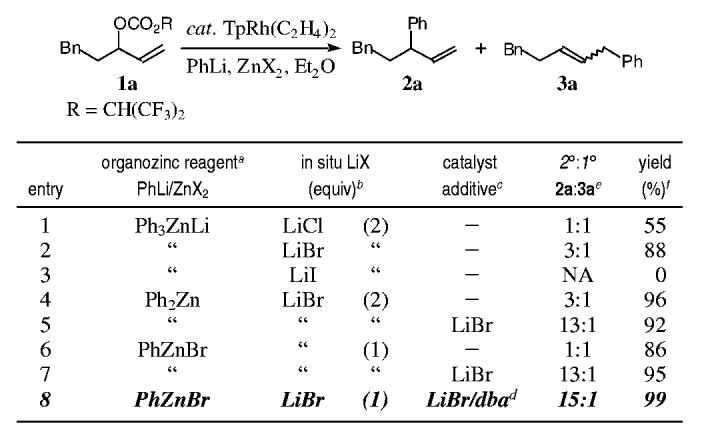
Effect of Lithium Halide Salts on the Nucleophile and Catalyst in the Regiospecific Rhodium-Catalyzed Allylic Alkylation a All the reactions were carried out on a 0.1 mmol reaction scale using 10 mol % of TpRh(C2H4)2 and 2.0 equiv of the arylzinc reagent at 0 °C for ≤ min. b The amount of lithium halide generated is a consequence of the phenyllithium/zinc halide stoichiometry. c The catalyst was modified with 200 mol % lithium bromide where indicated. d 20 mol % dibenzylideneacetone (dba). e Ratios of regioisomers were determined using capillary GLC on aliquots of the crude reaction mixture. f GLC yields.
Table 2 summarizes the application of the optimized reaction conditions (Table 1, entry 8)8 to a variety of racemic secondary allylic carbonates (vide supra). The allylic alkylation is clearly tolerant of electron-withdrawing and -donating substituents within the organozinc reagent, in which the former provides optimum selectivity (Table 2, entries 2-4). Additional studies demonstrated that linear and branched allylic carbonates serve as suitable substrates (entries 5-12), in which the α-branched derivatives afford the secondary alkylation products with optimum selectivity (entries 7 and 8) in sharp contrast to our previous studies.4 The allylic alkylation also proved feasible for the benzyl and acetate derivatives (entries 10 and 12), illustrating excellent substrate tolerance to the organozinc reagent. Hence, the regiospecific rhodium-catalyzed allylic alkylation with aryl zinc halides provides an important new method for the construction of ternary carbon stereogenic centers.
Table 2.
Scope of the Regiospecific9 Rhodium-Catalyzed Allylic Alkylation with Aryl Zinc Halides (eq 1; (Lg = CO2CH(CF3)2)a
| entry | allylic alcohol derivative, R = | 1 | nucleophile (Ar) | 2°:1° 2:3b,c | yield (%)d |
|---|---|---|---|---|---|
| 1 | Ph(CH2)2 | a | Ph- | 15:1 | 87 |
| 2 | “ | “ | p-MeO-C6H4- | 13:1 | 81 |
| 3 | “ | “ | p-Me-C6H4- | 15:1 | 84 |
| 4 | “ | “ | p-F-C6H4- | ≥19:1 | 85 |
| 5 | Me | b | Ph- | 13:1 | 76 |
| 6 | Me(CH2)8 | c | “ | 12:1 | 91 |
| 7 | iPr | d | “ | ≥19:1 | 74 |
| 8 | cHex | e | “ | ≥19:1 | 72 |
| 9 | iBu | f | “ | 9:1 | 91 |
| 10 | PhCH2 | g | “ | 13:1 | 91 |
| 11 | TBSO(CH2)5 | h | “ | 14:1 | 87 |
| 12 | AcO(CH2)5 | i | “ | ≥19:1 | 95 |
All reactions were carried out on a 0.1 mmol reaction scale.
Ratios of regioisomers were determined by 400 MHz 1H NMR.
The primary products 3 were prepared for comparison using Ph3ZnLi at room temperature.
Isolated yields.
The ability to obtain excellent regiospecificity prompted the examination of the enantiomerically enriched allylic carbonate (S)-1b with the aryl zinc bromide necessary for the synthesis of (S)ibuprofen 6,10 to determine the stereochemical course of this reaction. Treatment of (S)-1b (95%ee) under the optimized reaction conditions, furnished the 3-aryl propenyl derivatives (R)-4/5 in 90% yield (2°:1° = 10:1), with inversion of absolute configuration (100%cee). This result is consistent with direct addition of the nucleophile to the metal followed by reductive elimination and thereby indicates a significant departure from our previous studies.4 The synthesis of (S)-ibuprofen was then completed through the oxidative cleavage of the alkenes (R)-4/5 using catalytic ruthenium trichloride and sodium periodate at room temperature to afford 6 in 74% yield.11
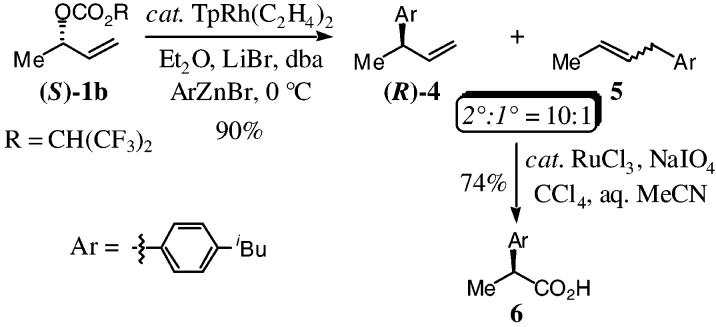
In conclusion, we have developed a new regio- and enantiospecific rhodium-catalyzed allylic alkylation of acyclic unsymmetrical chiral nonracemic allylic alcohol derivatives with aryl zinc bromides. This study demonstrates that the hydrotris(pyrazolyl)-borate rhodium catalyst and requisite zinc(II) halide salt are crucial for efficiency, while the addition of lithium bromide to the catalyst is necessary for obtaining optimal regiospecificity. The stereochemical course of this reaction was established through the synthesis of (S)-ibuprofen 6, which demonstrated that the alkylation proceeds with net inversion of absolute configuration consistent with direct addition of the nucleophile to the metal center followed by reductive elimination.
Supplementary Material
Acknowledgment
We sincerely thank the National Institutes of Health (GM58877) and the donors of the Petroleum Research Fund, administered by the American Chemical Society for generous financial support. We also thank Johnson and Johnson for a Focused Giving Award and Pfizer Pharmaceuticals for the Creativity in Organic Chemistry Award. We also acknowledge a Camille Dreyfus Teacher-Scholar Award (P.A.E.) from the Camille and Henry Dreyfus Foundation, and a JSPS Research Fellowship for Young Scientists (D.U.) from the Japan Society for the Promotion of Sciences.
Footnotes
Supporting Information Available: Representative experimental procedure for the preparation of 1 and the spectral data for 1-2, 4, and 6 (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Consiglio G, Waymouth RM. Chem. Rev. 1989;89:257. [Google Scholar]; (b) Tsuji J. Palladium Reagents and Catalysts. Wiley; New York: 1996. pp. 290–404. [Google Scholar]; (c) Trost BM, Lee C. In: Catalytic Asymmetric Synthesis. 2nd ed. Ojima I, editor. Wiley-VCH; New York: 2000. pp. 593–649. [Google Scholar]
- (2). For leading references on metal-catalyzed allylic arylation, see:; (a) Del Valle L, Stille JK, Hegedus LS. J. Org. Chem. 1990;55:3019. [Google Scholar]; (b) Legros J-Y, Flaud J-C. Tetrahedron Lett. 1990;31:7453. [Google Scholar]; (c) Kobayashi Y, Mizojiri R, Ikeda E. J. Org. Chem. 1996;61:5391. [Google Scholar]; (d) Matsuhashi H, Asai S, Hirabayashi K, Hatanaka Y, Mori A, Hiyama T. Bull. Chem. Soc. Jpn. 1997;70:1943. [Google Scholar]; (e) Uozumi Y, Danjo H, Hayashi T. J. Org. Chem. 1999;64:3384. doi: 10.1021/jo982438b. [DOI] [PubMed] [Google Scholar]; (f) Macsaári I, Hupe E, Szabo KJ. J. Org. Chem. 1999;64:9547. [Google Scholar]; (g) Chung K-G, Miyake Y, Uemura S. J. Chem. Soc., Perkin Trans. 2000;1:2725. [Google Scholar]; (h) Hoke ME, Brescia M-R, Bogaczyk S, DeShong P, King BW, Crimmins MT. J. Org. Chem. 2002;67:327. doi: 10.1021/jo010672n. [DOI] [PubMed] [Google Scholar]; (i) Kabalka GW, Dong G, Venkataiah B. Org. Lett. 2003;5:893. doi: 10.1021/ol034067d. [DOI] [PubMed] [Google Scholar]
- (3).Hayashi T, Yamamoto A, Hagihara T. J. Org. Chem. 1986;51:723. [Google Scholar]
- (4).(a) Evans PA, Nelson JD. J. Am. Chem. Soc. 1998;120:5581. [Google Scholar]; (b) Evans PA, Robinson JE, Nelson JD. J. Am. Chem. Soc. 1999;121:6761, 12214. [Google Scholar]; (c) Evans PA, Leahy DK. J. Am. Chem. Soc. 2000;122:5012. [Google Scholar]; (d) Evans PA, Kennedy LJ. J. Am. Chem. Soc. 2001;123:1234. doi: 10.1021/ja005689m. [DOI] [PubMed] [Google Scholar]; (e) Evans PA, Robinson JE. J. Am. Chem. Soc. 2001;123:4609. doi: 10.1021/ja015531h. [DOI] [PubMed] [Google Scholar]; (f) Evans PA, Leahy DK. J. Am. Chem. Soc. 2002;124:7882. doi: 10.1021/ja026337d. [DOI] [PubMed] [Google Scholar]
- (5). For a related example of an enantioselective rhodium-catalyzed allylic arylation, see:; Lautens M, Dockendorff C, Fagnou K. Malicki, A. Org. Lett. 2002;4:1311. doi: 10.1021/ol0256062. [DOI] [PubMed] [Google Scholar]
- (6). For a recent review of rhodium-tris(pyrazolyl)borate complexes, see:; Slugovc C, Padilla-Martinez I, Sirol S, Carmona E. Coord. Chem. Rev. 2001;213:129. [Google Scholar]
- (7). For a recent review on halide effects in transition metal catalysis, see: [DOI] [PubMed]; Fagnou K. Lautens, M. Angew. Chem., Int. Ed. 2002;41:26. doi: 10.1002/1521-3773(20020104)41:1<26::aid-anie26>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- (8). Representative experimental procedure: TpRh(C2H4)2 (3.7 mg, 0.01 mmol), lithium bromide (17.4 mg, 0.2 mmol) and dibenzylidenacetone (4.7 mg, 0.02 mmol) were dissolved in anhydrous diethyl ether (3 mL) at room temperature and stirred for ca. 1 h before being cooled to 0 °C. Zinc bromide (45 mg, 0.2 mmol) was dissolved in anhydrous diethyl ether (1 mL) and cooled with stirring to 0 °C. Phenyllithium (210 mL, 0.21 mmol, 1 M) was then added dropwise to the zinc bromide solution and the resulting mixture stirred for ca. 30 min. The allylic carbonate 1a (35.6 mg, 0.1 mmol) was then added via tared syringe to the catalyst solution, followed by the dropwise addition of the phenyl zinc bromide solution. The resulting reaction mixture then stirred at 0 °C for ≤15 min (TLC control). The reaction mixture was quenched (aq NH4Cl) and partitioned between saturated aqueous NH4Cl solution and pentane. The organic layers were combined, dried (Na2SO4), filtered, and concentrated in vacuo to afford a crude oil. Purification by flash chromatography (eluting with pentane) furnished the 3-phenyl propenyl derivatives 2a/3a (19.3 mg, 87%) as a colorless oil.
-
(9). Treatment of the primary allylic carbonate 1é under the analogous reaction conditions furnished the primary allylic alkylation product 3e as the major isomer. ;
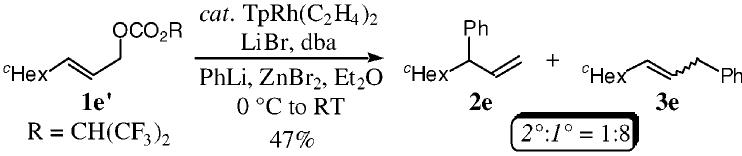
- (10). For recent enantioselective approaches to ibuprofen, see:; (a) Park H, RajanBabu TV. J. Am. Chem. Soc. 2002;124:734. doi: 10.1021/ja0172013. [DOI] [PubMed] [Google Scholar]; (b) Ishihara K, Nakashima D, Hiraiwa Y, Yamamoto H. J. Am. Chem. Soc. 2003;125:24. doi: 10.1021/ja021000x. [DOI] [PubMed] [Google Scholar]
- (11).Carlsen P-HJ, Katsuki T, Martin V-S, Sharpless KB. J. Org. Chem. 1981;46:3936. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.