

Abstract
Previous studies have documented that 0.1≈1% of input recombinant adeno-associated virus (rAAV) vectors could be stabilized and lead to transgene expression. To characterize the steps involving massive AAV DNA loss, we designed an“AAV footprinting” strategy that can track newly formed AAV dsDNA genomes. This strategy is based on an ROSA26R mouse model or cell line that carries a lacZ gene flanked by two loxP sites. When it is transduced by a rAAV vector carrying the Cre recombinase, the lacZ gene can be activated and remain active even when rAAV genomes are later lost. By using this sensitive AAV footprinting technique, we confirmed the existence of transient AAV dsDNA that went undetected by conventional DNA methods. Although these dsDNA intermediates could be efficiently formed in almost every cell and were competent for mRNA transcription and protein synthesis in vivo, they got lost continuously. Only a small fraction was eventually stabilized for sustained gene expression. Although both rAAV2 and rAAV8 can potentially have similar levels of dsDNA formation, AAV8 dsDNA was formed much faster than that of AAV2, which explains why rAAV8 is more efficient than rAAV2 in transducing the liver. Collectively, our studies suggested that rather than receptor binding, viral entry, and ssDNA to dsDNA conversion, the instability of newly formed AAV dsDNA was the primary contributing factor for the low rAAV transduction efficacy. The uncoating step significantly influenced the stability of AAV transient dsDNA. The identification of transient AAV dsDNA provided a new pathway for improving rAAV transduction.
Keywords: adeno-associated virus, DNA stability, gene therapy, vector, ROSA26R
Adeno-associated virus (AAV) is a nonpathogenic and replication-defective human parvovirus with a single-stranded (ss) DNA genome (1). AAV-based recombinant vectors (rAAVs) have been studied extensively in animal models, and the long-term transgene expression from rAAV vectors has been well documented (2–4). One distinct feature of rAAV transduction is the lack of T cell mediated immune responses to the vector and transgene in animal experiments (5, 6). To date, rAAV is the only ssDNA vector being tested in human clinical trials (7–10).
For long-term transgene expression from rAAV vectors, rAAV must successfully avoid degradation in the endosome and release its ssDNA genomes (11–17). The ssDNA genome will then be annealed or converted into the ds form (18–20). The ds form can serve as a template for RNA transcription, which can subsequently lead to translation of transgene products. Compared with other vectors, rAAV vectors posses a unique expression profile. For AAV serotype 2 (AAV2), after administration in vivo, significant transgene expression is not observed until 1–2 weeks, reaching a plateau at week 4–6. The slow rise of rAAV transduction was thought to be limited by the conversion from ssDNA to dsDNA (18–20). However, recent studies suggested that this expression delay is primarily the result of the uncoating efficacy of vector genomes in liver, which determines the ability of conversion from ssDNA to dsDNA (21, 22). Our previous studies of tracking free ssDNA genomes revealed that conversion from released ssDNA genome to dsDNA genome was a rather fast reaction because free ssDNA genomes could not be observed by a specially designed BrdU labeling technique (23). Therefore, steps before uncoating or after dsDNA formation may be the deciding factors for rAAV transduction efficacy.
Another unanswered issue is whether the initial dsDNAs formed after uncoating are stable forms leading to long-term gene expression. The circular intermediates of rAAV transduction have been studied extensively and were shown to be abundant after rAAV vector administration (24–28). Circular DNAs were eventually converted into high-molecular weight concatemers and were presumably stabilized. There are two major categories of stabilized AAV genomes that can be detected months after rAAV transduction: extrachromosomal concatemers and integrated AAV genomes (29, 30). Although wild-type AAV frequently underwent site-specific integration into human chromosome 19, similar integrations by rAAV were infrequent (31, 32). Even though a variety of integration junctions between rAAV and the host chromosome have been recovered, no evidence was found to support that these integrants are the primary sources for transgene expression. Instead, extrachromosomal concatemers were primarily responsible for stable hepatocyte transduction (32).
Many newly discovered AAV serotypes showed improved performance in transduction over AAV2 (33–37). The most notable differences are AAV1 and AAV6 in the muscle and AAV8 in the liver. Because the apparent differences among these serotypes are the capsid protein, viral entry and cellular receptors have been thought to be the primary determining factor for transduction efficacy. In this current work, we take advantage of ROSA26R mouse generated by Soriano (38) and use it to track the timing of AAV dsDNA conversion and the stability of newly formed AAV dsDNA genomes. In contrast to common belief, our results suggested that the amount of dsDNA formed was much more than what was observed previously. The timing of dsDNA formation, which was affected by AAV capsid rather than the total amount of dsDNA formed during rAAV transduction, is the critical determinant of its transduction efficacy.
Results
Differential Tracking of Stabilized rAAV Genomes and Transient Functional AAV dsDNA.
We speculate that not all formed AAV dsDNAs are stabilized and contributing to the long-term rAAV transduction. The most intuitive approach to address this problem is to compare the total amount of AAV dsDNA formed after rAAV infection with the total amount of dsDNA remaining after long-term rAAV transduction. In reality, it is virtually impossible to measure the total amount of dsDNA formed. The main reason is that AAV uncoating/intracellular processing is a slow process lasting for weeks, and the degradation of these monomers occurs simultaneously with synthesis. It is also difficult to define the time point at which the uncoating process and dsDNA formation are completed. A potential way to assess the loss of dsDNA is to track the cells that have been transduced with rAAV genomes but then lose dsDNA over time. However, it is equally difficult to identify these cells both in vivo and in vitro. To overcome this difficulty, we designed a strategy to estimate the cells that have been infected with rAAV genomes, harbored dsDNA genomes, and expressed transgene products but gradually lost their rAAV genomes. The key for this strategy is to imprint the cells that once harbored AAV dsDNA. Our strategy takes advantage of loxP-lacZ cell lines and mice, which carry an integrated inactive lacZ gene. This lacZ gene is under the control of loxP elements. When the stop element for translation is removed by Cre recombinase (Cre)-induced recombination, this lacZ gene becomes activated and leads to the expression of β-galactosidase, which can be detected by X-Gal staining. Because the excision is very efficient and the maintenance of LacZ expression does not require continuous expression of Cre protein, the specially designed rAAV can therefore be used to distinguish the host cells that have rAAV genomes from those that have been transduced by rAAV vectors but lose rAAV genomes. For tracking stabilized rAAV genomes, we used human placenta alkaline phosphatase (ALP) as the reporter gene. To avoid the complication of transgene inactivation, we chose a constitutive promoter, β-actin promoter with CMV enhancer (CB). The constitutive expression from this promoter has been demonstrated on many occasions. This strategy is illustrated in Fig. 1.
Fig. 1.

AAV genome footprinting strategy. (A) Schematic representation of the AAV-CB-ALP and AAV-CB-cre vectors used in this work. The locations of restriction enzymes and probe for Southern blotting are marked. PolyA, the human β-globin gene polyadenylation signal; s, stuffer sequence without function; B, BglII; E, EcoRI. (B) Schematic of lacZ activation by Cre in C2lacZ cells and ROSA26R (Gtrosa26tm1Sor) mice. (C) Strategy of AAV dsDNA footprinting. Key steps are shown: 1, rAAV entry; 2, uncoating, dsDNA synthesis, lacZ gene activation; 3, dsDNA get lost; 4, dsDNA stabilization. X stands for the stop sequence in the lacZ gene expression cassette.
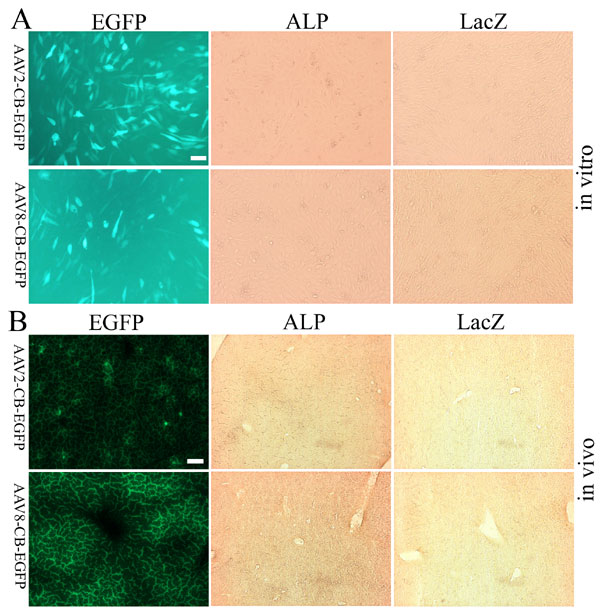
As shown in Fig. 1A, both AAV-CB-ALP and AAV-CB-cre vector DNA are similar in size. After ALP and cre vectors transduce the target cells and start to express ALP and Cre, there are two main consequences. As shown in Fig. 1B, the Cre expression may cause host chromosome rearrangement and activate the lacZ gene, which can be detected by LacZ staining. Once the lacZ gene is activated, it will remain active regardless of the status of AAV-cre genomes (Fig. 1C). However, ALP expression requires the presence of dsAAV-ALP genome. Thus, the transient AAV dsDNA, represented by LacZ expression, and stabilized genomes, represented by ALP expression, can be readily monitored. To rule out the potential activation of loxP recombination by rAAV itself, AAV-CB-EGFP was tested both in vitro and in vivo. There was no LacZ expression activated by the AAV-GFP vectors, confirming that lacZ can only get activated with the expression of Cre [supporting information (SI) Fig. 7].
Characterization of Transient AAV dsDNA Intermediates Formation in Vitro.
To assess rAAV genomes in vitro, we established the C2lacZ cell line. In C2lacZ cells, a lacZ reporter gene has been genetically engineered into the genomic DNA in a way that a loxP-flanked stop sequence prevents LacZ expression. When Cre is expressed in the cell, it mediates recombination between the loxP sites and splices out the stop sequence, thereby allowing constitutive LacZ expression in that cell (Fig. 1B). Because C2lacZ is derived from a mouse myoblast cell line, the cell division can be controlled by differentiation.
We infected C2lacZ cells with equal amounts of AAV-CB-ALP and AAV-CB-cre and monitored for ALP and LacZ expression. As shown in Fig. 2A, in nondividing cells, the amount of ALP- or LacZ-positive cells kept increasing after infection. The increasing profiles of ALP-positive cells and LacZ-positive cells were similar to each other. At day 1, there was no LacZ expression detected, which was probably caused by delay of the recombination reaction. Cre expression took ≈18–24 h after rAAV infection, and it corresponded to the lag of LacZ detection. At day 7, both ALP- and LacZ-positive cells were close to their peak levels. In contrast, it showed a very different ALP- and LacZ-staining profile in the dividing cells. The amount of LacZ-positive cells, representing all functional dsDNA synthesized, continued to increase. However, the number of ALP-positive cells was not only significantly less than LacZ-positive cells but also exhibited a peak at days 3–5 before falling off. Because ALP expression requires the presence of AAV dsDNA, whereas LacZ expression can be derived from transient expression from AAV dsDNA, synthesized AAV dsDNA was shown to be more stable under nondividing conditions, which confirmed that dsDNA genome existed as extrachromosomal episomes.
Fig. 2.

rAAV transduction in C2lacZ cells. C2lacZ cells were infected with AAV-CB-ALP and AAV-CB-cre (1:1) at a multiplicity of infection of 10,000. The expression of ALP or LacZ was analyzed at specified time points as indicated. (A) ALP or LacZ expression of C2lacZ cells infected with AAV2 vectors. (Upper) Nondividing C2lacZ cells. (Lower) Dividing C2lacZ cells. (Scale bar: 100 μm.) (B–E) Quantification of ALP- or LacZ-positive cells after AAV2 or AAV8 transduction of C2lacZ cells. The y axis stands for the percentage of cells positive for ALP or LacZ staining. Error bars indicate the SD values.
The amount of positive cells from ALP and LacZ staining at various time points was quantified with AAV2 (Fig. 2 B and C) or AAV8 (Fig. 2 D and E) vectors. In C2lacZ cells, both AAV2 and AAV8 showed a similar transduction pattern. The amount of LacZ-positive cells started to exceed ALP-positive cells ≈72 h after infection. The percentage of LacZ-positive cells was ≈5% more than ALP under nondividing conditions (P < 0.05). Under dividing conditions, there was a quick falling off in the amount ALP-positive cells after day 3, whereas LacZ-positive cells remained at similar level (P > 0.05) and decisively more than ALP-positive cells. This result confirmed the loss of newly formed AAV dsDNA under both dividing and nondividing conditions and that the instability of dsDNA intermediates may be a general issue for all of the AAV serotypes.
Characterization of Instability of Transient AAV dsDNA Intermediates in Vivo.
To investigate AAV dsDNA intermediates in vivo, ROSA26R mice were injected i.v. with an equal amount of AAV2- or AAV8-based vectors expressing ALP or cre reporter. Mouse livers were harvested and analyzed for ALP and LacZ expression at different time points after vector administration (Fig. 3A). Consistent with previous studies, AAV2-ALP exhibited a slow, increasing profile in transduction, reaching a plateau at week 6, transducing ≈5% of hepatocytes. In contrast, the amount of LacZ-positive liver cells continued to increase, and almost all cells in the entire liver expressed lacZ gene at the end of the experimental period. This result suggested that (i) AAV dsDNA was not synthesized in a short period but continuously over the time of transduction; (ii) nearly all liver cells were infected by AAV2 vectors and synthesized AAV dsDNA; (iii) the AAV dsDNA in all cells must be competent for transcription and protein translation; and (iv) only a small portion of synthesized dsDNA could be stabilized.
Fig. 3.

AAV dsDNA footprinting in the liver. AAV2 or AAV8 carrying ALP or cre was administered i.v. to ROSA26R mice at a dose of 1 × 1011 vector genomes. (A) Staining of ALP or LacZ expression in the liver section of mice receiving AAV2-based vectors at various time points after vector administration. Neg represents a negative control before vectors were administered. (Scale bar: 100 μm.) (B) Staining of ALP or LacZ expression in the liver section of mice receiving AAV8-based vectors at various time points after vector administration. (C) Southern blot analysis of rAAV genome after vector administration. Total cellular mouse liver DNA was extracted from liver samples of each time point. Twenty micrograms of BglII-, MfeI-, and EcoRV-digested genomic DNA was separated on 0.8% agarose gel. Hybridization was performed by using CB promoter probe recovered by EcoRI. Genome copy number standards are shown in the figure. Fragment size corresponding to the ALP or cre vector is also indicated. ss represents the ssDNA from the incoming vector.
In contrast, AAV8-mediated liver transduction exhibited a different pattern in ALP expression (Fig. 3B). The difference between AAV2 and AAV8 transduction lies mainly on two aspects. First, ALP- and LacZ-positive cells for AAV8-mediated transduction appeared faster than AAV2. At day 3 and day 7, the differences in both ALP and LacZ between AAV2 and AAV8 were remarkable, which suggested that AAV8 could synthesize its dsDNA at a faster pace. Because there is no difference in the vector DNA composition, this result suggested that AAV capsid contributed to these differences. Second, the differences of ALP expression between AAV2 and AAV8 remained remarkable at the end of the experimental period. However, the differences in LacZ-positive cells were reduced over time. A similar number of cells started to express lacZ gene at week 8, suggesting that the same amount of liver cells had received AAV2 and AAV8. Thus receptors are not the obstacles for cells to be transduced by rAAV vectors. The difference in ALP and lacZ gene expression suggested that the synthesized dsDNA degradation was the key factor.
To confirm that the differences between ALP and LacZ expression were not caused by the difference in viral genomes carrying ALP and cre vectors, we analyzed the dsDNA genome by Southern blotting. As shown in Fig. 3C, the amount of ds-ALP vector and ds-cre vectors remained similar to each other throughout the experimental period, which ruled out that there was a preference for stabilizing rAAV genomes carrying the cre gene in the liver. Consistent with the observation that AAV8 transduced more cells than AAV2 at days 3 and 7, the detected rAAV genomes from AAV8 at day 3 and day 7 were more than that of AAV2, which also confirmed that AAV8 released/uncoated AAV genomes at a much faster pace than AAV2.
AAV dsDNA Released from Self-Complementary AAV Vector (scAAV) Is Not Stable.
Because scAAV vectors release dsDNA instead of ssDNA, they bypass the step to convert ssDNA into dsDNA. To confirm that the instability of AAV dsDNA is the key factor affecting rAAV transduction, we constructed scAAV-TTR-ALP and scAAV-TTR-cre. The reporter genes are under the control of the liver-specific transthyretin (TTR) promoter. After AAV2 vectors were produced by using these two vector plasmids, they were administered i.v. to ROSA26R mice at a ratio of 1:1. The expression of ALP and LacZ was monitored and is presented in Fig. 4. Similar to what was observed with AAV2 carrying ss genomes, the expression of ALP increased slowly and reached a peak in ≈8–10% of liver cells at week 4–6. The LacZ expression was ≈10–20 times more than ALP expression at earlier time points and reached 100% transduction at 4–6 weeks. Because there was no interference of AAV ssDNA genomes, the results confirmed that the majority of released dsDNA were transient and were not stabilized.
Fig. 4.

Instability of scAAV genomes. Self-complementary AAV2 vectors carrying TTR-ALP or TTR-cre (1:1) were administered i.v. to the ROSA26R mice at a dose of 1 × 1011 vector genomes. Liver were harvested at various time points after vector administration, and the ALP and lacZ gene expression was analyzed by histochemical staining. Neg represents a negative control before vectors were administered. (Scale bar: 100 μm.)
Stabilized rAAV Genomes Remained as Episomes in Vivo.
To examine the dsDNA integration that led to ALP and lacZ gene expression, we performed partial hepatectomy on mice that had received AAV-CB-ALP and AAV-CB-cre at week 6 after vector administration. The partial hepatectomy is often used to stimulate liver regeneration. The dividing hepatocytes would keep the integrated AAV dsDNA and dilute/lose episomal AAV dsDNA. As shown in Fig. 5, there was a significant drop in the number of ALP-positive cells but not LacZ-positive cells. This result is consistent with the in vitro data of C2lacZ cells, confirming that most of the stabilized ALP gene from AAV dsDNA was present in nonintegrated episomal form. Because the lacZ gene was in the chromosome that was activated by Cre, it remained stable in nearly 100% of liver cells.
Fig. 5.

rAAV genomes remain mostly as episomes. ROSA26R mice were injected with AAV-CB-ALP and AAV-CB-cre (1:1) at a dose of 1 × 1011 vector genomes per animal through the tail vein. After 6 weeks, half of the mice (n = 3) underwent partial hepatectomy (PH). All animals were killed another 6 weeks later. ALP or LacZ staining of the liver sections before and after PH is shown. Control mice receiving saline were subjected to the same PH procedure. (Scale bar: 100 μm.)
Discussion
The rAAV DNA undergoes a series of changes during the transduction process. In the ssDNA stage, it exists as intact virion before uncoating or as free ssDNA after uncoating. After ssDNA conversion into dsDNA, it may exist as linear monomers, linear concatemers, circular monomer, circular concatemers, or integrated rAAV genomes. Here, we provide strong evidence on the existence of two classes of dsDNA: stabilized dsDNA and newly formed transient dsDNA. The transient form may come from annealing or second-stranded DNA synthesis.
It is documented that only <5–10% of hepatocytes are permissive to stable transduction with rAAV2 vectors, whereas rAAV8 could obtain almost 30% or more of stably transduced hepatocytes with a high dose (21, 39). The transduction usually resulted in an average of 0.5–2 copies of rAAV genomes per cell. Based on the conventional vector dose of 1 × 1011≈12 vector genomes per mouse, 99.9% of all input viral genomes were lost without contributing to rAAV transduction (40, 41). Viral DNA loss could occur at any step of its transduction, such as entry, translocation, uncoating, and conversion from ssDNA into dsDNA. There are no previous studies focusing on the stability of synthesized transient dsDNA genomes. In this work, we overcame the technical difficulties and specifically examined the stability of transient AAV dsDNA by using ROSA26R mice and rAAV vector expressing the cre gene.
The primary discovery of this work is the existence of transient functional dsDNA molecules in rAAV transduction. Because of their transient nature, conventional direct methods such as Southern blotting are not effective for monitoring them. In this work, we developed an assay to detect an often ignored AAV DNA species in the rAAV transduction life cycle. As shown in Fig. 3C, Southern blotting could not reveal the existence of this species because the average life of the transient dsDNA would be short. Based on the Southern blotting procedures, we estimated that the transient dsDNA genome would not be detected by Southern blotting if its half-life is only a couple of hours. In situ DNA hybridization is another method that may directly monitor AAV dsDNA in vivo. It has been demonstrated that only ≈5% of hepatocytes contained AAV dsDNA at 5 weeks after transduction. The dsDNA detected by this method was concomitant with the amount of cells expressing transgene (42). Therefore, in situ hybridization also only detected mostly stabilized dsDNA. Although the total amount of transient dsDNA formed could be abundant, the number of transient dsDNA would be rare compared with other intermediates of rAAV genomes at a specific time point. As shown in Fig. 3, the transient dsDNA formation is a dynamic process. It can be formed continuously depending on the intracellular trafficking, uncoating, and annealing/second-stranded DNA synthesis. The degradation/loss of dsDNA appears to be coupled with its formation. Thus, at any stage, either Southern blot analysis or in situ DNA hybridization can only detect the stabilized dsDNA in addition to transient dsDNA at that time point. Thus, only the “AAV dsDNA footprinting” technique can study the cumulative effects of transient dsDNA genomes.
Because stabilized rAAV genomes are represented by ALP expression, whether the amount of ALP expression is concomitant with dsDNA is critical. Silent dsDNA genomes would make the interpretation of the experiment results complicated. At similar dose, AAV2 was reported to transduce 5% of liver cells. The in situ DNA hybridization experiment reported that there were no silent AAV dsDNA templates, establishing that only 5% of cells actually harbored dsDNA (42), which is inconsistent with the ALP expression of our work. In addition, the promoter used for ALP expression is derived from constitutive β-actin or TTR promoter that has not been reported to switch off in vivo. Finally, the number of positive cells increased over the experiment period instead of decreasing, which is in contrast to what is observed for promoter silence. Taken together, promoter silence cannot be the prominent factor that would complicate our data interpretation.
Our results will help address a misconception that AAV receptor is the ultimate deciding factor for rAAV transduction efficacy. As shown in Fig. 3, there is no difference between AAV2 and AAV8 in LacZ expression in the liver, which indicates that the “cumulative” dsDNA formed are similar for AAV2 and AAV8. This finding implies that the AAV-CB-cre vector has successfully gained entry to almost all liver cells, uncoated, converted ssDNA into dsDNA, and synthesized Cre protein, which subsequently activated the lacZ gene. Even with a 10-fold lower dose, similar phenomena were also observed (SI Fig. 8). This result is also in agreement with a previous study showing a high percentage of liver cells infected by rAAV vectors using DNA in situ hybridization (42). As a further step, we demonstrated that all cells have functional rAAV genomes synthesized, which were then later lost. Furthermore, our results also confirmed that dsDNA were formed much faster for AAV8, consistent with previous observations (21). Infection kinetics is noticeably different between AAV2 and AAV8. How AAV ssDNA is released is probably the key leading to difference in transduction level between AAV2 and AAV8. Thus, the uncoating process may have significantly impacted the stability of subsequently formed dsDNA.
Our assays further confirmed that the majority of rAAV genomes remained as episomal DNA after transduction. Both in vitro and in vivo results (Figs. 2 and 5) supported this conclusion. There might be only a small amount of cells harboring integrated rAAV genomes, which did not get lost during the host cell divisions. The faster host cells divide, the more functional episomal dsDNA are lost, which is also why rAAV often performs better in differentiated cells (3, 4, 43). The combined results of this work and other studies for rAAV transduction prompted us to propose a working model for the rAAV transduction pathway (Fig. 6). As illustrated, there are potential losses of rAAV genomes at all stages of rAAV transduction: entry, intracellular trafficking, uncoating, and ssDNA to dsDNA conversion. Because 100% cells showed LacZ expression even at doses of 1 × 1010 vector genomes per mouse, these steps appear not to be the deciding factor for rAAV transduction efficiency. The ultimate deciding factor for transduction efficiency is the amount of dsDNA intermediates stabilized. The loss of dsDNA intermediates contributes to the differences in transduction between AAV2 and AAV8. Although we emphasize the importance of dsDNA stability, the other steps such as vector receptor binding, intracellular trafficking, and uncoating may still be different and contribute to the kinetics of transient dsDNA, which ultimately determines whether these transient dsDNA are stabilized or degraded.
Fig. 6.

Theoretical model for transient AAV dsDNA in rAAV transduction life cycle. (A) Transient dsDNA status during AAV2 transduction. (B) Transient dsDNA during AAV8 transduction. The difference between AAV2 and AAV8 in uncoating is noted in the figure as “slow” and “fast.” The traditional rate-limiting step is also noted in the figure. **, all of the steps that may involve degradation. The vector DNA illustrated by dotted line represents lost AAV genomes.
Our model is also supported by experimental evidence from the most recently developed scAAV vectors (24, 44–48). Instead of releasing ssDNA from uncoating, scAAV vectors release dsDNA genomes. As shown in Fig. 4, releasing dsDNA genomes improved the percentage of cells expressing ALP from 2–5% to 8–10%. But there was still a significant difference between ALP and LacZ expression. For ssAAV vector, >95% cells lost transient dsDNA molecules. For scAAV vectors, 90% of cells lost dsDNA genomes. Because ssAAV and scAAV vectors showed only 5% difference in the transduction whereas AAV2 and AAV8 vectors showed 30–80% difference, fast uncoating is confirmed to be a more efficient way for stabilizing rAAV genomes.
It has been recognized that the ssDNA to dsDNA conversion is a rate-limiting step for rAAV transduction (19, 20). Our previous experiments demonstrated that second-stranded DNA synthesis was a fast reaction (23). AAV footprinting confirmed that rather than viral entry and transient dsDNA formation, the instability of AAV transient dsDNA is the determinant for efficient rAAV transduction. These seemly contradictive observations were reconciled in Fig. 6. Because transient dsDNA was unknown in the previous studies on second-stranded DNA synthesis, and Southern blotting and conventional molecular biology techniques only detected stabilized AAV dsDNA, a modified conclusion should be that stabilized dsDNA formation is the rate-limiting step for rAAV transduction. The stabilized dsDNA genomes were the combined results from viral entry, translocation, uncoating, and viral DNA degradation. Taken together, transient dsDNA is the decisive intermediate in the rate-limiting step, and its stability dictates the performance of rAAV transduction. Control of transient dsDNA degradation will be the key pathway for improving rAAV transduction efficacy.
Materials and Methods
Recombinant AAV Vector Production, Purification.
All rAAV vectors were produced by the triple-transfection method, which has been described previously (49). Briefly, a vector plasmid (with AAV2 inverted terminal repeats), a helper plasmid (with the rep and cap genes of AAV2 or AAV8), and mini adenovirus helper plasmid (pFΔ6, with essential regions from the adenovirus genome) were cotransfected into 293 cells by calcium phosphate precipitation. AAV particles were purified by cesium chloride gradient ultracentrifugation. The physical particle titers were determined by silver staining and quantitative dot-blot assay.
AAV2 or AAV8 vectors carrying human placenta ALP or the cre recombinase gene were cross-packaged with AAV2 or AAV8 helper plasmids by using AAV-CB-ALP or AAV-CB-cre as the vector plasmid, respectively. Both ALP and cre genes are under the control of CB. Both vectors are similar in size. Self-complementary AAV2 vector carrying ALP or cre gene was produced by cross-packaging AAV2 helper plasmid with scAAV-TTR-ALP or scAAV-TTR-cre. ALP and cre gene are under the control of a ≈230-bp liver-specific TTR promoter because of the limited package capacity of scAAV2.
Establishment of C2lacZ Stable Cell Line.
The plasmid pPGK-neotpalox2-lacZ was a gift from Philippe Soriano (Fred Hutchinson Cancer Research Center, Seattle, WA). Mouse myoblast cell line C2C12 was purchased from American Type Culture Collection and maintained in DMEM/10% FBS. Stable C2C12 cells carrying the loxP-regulated lacZ gene were generated by transfecting pPGK-neotpalox2-lacZ into C2C12 cells, and they were selected under 400 μg/ml G418. Resulting colonies were screened for LacZ expression after pAAV-CB-cre plasmid transfection. The positive clone with the highest level of LacZ expression was characterized further and renamed as C2lacZ.
Histochemical Staining.
To stain LacZ expression, cells or frozen liver sections were fixed in PBS containing 2% formaldehyde and 0.2% glutaraldehyde at 4°C for 5 min. After being washed with PBS three times, cells or liver sections were incubated at 37°C overnight in PBS/5 mM potassium ferricyanide/5 mM potassium ferrocyanide/2 mM MgCl2/1 mg/ml X-Gal.
To detect human placenta ALP activity, fixed cells or liver sections were washed once in PBS, heat-inactivated in PBS at 70°C for 30 min, and equilibrated in PBS containing 100 mM Tris·HCl, pH 9.5/100 mM NaCl/10 mM MgCl2 for 10 min. Subsequently, cells or sections were stained at room temperature for 30 min with nitroblue tetrazolium–5-bromo-4-chloro-3-indolyl phosphate ready-to-use tablets (Roche, Indianapolis, IN).
rAAV Vector Infection in Vitro.
C2lacZ cells were seeded at 50% confluence in a 6-well plate. Twenty four hours later, AAV-CB-ALP and AAV-CB-cre (1:1), at a dose of 5 × 109 vector genomes, were mixed together and added to the wells. Histochemical staining was performed at various time points. C2lacZ cells can be maintained under either dividing or differentiation conditions. Under the differentiation condition (nondividing), cells were maintained as confluent cell monolayers in reduced horse serum. Under the dividing condition, cells were maintained in 10% FBS and passed every other day. The cells maintained their active growth during the experiment period. The ALP- or LacZ-positive cells were quantified from five random fields under ×10 magnification with a DIAPHOT 200 microscope (Nikon, Melville, NY).
Animal Procedures.
ROSA26R (Gtrosa26tm1Sor) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Six- to eight-week-old male mice were used for the experiments. All of the animal experiments were performed according to the Children's Hospital of Philadelphia animal guidelines.
AAV-CB-ALP and AAV-CB-cre (1:1), at a dose of 1 × 1011 vector genomes, were mixed together in 0.9% NaCl to a final volume of 200 μl and injected into the mouse through the tail vein. Livers were harvested and analyzed at day 3, day 7, week 2, week 4, week 6, and week 8 after vector administration. The harvested liver tissues were embedded in Tissue-Tek optimal cutting temperature compound (Sakura Finetek, Torrance, CA) and frozen in dry ice. Twenty-micrometer sections were cut and stained to detect LacZ and ALP expression as described above. After staining, sections were dehydrated with xylene, mounted with Cytoseal 60 mounting medium (Richard-Allan Scientific, Kalamazoo, MI), and protected with covering slides. Pictures were taken under an Eclipse E800 microscope (Nikon).
For partial hepatectomy, animals receiving vectors after 6 weeks were anesthetized by 2 liters of oxygen/2% Forane per min. Partial hepatectomy was performed according to the method of Higgins and Anderson (50), and animals were killed 6 weeks after the procedure. Livers were harvested, and ALP or LacZ staining was performed as described above.
Southern Blot Analysis.
Twenty micrograms of total cellular DNA from AAV-transduced cells or livers was digested with BglII, MfeI, and EcoRV, and Southern blotting was performed by using CB DNA probe, which was an 800-bp EcoRI fragment labeled with [α-32P]dCTP. The vector genome copy number standards were prepared by adding an equivalent number of corresponding plasmid molecules to 20 μg of total DNA extracted from control cells.
Statistical Analysis.
Data were described by using mean ± SD whenever appropriate. ANOVA was performed to analyze differences among the groups. A P value of <0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank Junwei Sun, Marlene Webber, and Daniel Hui for critical reading of the manuscript. W.X. is supported by National Institutes of Health (NIH) Grant R01HL080789. R.J.S. is supported by NIH Grants P01GM059299 and P01HL051818.
Abbreviations
- AAV
adeno-associated virus
- ALP
alkaline phosphatase
- CB
β-actin promoter with CMV enhancer
- Cre
Cre recombinase
- rAAV
recombinant AAV
- scAAV
self-complemetary AAV
- TTR
transthyretin.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0702778104/DC1.
References
- 1.Muzyczka N, Berns KI. In: Fields Virology. Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Philadelphia: Lippincott Williams & Wilkins; 2002. pp. 2327–2359. [Google Scholar]
- 2.Flotte TR, Berns KI. Hum Gene Ther. 2005;16:401–407. doi: 10.1089/hum.2005.16.401. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, Xiao X. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 4.Herzog RW, Yang EY, Couto LB, Hagstrom JN, Elwell D, Fields PA, Burton M, Bellinger DA, Read MS, Brinkhous KM, et al. Nat Med. 1999;5:56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- 5.Fisher KJ, Jooss K, Alston J, Yang Y, Haecker SE, High K, Pathak R, Raper SE, Wilson JM. Nat Med. 1997;3:306–312. doi: 10.1038/nm0397-306. [DOI] [PubMed] [Google Scholar]
- 6.Jooss K, Yang Y, Fisher KJ, Wilson JM. J Virol. 1998;72:4212–4223. doi: 10.1128/jvi.72.5.4212-4223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.High KA. Semin Thromb Hemostasis. 2004;30:257–267. doi: 10.1055/s-2004-825639. [DOI] [PubMed] [Google Scholar]
- 8.Jiang H, Pierce GF, Ozelo MC, de Paula EV, Vargas JA, Smith P, Sommer J, Luk A, Manno CS, High KA, et al. Mol Ther. 2006;14:452–455. doi: 10.1016/j.ymthe.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Brantly ML, Spencer LT, Humphries M, Conlon TJ, Spencer CT, Poirier A, Garlington W, Baker D, Song S, Berns KI, et al. Hum Gene Ther. 2006;17:1177–1186. doi: 10.1089/hum.2006.17.1177. [DOI] [PubMed] [Google Scholar]
- 10.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, Ozelo MC, Hoots K, Blatt P, Konkle B, et al. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 11.Ding W, Zhang L, Yan Z, Engelhardt JF. Gene Ther. 2005;12:873–880. doi: 10.1038/sj.gt.3302527. [DOI] [PubMed] [Google Scholar]
- 12.Seisenberger G, Ried MU, Endress T, Buning H, Hallek M, Brauchle C. Science. 2001;294:1929–1932. doi: 10.1126/science.1064103. [DOI] [PubMed] [Google Scholar]
- 13.Hansen J, Qing K, Srivastava A. J Virol. 2001;75:4080–4090. doi: 10.1128/JVI.75.9.4080-4090.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douar AM, Poulard K, Stockholm D, Danos O. J Virol. 2001;75:1824–1833. doi: 10.1128/JVI.75.4.1824-1833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan D, Yue Y, Yan Z, Yang J, Engelhardt JF. J Clin Invest. 2000;105:1573–1587. doi: 10.1172/JCI8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartlett JS, Wilcher R, Samulski RJ. J Virol. 2000;74:2777–2785. doi: 10.1128/jvi.74.6.2777-2785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding W, Zhang LN, Yeaman C, Engelhardt JF. Mol Ther. 2006;13:671–682. doi: 10.1016/j.ymthe.2005.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakai H, Storm TA, Kay MA. J Virol. 2000;74:9451–9463. doi: 10.1128/jvi.74.20.9451-9463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher KJ, Gao GP, Weitzman MD, DeMatteo R, Burda JF, Wilson JM. J Virol. 1996;70:520–532. doi: 10.1128/jvi.70.1.520-532.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrari FK, Samulski T, Shenk T, Samulski RJ. J Virol. 1996;70:3227–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas CE, Storm TA, Huang Z, Kay MA. J Virol. 2004;78:3110–3122. doi: 10.1128/JVI.78.6.3110-3122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong L, Li W, Yang Z, Qing K, Tan M, Hansen J, Li Y, Chen L, Chan RJ, Bischof D, et al. Hum Gene Ther. 2004;15:1207–1218. doi: 10.1089/hum.2004.15.1207. [DOI] [PubMed] [Google Scholar]
- 23.Hauck B, Zhao W, High K, Xiao W. J Virol. 2004;78:13678–13686. doi: 10.1128/JVI.78.24.13678-13686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi VW, McCarty DM, Samulski RJ. J Virol. 2006;80:10346–10356. doi: 10.1128/JVI.00841-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnepp BC, Jensen RL, Chen CL, Johnson PR, Clark KR. J Virol. 2005;79:14793–14803. doi: 10.1128/JVI.79.23.14793-14803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan Z, Zak R, Zhang Y, Engelhardt JF. J Virol. 2005;79:364–379. doi: 10.1128/JVI.79.1.364-379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan D, Yan Z, Yue Y, Engelhardt JF. Virology. 1999;261:8–14. doi: 10.1006/viro.1999.9821. [DOI] [PubMed] [Google Scholar]
- 28.Duan D, Sharma P, Yang J, Yue Y, Dudus L, Zhang Y, Fisher KJ, Engelhardt JF. J Virol. 1998;72:8568–8577. doi: 10.1128/jvi.72.11.8568-8577.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakai H, Wu X, Fuess S, Storm TA, Munroe D, Montini E, Burgess SM, Grompe M, Kay MA. J Virol. 2005;79:3606–3614. doi: 10.1128/JVI.79.6.3606-3614.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kay MA, Nakai H. Nature. 2003;424:251. doi: 10.1038/424251b. [DOI] [PubMed] [Google Scholar]
- 31.Young SM, Jr, McCarty DM, Degtyareva N, Samulski RJ. J Virol. 2000;74:3953–3966. doi: 10.1128/jvi.74.9.3953-3966.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. J Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Proc Natl Acad Sci USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao W, Chirmule N, Berta SC, McCullough B, Gao G, Wilson JM. J Virol. 1999;73:3994–4003. doi: 10.1128/jvi.73.5.3994-4003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA, Agbandje-McKenna M. J Virol. 2006;80:11556–11570. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiorini JA, Kim F, Yang L, Kotin RM. J Virol. 1999;73:1309–1319. doi: 10.1128/jvi.73.2.1309-1319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soriano P. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 39.Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay MA. J Virol. 2005;79:214–224. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao W, Berta SC, Lu MM, Moscioni AD, Tazelaar J, Wilson JM. J Virol. 1998;72:10222–10226. doi: 10.1128/jvi.72.12.10222-10226.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miao CH, Snyder RO, Schowalter DB, Patijn GA, Donahue B, Winther B, Kay MA. Nat Genet. 1998;19:13–15. doi: 10.1038/ng0598-13. [DOI] [PubMed] [Google Scholar]
- 42.Miao CH, Nakai H, Thompson AR, Storm TA, Chiu W, Snyder RO, Kay MA. J Virol. 2000;74:3793–3803. doi: 10.1128/jvi.74.8.3793-3803.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo J, Kaplitt MG, Fitzsimons HL, Zuzga DS, Liu Y, Oshinsky ML, During MJ. Science. 2002;298:425–429. doi: 10.1126/science.1074549. [DOI] [PubMed] [Google Scholar]
- 44.Wu J, Zhao W, Zhong L, Han Z, Li B, Ma W, Weigel-Kelley KA, Warrington KH, Srivastava A. Hum Gene Ther. 2007;18:171–182. doi: 10.1089/hum.2006.088. [DOI] [PubMed] [Google Scholar]
- 45.Zhao W, Zhong L, Wu J, Chen L, Qing K, Weigel-Kelley KA, Larsen SH, Shou W, Warrington K. H., Jr, Srivastava A. Virology. 2006;353:283–293. doi: 10.1016/j.virol.2006.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN, Tuddenham EG, Kemball-Cook G, McIntosh J, Boon-Spijker M, et al. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong L, Chen L, Li Y, Qing K, Weigel-Kelley KA, Chan RJ, Yoder MC, Srivastava A. Mol Ther. 2004;10:950–957. doi: 10.1016/j.ymthe.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 48.McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- 49.Hauck B, Xu RR, Xie J, Wu W, Ding Q, Sipler M, Wang H, Chen L, Wright JF, Xiao W. Hum Gene Ther. 2006;17:46–54. doi: 10.1089/hum.2006.17.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Higgins GM, Anderson RM. Arch Pathol. 1931;12:186–202. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}